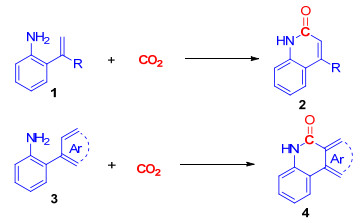
图式 1.
二氧化碳与邻烯基/芳基苯胺的内酰胺化反应
Scheme 1.
Lactamization of 2-alkenyl and 2-aryl anilines with CO2

随着工业化进程的发展及化石能源的过度消耗, 大量温室效应气体——二氧化碳(CO2)被直接排放到大气之中, 对环境造成了严重的影响[1].从合成化学角度考虑, CO2具有廉价易得、储量丰富和无毒等优点, 是有机合成反应中一种理想的C1合成子.然而, CO2固有的热力学及动力学稳定性限制了其作为C1合成子的广泛应用[2].近年来, 人们已经开发出较多的合成方法, 将CO2转化成精细化学合成品、聚合物及燃料等[3].
另一方面, 以简单易得的化合物为起始原料的环化反应是构建功能化的有机杂环化合物如药物、天然产物以及生物活性分子等的有效途径[4].随着金属有机化学的发展, 近年来, 以二氧化碳作为羰基/羧基源, 通过环化反应构建含羰基杂环化合物, 越来越引起人们的研究兴趣[5].这主要由于传统的羰基化反应, 常以一氧化碳和光气等具有较高毒性的化合物为羰基源, 在实际生产中具有较高的危险性[6].而CO2具有无毒、廉价及储量巨大等特点, 是一种理想的羰基/羧基化试剂.在这方面, 吕小兵[7]、何良年[8]、江焕峰[9]、韩布兴[10]、余达刚[11]和Yamada[12]等小组均取得了很多系统性的研究成果, 相关的工作已有很多综述从不同角度做了概括总结[13], 本文将不再介绍.
本文主要总结了我们及其他课题组在利用含N-、O-亲核原子的试剂在常压条件下与二氧化碳, 通过碱促进或者过渡金属催化的环化反应, 特别是多组分环化反应, 构建苯并噁嗪、环碳酸酯、内酰胺及2, 4-二噁唑啉酮等含羰基杂环结构的工作.其次, 也总结了原位生成的含C-亲核原子的试剂与二氧化碳羧基化反应的相关工作.
2-喹啉酮是一类重要的内酰胺化合物, 广泛存在于药物、天然产物以及光电材料中.该类化合物也是一类重要的有机合成中间体[14].邻氨基烯烃及芳烃与一氧化碳的羰基化/环化反应, 是构建该类化合物的主要方法[15].余达刚课题组和席婵娟课题组[16]报道了碱促进二氧化碳与邻氨基烯烃、芳烃的环化反应, 利用无毒的CO2作为羰基源, 实现了一系列2-喹啉酮的构建(Scheme 1).
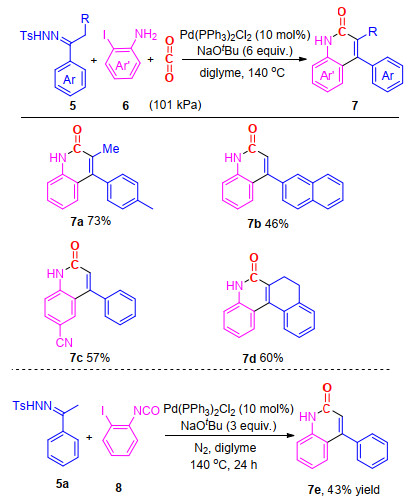
然而, 在上述反应中都需要首先合成邻氨基烯烃及芳烃等反应前体, 这无疑会添加繁琐的合成、分离步骤.在已有工作基础上, 我们[17]报道了钯催化的N-对甲苯磺酰基腙、邻碘芳胺与CO2的三组分反应, 一步实现了4-芳基-2-喹啉酮的构建, 该反应对于基团的适应性较好, 含有吸电子及供电子基的邻碘芳基胺及N-对甲苯磺酰基腙都能很好地参与反应, 并能够以中等至较高的收率得到目标产物(Scheme 2).该三组分反应的优势在于, 通过一步反应实现了两根C—C单键、一根C=C双键和一根C—N单键共四组新的化学键的快速构建, 具有较大的合成意义.
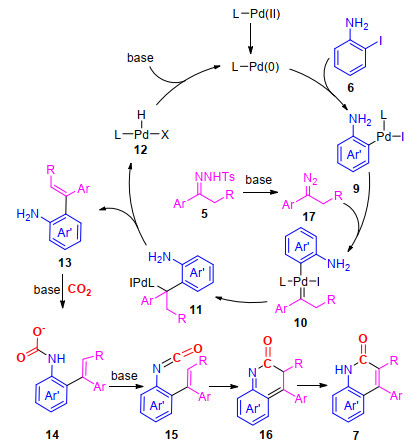
经过相关控制实验的研究证明该反应可能经历了以下的反应过程(Scheme 3):首先, 邻碘苯胺与N-对甲苯磺酰基腙在钯与碱的作用下, 依次经过氧化加成、卡宾迁移插入、迁移和β-H消除反应, 生成邻氨基烯烃中间体13; 随后, 在碱的作用下, 中间体13与二氧化碳发生羧基化反应, 生成氨基甲酸盐14; 再经过脱水和6-π电环化反应, 生成最终的产物7.此外, 邻碘苯胺先和二氧化碳反应, 生成邻碘苯基异氰酸酯8, 然后再与N-对甲苯磺酰腙进行迁移插入/内酰胺化, 这一反应途径也不能完全被排除(Scheme 2).
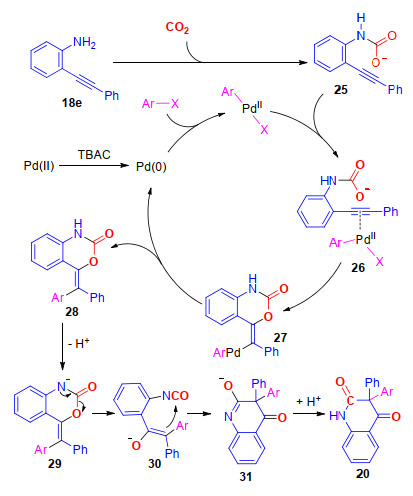
2, 4-喹啉二酮也是一类重要的杂环化合物, 具有优良的生物活性, 是一类良好的生物抑制剂[18]. 2017年, 我们课题组[19]报道了钯催化的邻炔基苯胺、碘代芳烃与CO2的多组分反应, 以中等到较高的产率合成了一系列具有季碳中心的3, 3-二芳基-2, 4-喹啉二酮(Scheme 4).研究结果表明, 该反应对于底物适应性较好, 无论是连有吸电子基还是供电子基的炔基芳香胺和碘代芳烃都能以较好的收率得到目标化合物.该反应的特点在于通过“一锅煮”的方法实现了四根化学键的构建.
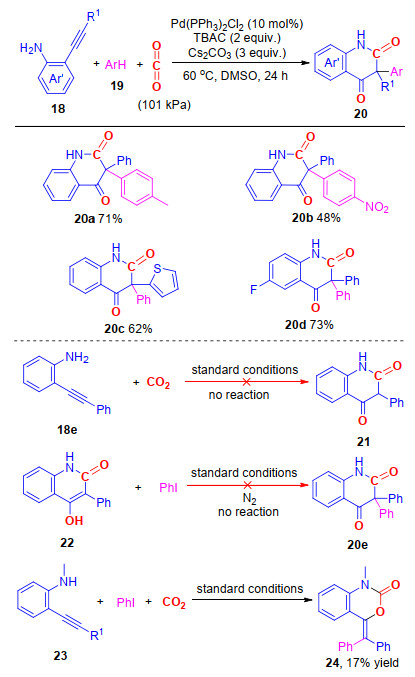
通过相关控制实验, 我们对反应机理进行了研究.在没有碘苯的情况下, 化合物18e不能与CO2反应生成目标化合物21, 这表明碘苯与钯的氧化加成物种对于目标化合物的生成具有至关重要的作用(Scheme 4); 其次, 化合物22(化合物21的异构体)没能在标准条件下与碘苯反应, 生成目标化合物20e, 排除了该反应先生成22, 再与碘苯进行芳基化反应生成最终产物20e的可能性(Scheme 4); 最后, 当在底物的氮原子上引入甲基后(化合物23), 反应只得到化合物24, 这说明氮原子上的甲基抑制了产物24发生重排反应的可能性(Scheme 4).
因此, 我们推测反应可能经历了以下的步骤(Scheme 5).首先, 碘代芳烃与零价钯发生氧化加成反应, 得到二价钯物种; 同时, 在碱的作用下, 邻炔基苯胺化合物18e与CO2发生亲核加成反应, 生成中间体25; 接着, 中间体25中的炔基与二价钯物种发生配位; 随后, 发生氧-钯化反应, 生成中间体27, 27再经还原消除生成中间体28, 最后, 在碱的作用下, 中间体28失去一个质子, 生成中间体29, 再经历分子内的重排反应, 得到最终产物20.
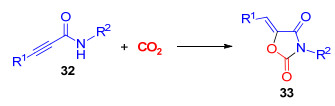
此外, 四取代乙烯基噁唑烷-2, 4-二酮是一类重要的结构单元, 广泛存在于多种具有生物活性的天然产物、医药及农药分子中[20].在已报道的合成方法中, 麻生明与吕小兵课题组[21]利用廉价易得的CO2为羧基源, 成功高效地构建了三取代乙烯基噁唑烷-2, 4-二酮(Scheme 6).然而, 还没有以CO2与炔丙基酰胺构建具有四取代乙烯基噁唑烷-2, 4-二酮的报道.
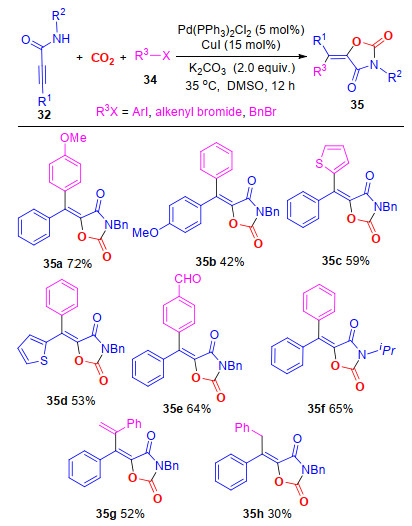
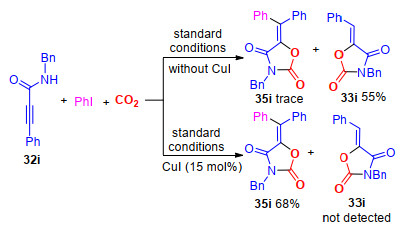
2019年, 我们[22]首次实现了在钯催化及铜盐促进作用下炔丙基酰胺、卤代烃和CO2的三组分反应, 制备出了一系列含四取代乙烯基结构的噁唑烷-2, 4-二酮.该反应对于卤代烃和炔丙基酰胺的底物适用性广.值得指出的是, 通过调控反应原料的结构, 能够选择性地得到不同构型的烯烃结构, 且产率较高(Scheme 7).
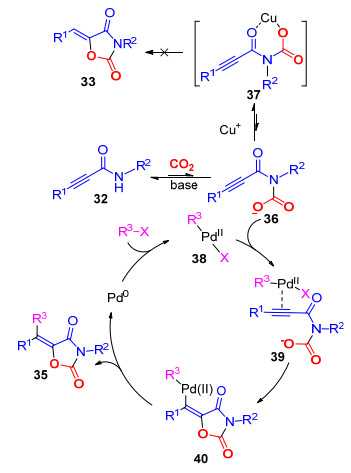
通过相关的控制实验, 我们发现未加15 mol%碘化亚铜的反应体系的主要产物是三取代烯烃产物33i, 四取代烯烃化合物35i几乎检测不到(Scheme 8); 然而, 当往反应中加入15 mol%碘化亚铜时, 反应结果正好相反, 反应以生成四取代烯烃结构产物35i为主, 而三取代产物33i几乎检测不到(Scheme 8).这显示碘化亚铜对于抑制三取代烯烃产物33i的生成具有关键作用.
对于可能经历的反应历程, 我们进行了如下推测(Scheme 9):首先, 在碱的作用下, 化合物32与CO2发生羧基化反应, 生成中间体36, 在铜离子的作用下, 可能生成螯合物37, 由于中间体37的特殊结构, 限制了羧基中的氧负离子对于叁键的加成反应, 进而抑制了三取代烯烃结构产物33的生成.另一方面, 钯与卤代烃发生氧化加成反应, 生成中间体38, 接着与中间体38的叁键发生配位作用, 生成中间体39; 随后, 发生氧-钯化反应生成40, 经过一步还原消除反应, 生成目标化合物35, 并完成催化剂再生.
近年来, 利用光化学固定转化二氧化碳已经取得了很大的研究进展, 相比于传统利用高热量来引发反应而言, 利用可见光作为能量源来诱发反应显得更加绿色环保, 通过可见光促进的方式来固定转化二氧化碳就更加有意义了.余达刚等[23]相继报道了多种可见光促进的二氧化碳和烯丙基胺41参与的多组分反应, 并构建了一系列的官能团化的噁唑啉化合物42 (Scheme 10).
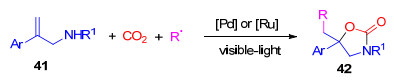
与烷基胺相比, 芳基胺的亲核性较低, 在可见光驱动下, 利用芳胺来与二氧化碳反应更具难度.另一方面, 1, 3-苯并噁嗪-2(4H)-酮及其衍生物也是一类重要的结构单元, 广泛存在于多种具有生物活性的天然产物以及药物分子中[24].邻羟甲基芳胺与光气或者四溴化碳的环化反应是该类化合物的主要合成方法[25].然而, 光气和四溴化碳的毒性较高, 在生产过程中, 有较大的安全隐患.因此, 进一步寻找安全的羰基源合成该类化合物具有重要的研究价值.近期, 我们[26]实现了在蓝光照射条件下, 以钯为催化剂促进的2-(α-芳烯基)芳胺、溴代烷烃合CO2的三组分反应, 制备出了一系列的4-亚甲基烷基-4-芳基-1, 3-苯并噁嗪-2(4H)-酮(45).该反应的底物适应性较好, 一级、二级和三级溴代烷烃都能很好地参与反应, 且2-(α-芳烯基)芳胺上的取代基的电子效应对反应没有较大影响, 都以较好的收率得到相应的目标化合物(Scheme 11).
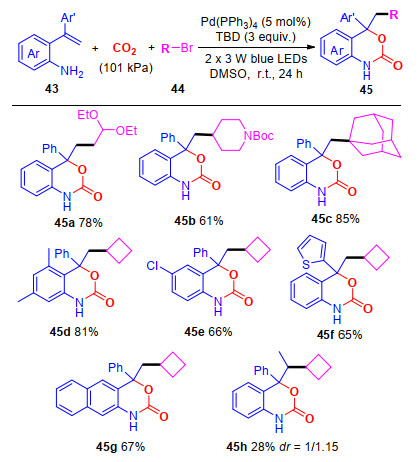
通过相关的控制实验, 我们对可能的反应机理进行推测(Scheme 12):首先, 在可见光促进下, 处于基态的Pd(0)被活化成激发态Pd(0)*; 接着, 钯与溴代烷烃发生单电子转移反应, 生成的烷基自由基46; 同时, 在碱的作用下, CO2与氨基发生羧基化反应, 生成中间体47; 然后, 烷基自由基对中间体47的双键进行自由基加成, 得到中间体48; 随后, 中间体48再与Pd(I)发生单电子氧化反应生成碳正离子中间体49, 最后发生分子内环化反应, 得到最终产物45.
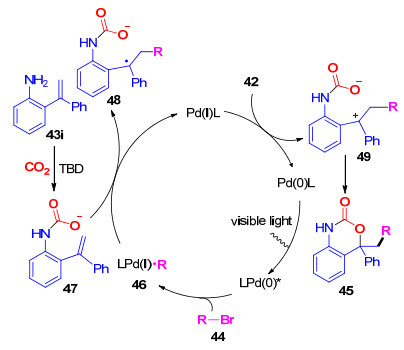
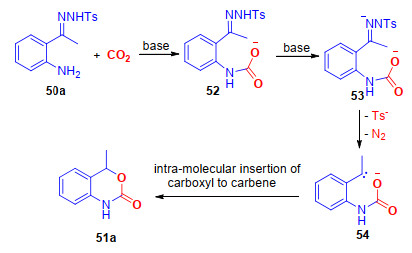
与此同时, 我们[27]也实现了无过渡金属参与的, 仅需Cs2CO3促进的邻氨基芳基腙与CO2的分子内羧基化/环化反应, 能够以中等到良好的收率得到一系列1, 3-苯并噁嗪-2(4H)-酮.在最优的反应条件下, 底物的官能团适应性较好, 对于邻氨基N-对甲基苯磺酰基酮腙和醛腙, 都能够以较高的收率得到目标化合物.此外, 利用上述方法, 可以方便地实现两种具有生物活性的药物分子结构51g和51h的快速构建[28].由于该反应不需要过渡金属催化剂的参与, 可以有效避免重金属残留, 因此是一个绿色的合成该类结构的方法(Scheme 13).
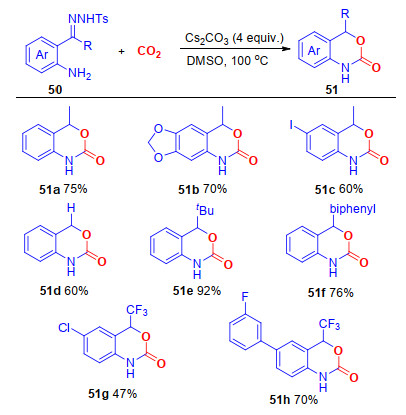
该反应可能经历了以下的反应步骤(Scheme 14):首先, 在碱的作用下, 氨基发生羧基化反应, 生成中间体52; 同时, 在碱的作用下, 腙发生脱苯磺酰基和氮气的反应, 生成中间体54, 随后分子内的环化反应, 生成最终的产物51a.
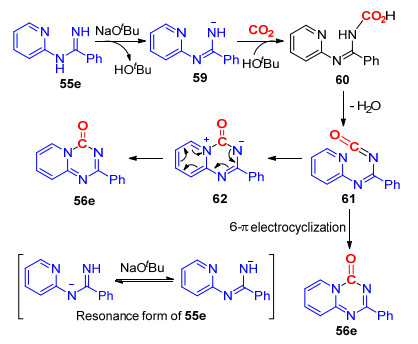
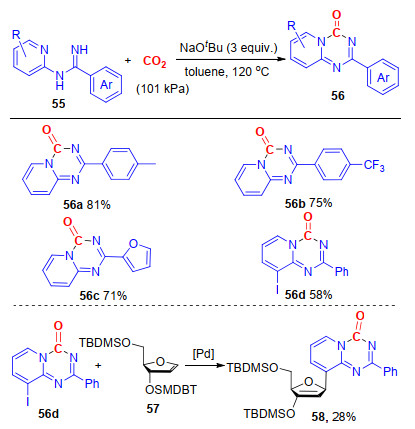
同样在无过渡金属催化作用下, 我们[29]实现了叔丁醇钠促进的N-2-吡啶基脒与CO2的去芳香性环化反应, 以中等到良好的收率得到了一系列吡啶并[1, 2-a]- 1, 3, 5-三嗪酮.在该反应中, CO2作为羰基源, 唯一副产物为H2O, 不需要脱氢试剂以及过渡金属催化剂参与, 是一种绿色、可持续的合成该类结构的方法(Scheme 15).所得化合物可经过一步简单的偶联反应, 快速构建核苷类化合物58, 与该类化合物的传统合成方法相比, 我们所发展的合成路线更简单、更具经济性[30].
该反应可能经历了一下的反应历程(Scheme 16):首先, 在碱的作用下, 二氧化碳与生成的氮负离子发生亲核加成反应, 生成氨基甲酸中间体60, 接着中间体60发生脱水反应, 生成含异氰酸酯基团的中间体61; 随后, 中间体61经历6-π电环化反应, 生成产物56e; 另一方面, 该反应也可能经历了吡啶环上氮原子对NCO基团的亲核加成反应, 再经历电子重排反应, 生成最终的目标化合物56e.
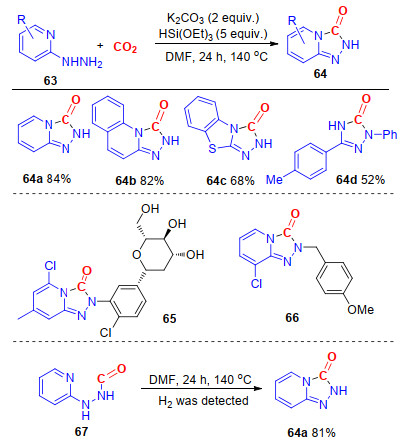
三氮唑酮结构广泛存在于许多天然产物以及具有生物活性的分子中, 该类化合物及其衍生物还具有良好的抗菌、抗病毒、抗肿瘤及镇喘等药效[31]. 2017年, 我们[32]实现了碳酸钾促进的2-肼基吡啶与CO2的环化反应, 以中等到良好的收率得到一系列取代三唑酮.反应以硅烷为还原剂[33], CO2作为羰基来源.进一步研究发现, 该反应的底物适应性较好, 含供电子基和吸电子基的底物都能很好地兼容反应; 其次, 底物的位阻对该反应几乎没有影响.此外, 芳甲亚胺酸酰肼也能很好地参与反应, 得到相应的3-芳基三唑酮化合物, 显示出该方法的良好适用性.该反应以二氧化碳作为羰基源, 是一类绿色、可持续的直接合成三唑酮的方法.所得三唑酮可以进一步用于合成抗惊厥药物65[34]及血管紧张素Ⅱ AT1受体拮抗剂66[35](Scheme 17).
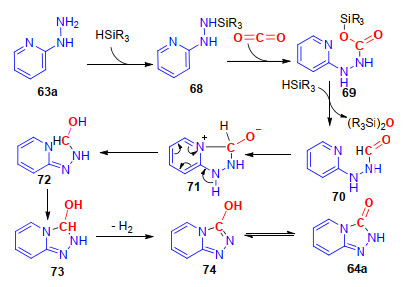
该反应可能首先经历了2-肼基吡啶与硅烷的反应, 生成中间体68; 接着二氧化碳插入到中间体68的N—Si键中, 生成中间体69; 中间体69再与另一分子硅烷反应, 生成中间体70, 并释放出一分子的硅醚; 随后, 吡啶环上的氮原子对于羰基的亲核加成反应, 生成中间体71; 再经历分子内电子转移, 生成中间体72, 最后, 中间体72发生芳构化反应, 生成最终的产物64a, 并释放出一分子氢气, 并通过氢气检测试纸证实了在反应体系中有H2产生(Scheme 18).
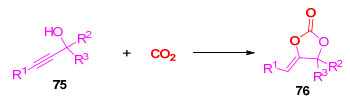
环碳酸酯是一类重要的有机合成中间体.利用Lewis酸催化炔丙醇与二氧化碳的环化反应是构建该类化合物的主要方法[36].然而, 在已报道的工作中, 所得的α-亚烷基碳酸酯76中的烯基大多数是三取代结构(Scheme 19).
合成含四取代烯烃结构的α-亚烷基碳酸酯, 可进一步丰富该类化合物的用途.近期, 我们实现了炔丙醇、CO2以及芳基卤代烃的芳基化/羧基化反应, 得到一系列含四取代烯烃结构的α-亚烷基碳酸酯(Scheme 20)[37].
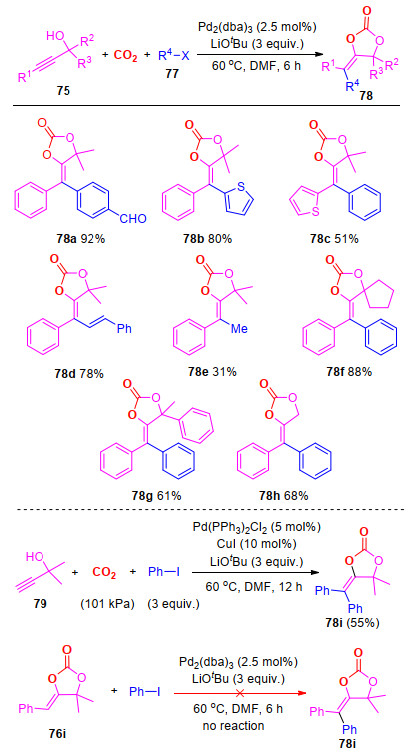
该反应的底物适应性较好, 底物中取代基的电子效应对反应没有明显影响, 且1°, 3°炔丙醇都能很好地参与反应, 并以较高的收率得到相应的目标化合物, 所得产物的构型也通过了单晶衍射进行结构确定.此外, 一些反应性基团, 例如Cl, COOEt, CHO和Ac等能够很好地兼容反应, 这为产物的进一步转化提供了方便; 另外, 2-碘噻吩、β-溴代苯乙烯和碘甲烷也能参与反应.值得注意的是, 可以通过调控反应底物的结构, 实现含不同构型烯烃的α-亚烷基碳酸酯的选择性构建(如化合物78b vs. 78c).该反应的特点在于一步实现四个新化学键的构建, 并且拓展了CO2在合成新的结构杂环中的适用范围.
此外, 在标准条件下, 端炔79也能参与反应, 得到目标化合物78i (Scheme 20).化合物76i并不能在标准条件下, 与碘苯发生Heck反应, 得到化合物78i, 这就排除了反应中先生成化合物76i, 再通过Heck反应得到目标化合物的可能性(Scheme 20).
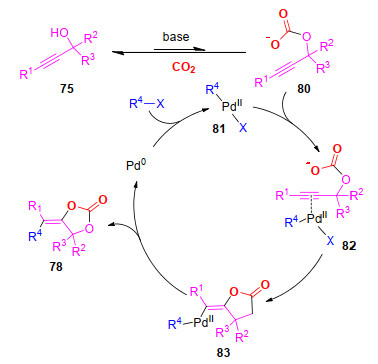
该反应可能经历了以下反应步骤(Scheme 21):首先, 在碱的作用下, 炔丙醇75与CO2发生亲核加成反应, 生成碳酸酯中间体80; 同时, 芳基碘与Pd(0)发生氧化加成反应, 生成中间体81; 随后, 二价钯中间体81与中间体80中的碳碳叁键发生配位作用, 活化叁键; 接着, 发生反式的氧-钯化反应, 生成中间体82; 最后, 中间体82发生还原消除反应, 生成最终产物78, 并释放出Pd(0), 完成催化循环.
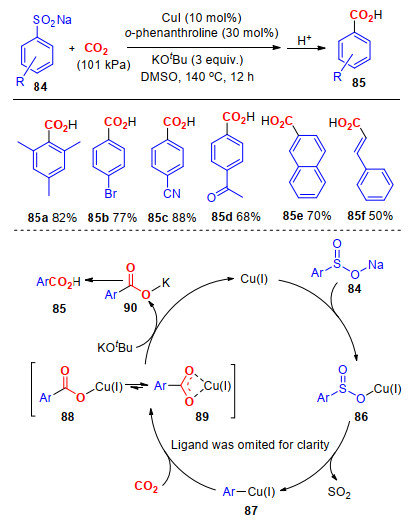
二氧化碳是一种弱的亲电试剂, 能够与亲核性较强的格氏试剂及有机锂试剂发生亲核加成反应, 生成羧酸.然而, 亲核性较强的试剂在反应中的官能团适应性较差, 因而限制了其在CO2固定转化反应中的应用.研究发现, 在过渡金属的催化作用下, 一些亲核性较弱的试剂也能与CO2发生亲核加成反应, 生成羧酸, 且反应具有较好的官能团适应性[38]. 2015年, 我们课题组[39]报道了以碘化亚铜作为催化剂, 1, 10-啡啰啉作为配体, 叔丁醇钾作为碱的条件下, 芳基亚磺酸钠发生脱二氧化硫羧基化反应, 生成芳基甲酸(Scheme 22).该反应可能经历了一下反应历程:首先, 铜盐与芳基亚磺酸钠84作用, 生成相应的亚磺酸铜86; 接着, 发生脱SO2作用, 生成含有碳-铜键的芳基铜配合物87; 随后, 二氧化碳插入到芳基铜配合物87中, 生成羧酸基铜配合物88; 最后, 在叔丁醇钾的转金属作用下, 生成羧酸钾盐90, 并完成催化剂再生, 实现催化循环, 最后, 中间体90经由酸化得到芳基羧酸85.在最优的反应条件下, 连有吸电子基和供电子基的底物都能以较高的收率得到相应的产物; 其次, 一些官能团, 例如甲基、卤素、氰基和酰基等也能很好地兼容反应.
此外, α-芳基丙烯酸不仅是一种重要的有机合成片断, 而且广泛存在于多种天然化合物以及药物中.例如, α-芳基丙烯酸及其衍生物可以作为重要的反应前体合成多种抗菌药如萘普生、布洛芬和非诺洛芬等[40].近年来, α-芳基丙烯酸及其衍生物合成新方法的研究越来越引起人们的重视.其中, 以CO2为羧基源, 在正丁基锂的作用下, 通过磺酰基腙与CO2的Shapiro反应, 可以实现α-芳基丙烯酸的合成.然而, 该反应需要使用强碱性的正丁基锂以及-78 ℃的反应条件, 苛刻的反应条件以及较低的官能团适应性严重降低了该反应的实际应用价值[41]. 2015年, 我们[42]对传统的Shapiro反应进行了改进, 研究发现, 仅需Cs2CO3作碱, 即可实现N-甲苯磺酰腙与一个大气压力的CO2发生羧基化反应, 能够以较高的收率得到α-芳基丙烯酸(Scheme 23).与现有的制备该类化合物的方法相比, 该方法反应条件温和, 官能团适应性广, 使用无毒的CO2替代了有毒有害的一氧化碳作为羰基源, 而且避免了使用过渡金属催化剂.
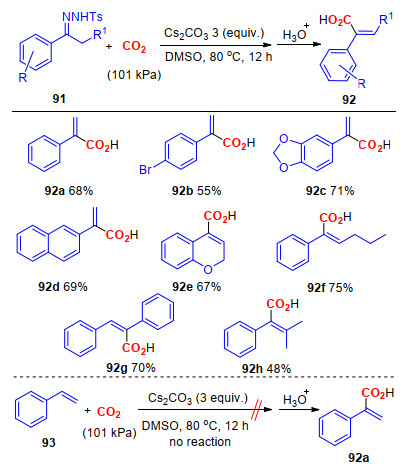
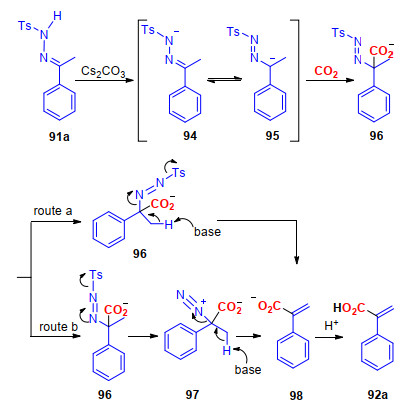
通过相关的控制实验发现, 苯乙烯(93)并不能在标准条件下与CO2反应, 生成目标化合物92a, 这就排除了苯乙烯作为反应中间体的可能性.由此, 我们推测该反应经历了以下反应步骤(Scheme 24):首先, 在Cs2CO3的作用下, 腙91a发生脱质子反应, 生成中间体94; 随后, 中间体94经过异构化作用, 生成中间体95, 然后中间体95中的碳负离子与CO2发生亲核加成反应, 生成中间体96; 接着, 在碱的作用下, 中间体96经历脱质子、脱Ts和N2等反应, 生成中间体98.另一方面, 中间体先脱去Ts, 再发生脱N2和脱质子的步骤, 生成中间体98, 这条反应路线也不能完全被排除.最后, 中间体98经过酸化, 得到最终的产物92a.
二氧化碳具有价廉易得、储量丰富及无毒等优点, 在有机合成反应中是一种理想的C1合成子.以二氧化碳为羰基/羧基源, 在温和条件下, 二氧化碳可以直接与简单易得的试剂通过羧基化反应得到羧酸; 也可以与其它的试剂通过多组分环化反应得到三唑酮、吡啶并[1, 2-a]-1, 3, 5-三嗪-4-酮、芳基噁嗪-2-酮、芳基喹啉酮、环状碳酸酯和噁唑啉酮等重要的杂环化合物, 上述反应在替代传统的羰基化/羧基化方法上具有巨大潜力.然而, 目前基于含氮-、氧-、碳-亲核原子试剂参与的CO2的固定转化反应, 还存在着底物类型和反应模式单一等缺点.其次, 所开发出来的二氧化碳固定转化反应距离工业化生产还具有相当的距离, 其主要局限在于: (1)反应中需要使用大量的碱作为转金属试剂; (2)过渡金属催化剂的催化效率还不高, 价格昂贵且难以回收利用; (3)绝大多数反应还需在高温高压下下进行, 这就极大地提高了反应成本, 也达不到绿色化学的要求.
因此, 通过对反应机理进行深入研究, 设计出新型结构的底物分子和催化剂, 提高二氧化碳的固定转化效率, 是二氧化碳固定转化反应设计的努力方向.另外, 借助于合适的手性配体, 实现具有对映选择性产物的构建, 将会极大地提高目标产物的附加值.其次, 到目前为止, 绝大多数反应都是基于底物的亲核位点与二氧化碳的亲核加成来实现二氧化碳的固定转化.相比而言, 通过将二氧化碳还原生成二氧化碳自由基阴离子进而更高效地进行自由基加成的策略却鲜有报道.因此, 进一步开发通过光化学和电化学的途径, 实现二氧化碳的固定转化, 让资源化利用二氧化碳的反应变得更加绿色环保, 所得产物的结构更加丰富多样, 也是需要努力的方向.
Le Quere, C.; Andrew, R. M.; Friedlingstein, P.; Sitch, S.; Hauck, J.; Pongratz, J.; Pickers, P. A.; Korsbakken, J. I.; Peters, G. P.; Canadell, J. G.; Arneth, A.; Arora, V. K.; Barbero, L.; Bastos, A.; Bopp, L.; Chevallier, F.; Chini, L. P.; Ciais, P.; Doney, S. C.; Gkritzalis, T.; Goll, D. S.; Harris, I.; Haverd, V.; Hoffman, F. M.; Hoppema, M.; Houghton, R. A.; Hurtt, G.; Ilyina, T.; Jain, A. K.; Johannessen, T.; Jones, C. D.; Kato, E.; Keeling, R. F.; Goldewijk, K. K.; Landschutzer, P.; Lefevre, N.; Lienert, S.; Liu, Z.; Lombardozzi, D.; Metzl, N.; Munro, D. R.; Nabel, J. E. M. S.; Nakaoka, S.; Neill, C.; Olsen, A.; Ono, T.; Patra, P.; Peregon, A.; Peters, W.; Peylin, P.; Pfeil, B.; Pierrot, D.; Poulter, B.; Rehder, G.; Resplandy, L.; Robertson, E.; Rocher, M.; Rodenbeck, C.; Schuster, U.; Schwinger, J.; Seferian, R.; Skjelvan, I.; Steinhoff, T.; Sutton, A.; Tans, P. P.; Tian, H. Q.; Tilbrook, B.; Tubiello, F. N.; van der Laan-Luijkx, I. T.; van der Werf, G. R.; Viovy, N.; Walker, A. P.; Wiltshire, A. J.; Wright, R.; Zaehle, S.; Zheng, B. Earth Syst. Sci. 2018, 10, 2141. doi: 10.5194/essd-10-2141-2018
(a) Hulla, M.; Dyson, P. J. Angew. Chem., Int. Ed. 2020, 59, 1002.
(b) Yeung, C. S. Angew. Chem., Int. Ed. 2019, 58, 5492.
(c) Luo, J.; Larrosa, I. ChemSusChem 2017, 10, 3317.
(d) Gui, Y.-Y.; Zhou, W.-J.; Ye, J.-H.; Yu, D.-G. ChemSusChem 2017, 10, 1337.
(e) Lu, X. Carbon Dioxide and Organometallics, Springer International Publishing, Switzerland, 2016.
(f) Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Nat. Commun. 2015, 6, 5933.
(g) Bhanage, B. M.; Arai, M. Transformation and Utilization of Carbon Dioxide, Springer-Verlag, Berlin, Heidelberg, 2014.
(h) Zhang, L.; Hou, Z. Chem. Sci. 2013, 4, 3395.
(i) Cokoja, M.; Bruckmeier, C.; Rieger, B.; Herrmann, W. A.; Kühn, F. E. Angew. Chem., Int. Ed. 2011, 50, 8510.
(j) Huang, K.; Sun, C.-L.; Shi, Z.-J. Chem. Soc. Rev. 2011, 40, 2435.
(k) Aresta, M. Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010.
(l) Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365.
(a) Aresta, M.; Dibenedetto, A.; Angelini, A. Chem. Rev. 2014, 114, 1709.
(b) Hua, B.; Guild, C.; Suib, S. L. J. CO2 Util. 2013, 1, 18.
(a) Parvatkar, P. T.; Parameswaran, P. S.; Tilve, S. G. Chem.-Eur. J. 2012, 18, 5460.
(b) Weibel, J.-M.; Blanc, A.; Pale, P. Chem. Rev. 2008, 108, 3149.
(a) Peshkov, V. A.; Pereshivko, O. P.; Nechaev, A. A.; Peshkovc, A. A.; Van der Eycken, E. V. Chem. Soc. Rev. 2018, 47, 3861.
(b) Liu, X.; He, L.-N. Top. Curr. Chem. 2017, 375, 21.
(c) Zhang, Z.; Ju, T.; Ye, J.-H.; Yu, D.-G. Synlett 2017, 28, 741.
(d) Sekine, K.; Yamada, T. Chem. Soc. Rev. 2016, 45, 4524.
Wu, L.; Liu, Q.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2014, 53, 6310. doi: 10.1002/anie.201400793
(a) Guo, C.-X.; Zhang, W.-Z.; Zhang, N.; Lu, X.-B. J. Org. Chem. 2017, 82, 7637.
(b) Zhang, W.-Z.; Yang, M.-W.; Lu, X.-B. Green Chem. 2016, 18, 4181.
(c) Zhang, W.-Z.; Yang, M.-W.; Yang, X.-T.; Shi, L.-L. Wang, H.-B.; Lu, X.-B. Org. Chem. Front. 2016, 3, 217.
(d) Zhang, W.-Z.; Xia, T.; Yang, X.-T.; Lu, X.-B. Chem. Commun. 2015, 51, 6175.
(e) Wang, Y.-B.; Wang, Y.-M.; Zhang, W.-Z.; Lu, X.-B. J. Am. Chem. Soc. 2013, 135, 11996.
(a) Lang, X.-D.; You, F.; He, X.; Yu, Y.-C.; He, L.-N. Green Chem. 2019, 21, 509.
(b) He, X.; Yao, X.-Y.; Chen, K.-H.; He, L.-N. ChemSusChem 2019, 12, 5081.
(c) Lang, X.-D.; You, F.; He, X.; Yu, Y.-C.; He, L.-N. Green Chem. 2019, 21, 509.
(d) Wang, M.-Y.; Cao, Y.; Liu, X.; Wang, N.; He, L.-N.; Li, S.-H. Green Chem. 2017, 19, 1240.
(e) Wang, M.-Y.; Song, Q.-W.; Ma, R.; Xie, J.-N.; He, L.-N. Green Chem. 2016, 18, 282.
(f) Xie, J.-N.; Yu, B.; Guo, C.-X.; He, L.-N. Green Chem. 2015, 17, 4061.
(a) Ouyang, L.; Tang, X.; He, H.; Qi, C.; Xiong, W.; Ren, Y.; Jiang, H. Adv. Synth. Catal. 2015, 357, 2556.
(b) Qi, C.; Jiang, H.; Huang, L.; Yuan, G.; Ren, Y. Org. Lett. 2013, 13, 5520.
(c) Zhao, J.; Huang, H.; Qi, C.; Jiang, H. Eur. J. Org. Chem. 2012, 5665.
(a) Hu, J.; Ma, J.; Zhu, Q.; Zhang, Z.; Wu, C.; Han, B. Angew. Chem., Int. Ed. 2015, 54, 5399.
(b) Gao, X.; Yu, B.; Yang, Z.; Zhao, Y.; Zhang, H.; Hao, L.; Han, B.; Liu, Z. ACS Catal. 2015, 5, 6648.
(a) Ye, J.-H.; Song, L.; Zhou, W.-J.; Ju, T.; Yin, Z.-B.; Yan, S.-S.; Zhang, Z.; Li, J.; Yu, D.-G. Angew. Chem., Int. Ed. 2016, 55, 10022.
(b) Song, L.; Zhu, L.; Zhang, Z.; Ye, J.-H.; Yan, S.-S.; Han, J.-L.; Yin, Z.-B.; Lan, Y.; Yu, D.-G.; Org. Lett. 2018, 20, 3776.
(c) Zhang, Z.; Ju, T.; Miao, M.; Han, J.-L.; Zhang, Y.-H.; Zhu, X.-Y.; Ye, J.-H.; Yu, D.-G.; Zhi, Y.-G. Org. Lett. 2017, 19, 396.
(d) Sun, L.; Ye, J.-H.; Zhou, W.-J.; Zeng, X.; Yu, D.-G. Org. Lett. 2018, 20, 3049.
(a) Sadamitsu, Y.; Komatsuki, K.; Saito, K.; Yamada, T. Org. Lett. 2017, 19, 3191.
(b) Ishida, T.; Kikuchi, S.; Tsubo, T.; Yamada, T. Org. Lett. 2013, 15, 848.
(c) Ishida, T.; Kikuchi, S.; Yamada, T. Org. Lett. 2013, 15, 3710.
(d) Kikuchi, S.; Sekine, K.; Ishida, T.; Yamada, T. Angew. Chem., Int. Ed. 2012, 51, 6989.
(a) Wang, S.; Xi, C. Chem. Soc. Rev. 2019, 48, 382.
(b) Wang, L.; Sun, W.; Liu, C. Chin. J. Chem. 2018, 36, 353.
(c) Zhang, W.; Zhang, N.; Guo, C.; Lu, X.-B. Chin. J. Org. Chem. 2017, 37, 1309(in Chinese).
(张文珍, 张宁, 郭春晓, 吕小兵, 有机化学, 2017, 37, 1309.)
(d) Zhu, Q.; Wang, L.; Xia, C.; Liu, C. Chin. J. Org. Chem. 2016, 36, 2813(in Chinese).
(朱庆, 王露, 夏春谷, 刘超, 有机化学, 2016, 36, 2813.)
(e) Rintjema, J.; Kleij, A. W. Synthesis 2016, 48, 3863.
(f) Yu, B.; He, L.-N. ChemSusChem 2015, 8, 52.
(g) Kielland, N.; Whiteoak, C.; Kleij, A. W. Adv. Synth. Catal. 2013, 355, 2115.
(h) Lu, X.-B.; Darensbourg, D. Chem. Soc. Rev. 2012, 41, 1462.
(a) Miyashiro, J.; Woods, K. W.; Park, C. H.; Liu, X.; Shi, Y.; Johnson, E. F.; Bouska, J. J.; Olson, A. M.; Luo, Y.; Fry, E. H.; Giranda, V. L.; Penning, T. D. Bioorg. Med. Chem. Lett. 2009, 19, 4050.
(b) Angibaud, P. R.; Venet, M. G.; Filliers, W.; Broeckx, R.; Ligny, Y. A.; Muller, P.; Poncelet, V. S.; End, D. W. Eur. J. Org. Chem. 2004, 479.
(a) Ferguson, M J.; Zeng, F.; Alwis, N.; Alper, H. Org. Lett. 2013, 15, 1998.
(b) Liang, Z.; Zhang, J.; Liu, Z.; Wang, K.; Zhang, Y. Tetrahedron 2013, 69, 6519.
(a) Zhang, Z.; Liao, L.-L.; Yan, S.-S.; Wang, L.; He, Y.-Q.; Ye, J.-H.; Li, J.; Zhi, Y.-G.; Yu, D.-G. Angew. Chem., Int. Ed. 2016, 55, 7068.
(b) Wang, S.; Shao, P.; Du, G.; Xi, C. J. Org. Chem. 2016, 81, 6672.
Sun, S.; Hu, W.-M.; Gu, N.; Cheng, J. Chem.-Eur. J. 2016, 22, 18729. doi: 10.1002/chem.201604256
(a) Ishida, T.; Kikuchi, S.; Tsubo, T.; Yamada, T. Org. Lett. 2013, 15, 848.
(b) Childers, W. E. Jr.; Baudy, R. B. J. Med. Chem. 2007, 50, 2557.
Wang, B.; Sun, S.; Yu, J.-T.; Jiang, Y.; Cheng, J. Org. Lett. 2017, 19, 4319. doi: 10.1021/acs.orglett.7b01989
(a) Pastore, V.; Sabatier, L.; Enrique, A.; Marder, M.; Bruno-Blanch, L. E. Bioorg. Med. Chem. 2013, 21, 841.
(b) Heerding, D. A.; Christmann, L. T.; Clark, T. J.; Holmes, D. J.; Rittenhouse, S. F.; Takata, D. T.; Venslavsky, J. W. Bioorg. Med. Chem. Lett. 2003, 13, 3771.
(a) Zhou, H.; Mu, S.; Ren, B.-H.; Zhang, R.; Lu, X.-B. Green Chem. 2019, 21, 991.
(b) Chen, G.; Fu, C.; Ma, S. Org. Biomol. Chem. 2011, 9, 105.
Zhou, C.; Dong, Y.; Yu, J.-T.; Sun, S.; Cheng, J. Chem. Commun. 2019, 55, 13685. doi: 10.1039/C9CC07027C
(a) Yin, Z.; Ye, J.; Zhou, W.; Zhang, Y.; Ding, L.; Gui, Y.; Yan, S.; Li, J.; Yu, D. Org. Lett. 2018, 20, 190.
(b) Sun, L.; Ye, J.; Zhou, W.; Zeng, X.; Yu, D. Org. Lett. 2018, 20, 3049.
(a) Commons, T. J.; Jenkins, D. J.; Trybulski, E. J.; Fensome, A. US 20090197878, 2009.
(b) Collins, M. A.; Hudak, V.; Bender, R.; Fensome, A.; Zhang, P.; Miller, L.; Winneker, R. C.; Zhang, Z.; Zhu, Y.; Cohen, J. Bioorg. Med. Chem. Lett. 2004, 14, 2185.
(a) Zhao, Y.; Huang, B.; Yang, C.; Chen, Q.; Xia, W. Org. Lett. 2016, 18, 5572.
(b) Hernandez, E.; Velez, J. M.; Vlaar, C. P. Tetrahedron Lett. 2007, 48, 8972.
(c) Lagu, B.; Pio, B.; Lebedev, R.; Yang, M.; Pelton, P. D. Bioorg. Med. Chem. Lett. 2007, 17, 3497.
Sun, S.; Zhou, C.; Dong, Y.; Yu, J.-T.; Cheng, J. Org. Lett. 2019, 21, 6579. doi: 10.1021/acs.orglett.9b02700
Xiong, H.; Wu, X.; Wang, H.; Sun, S.; Yu, J.; Cheng, J. Adv. Synth. Catal. 2019, 361, 3538. doi: 10.1002/adsc.201900341
Frantz, M.-C.; Rodrigo, J.; Boudier, L.; Durroux, T.; Mouillac, B.; Hibert, M. J. Med. Chem. 2010, 53, 1546.
(b) Collins, M. A.; Hudak, V.; Bender, R.; Fensome, A.; Zhang, P.; Miller, L.; Winneker, R. C.; Zhang, Z.; Zhu, Y.; Cohen, J. Bioorg. Med. Chem. Lett. 2004, 14, 2185.
Xia, M.; Hu, W.; Sun, S.; Yu, J.-T.; Cheng, J. Org. Biomol. Chem. 2017, 15, 4064. doi: 10.1039/C7OB00777A
(a) Holtwick, J. B.; Leonard, N. J. J. Org. Chem. 1981, 46, 3681.
(b) Holtwick, J. B.; Golankiewicz, B.; Holmes, B. N.; Leonard, N. J. J. Org. Chem. 1979, 44, 3835.
(a) Kahveci, B.; Yilmaz, F.; Mentese, E.; Ulker, S. Chem. Heterocycl. Compd. 2015, 51, 447.
(b) Han, S.; Zhang, F.-F.; Xie, X.; Chen, J.-Z. Eur. J. Med. Chem. 2014, 74, 73.
Wu, X.; Sun, S.; Wang, B.; Cheng, J. Adv. Synth. Catal. 2017, 359, 3855. doi: 10.1002/adsc.201700869
Ren, X.; Zheng, Z.; Zhang, L.; Wang, Z.; Xia, C.; Ding, K. Angew. Chem., Int. Ed. 2017, 56, 310. doi: 10.1002/anie.201608628
Kane, J. M.; Baron, B. M.; Dudley, M. W.; Sorensen, S. M.; Staeger, M. A.; Miller, F. P. J. Med. Chem. 1990, 33, 2772. doi: 10.1021/jm00172a015
(a) Sharma, M. C.; Sharma, S.; Sharma, P.; Kumar, A.; Bhadoriya, K. S. Med. Chem. Res. 2014, 23, 2486.
(b) Pai, N. R.; Dubhashi, D. S.; Pusalkar, D. A. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 180.
(a) Song, Q.-W.; Yu, B.; Li, X.-D.; Ma, R.; Diao, Z.-F.; Li, R.-G.; Li, W.; He, L.-N. Green Chem. 2014, 16, 1633.
(b) Yoshida, S.; Fukui, K.; Kikuchi, S.; Yamada, T. J. Am. Chem. Soc. 2010, 132, 4072.
(c) Kayaki, Y.; Yamamoto, M.; Ikariya, T. Angew. Chem., Int. Ed. 2009, 48, 419.
(d) Jiang, H.-F.; Wang, A.-Z.; Liu, H.-L.; Qi, C.-R. Eur. J. Org. Chem. 2008, 2309.
(a) Sun, S.; Wang, B.; Gu, N.; Yu, J.-T.; Cheng, J. Org. Lett. 2017, 19, 1088.
(b) Iritani, K.; Yanagihara, N.; Utimoto, K. J. Org. Chem. 1986, 51, 5499.
(a) León, T.; Correa, A.; Martin, R. J. Am. Chem. Soc. 2013, 135, 1221.
(b) Williams, C. M.; Johnson, J. B.; Rovis, T. J. Am. Chem. Soc. 2008, 130, 14936.
Sun, S.; Yu, J.-T.; Jiang, Y.; Cheng, J. Adv. Synth. Catal. 2015, 357, 2022. doi: 10.1002/adsc.201500101
(a) Windsor, M. A.; Hermanson, D. J.; Kingsley, P. J.; Xu, S.; Crews, B. C.; Ho, W.; Keenan, C. M.; Banerjee, S.; Sharkey, K. A.; Marnett, L. J. ACS Med. Chem. Lett. 2012, 3, 759.
(b) Zhu, S.-F.; Yu, Y.-B.; Li, S.; Wang, L.-X.; Zhou, Q.-L. Angew. Chem., Int. Ed. 2012, 51, 8872.
(c) Hu, W.; Chen, C.-C.; Xue, G.; Chan, A. S. C. Tetrahedron: Asymmetry 1998, 9, 4183.
Stemke, J. E.; Chamberlin, A. R.; Bond, F. T. Tetrahedron Lett. 1976, 17, 2947. doi: 10.1016/S0040-4039(01)85496-4
Sun, S.; Yu, J.-T.; Jiang, Y.; Cheng, J. J. Org. Chem. 2015, 80, 2855. doi: 10.1021/jo502908v

图式 1 二氧化碳与邻烯基/芳基苯胺的内酰胺化反应
Scheme 1 Lactamization of 2-alkenyl and 2-aryl anilines with CO2

图式 2 钯催化N-对甲苯磺酰基腙、邻碘芳胺与CO2的三组分反应合成4-芳基-2-喹啉酮
Scheme 2 Palladium-catalyzed multi-component reactions of N-tosylhydrazones, 2-iodoanilines and CO2 towards 4-aryl-2- quinolinones

图式 3 合成4-芳基-2-喹啉酮可能的反应机理
Scheme 3 Proposed mechanism for the synthesis of 4-aryl-2- quinolinones

图式 4 钯催化邻炔基芳胺、芳基碘和二氧化碳的多组分反应合成3, 3-二芳基-2, 4-喹啉二酮
Scheme 4 Palladium-catalyzed multi-component reactions of o-alkynylanilines, aryl iodides and CO2 toward 3, 3-diaryl 2, 4-quinolinediones

图式 5 合成3, 3, -二芳基-2, 4-喹啉二酮可能的反应机理
Scheme 5 Proposed mechanism of the synthesis of 3, 3-diaryl 2, 4-quinolinediones

图式 6 炔丙基酰胺与CO2反应生成噁唑烷-2, 4-二酮
Scheme 6 Reaction of propargyl amides with CO2 toward oxazolidine-2, 4-diones

图式 7 钯/铜催化炔丙基酰胺、卤代烃和CO2的多组分反应合成多取代噁唑烷-2, 4-二酮
Scheme 7 Palladium/copper-catalyzed multicomponent reactions of propargylic amides, halohydrocarbons and CO2 toward functionalized oxazolidine-2, 4-diones

图式 9 合成多取代噁唑烷-2, 4-二酮可能的反应机理
Scheme 9 Proposed mechanism of the synthesis of functionalized oxazolidine-2, 4-diones

图式 10 可见光促进CO2与烯丙基胺多组分反应
Scheme 10 Visible light promoted the multi-component reaction of allylamines with CO2

图式 11 可见光促进/钯催化邻烯基芳胺、溴代烷烃和CO2的多组分反应
Scheme 11 Visible-light-driven palladium-catalyzed multi- component reaction of 2-(1-arylvinyl)anilines, alkyl bromides and CO2

图式 12 合成多取代苯并噁嗪-2-酮的可能机理
Scheme 12 Proposed mechanism for the synthesis of functionalized benzoxazin-2-ones

图式 13 碳酸铯促进邻氨基-N-对甲苯磺酰基腙与CO2
Scheme 13 Cs2CO3-Promoted the annulation of o-amino-N- tosylhydrazones and CO2

图式 14 碳酸铯促进邻氨基-N-对甲苯磺酰基腙与CO2环化反应的可能反应机理
Scheme 14 Proposed mechanism of Cs2CO3-promoted the annulation of o-amino-N-tosylhydrazones and CO2

图式 16 N-2-吡啶基脒与CO2的环化反应机理
Scheme 16 Proposed mechanism of the annulation of N-2- pyridylamidine and CO2

图式 17 碱促进2-肼基吡啶、芳甲亚胺酸酰肼与CO2的环化反应
Scheme 17 Base-promoted annulation of 2-hydrazinyl pyridines, arylzamidrazones and CO2

图式 18 碱促进2-肼基吡啶与CO2的环化反应机理
Scheme 18 Proposed mechanism of base-promoted annulation of 2-hydrazinyl pyridines and CO2

图式 19 炔丙醇与CO2反应生成环碳酸酯
Scheme 19 Reaction of propargylic alcohols with CO2 toward cyclic carbonates

图式 20 钯催化炔丙醇与芳基碘以及CO2的碳-芳基化反应
Scheme 20 Palladium-catalyzed arylcarboxylation of propargylic alcohols with CO2 and aryl halides

图式 21 合成官能团化环碳酸酯可能的反应机理
Scheme 21 Proposed mechanism of the synthesis of functionalized α-alkylidene cyclic carbonates

图式 22 铜催化亚磺酸钠与CO2的羧基化反应
Scheme 22 Copper-catalyzed carboxylation of sodium sulfinates with CO2

图式 23 Cs2CO3促进N-对甲苯磺酰腙与CO2的羧基化反应生成α-芳基丙烯酸
Scheme 23 Cs2CO3-promoted carboxylation of N-tosylhydra- zones with CO2 towards α-arylacrylic acids

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:


