Scheme 1.
Os-catalyzed asymmetric electrochemical dihydroxylation of olefins
Advances in Asymmetric Organotransition Metal-Catalyzed Electrochemistry
Xiangyang Wang , Xuetao Xu , Zhenhua Wang , Ping Fang , Tiansheng Mei

Kolbe[1] and Baizer[2] successful introduced electricity into organic synthesis by dimerizing carboxylic acids (in 1847) and acrylonitrile (in 1964), respectively. In recent decades, electrochemistry has made tremendous advances as a platform for the discovery of novel synthetic transformations.[3] Through electrochemistry, challenging transformations are made possible by the ability to generate free radical intermediates under mild conditions.[4] At the same time, controlling chemoselectivity, regioselectivity, and stereoselectivity of a given electrochemical transformation can be difficult, particularly converting achiral compounds into chiral ones.
Significant effort has been made to develop efficient enantioselective electrochemical protocols, [5] including via the use of chiral solvents, [6] chiral auxiliaries, [7] chiral supporting electrolytes, [8] chiral mediators, [9] chiral electrodes, [10] or chiral catalysts.[5, 11]
Organotransition-metal catalysts provide a particular promising means to realizing high chemoselectivity, regioselectivity, and stereoselectivity within an electrochemical process. Three specific advantages of an organotransition metal-catalyzed approach are: (1) the redox potential of the transition-metal catalyst may be tuned by changing substituents on the ligand, (2) modification of the ligand can dictate the reaction stereoselectivity (and chemoselectivity and regioselectivity), (3) well-established thermochemical organotransition metal-catalyzed methodologies provide a rigorous foundation for the development of electrochemical variants.
In this review, we emphasize the recent advances in asymmetric organotransition metal-catalyzed electrochemistry (AOMCE), which we defined as reactions that require the combination of transition-metal catalyst, chiral ligand, and electricity. AOMCE can be divided into two types: asymmetric electrochemical oxidation and asymmetric electrochemical reduction. In the asymmetric electrochemical oxidation section, olefin functionalization, kinetic resolution of secondary alcohols or aldehydes, and C—H functionalization are discussed. In the reductive section, asymmetric electroreductive carboxylation with carbon dioxide, electrochemical decarboxylation, and reductive couplings are highlighted.
The osmium tetroxide-catalyzed oxidation of olefins to produce vicinal diols is a well-known and reliable synthetic method in organic chemistry.[12] Due to the high cost and toxicity of osmium, it is employed catalytically for alkene oxidations in the presence of a less expensive and toxic terminal oxidant.[13]
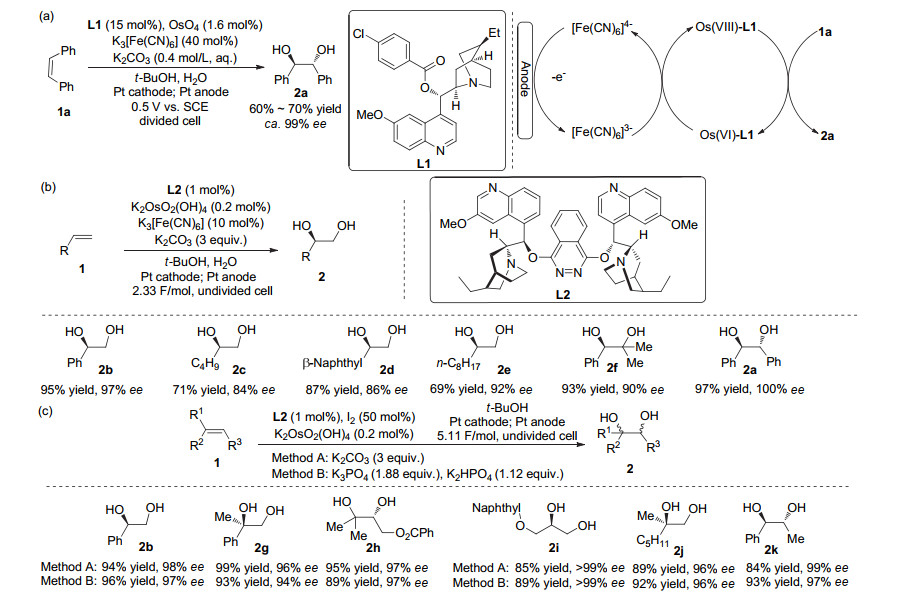
In 1992, Amundsen and co-workers[14] reported the synthesis of a number of chiral diols using a modification of the Sharpless process in an electrolytically regenerated system (Scheme 1a). Osmium tetroxide bearing chiral ligand L1 (hydroquinidine 4-chlorobenzoate) was employed as catalyst, and a catalytic amount of potassium ferricyanide was used as mediating oxidant. A divided cell was necessary to prevent cathodic reduction of ferricyanide oxidant and electrodeposition of osmium. Enantiomeric selectivity was predicated on having bulky alkene substituents, namely phenyls.[15]
In 1995, Torii and co-workers[16] improved the process of asymmetric electrodihydroxylation of olefins under constant current in an undivided cell using dihydroquinidine phthalazine ligand L2 and less osmate catalyst and potassium ferricyanide (Scheme 1b). A year later Torii and co-workers[17] developed a variant employing iodine as an oxidizing mediator (Scheme 1c). The desired diols were obtained in high ee values and high yields in each of these optimized processes.
Optically active epoxides are versatile intermediates in the synthesis of pharmaceuticals, agrochemicals, and functional materials.[18] Electrochemical asymmetric epoxidation has been investigated as an alternative process avoiding the use of a stoichiometric amount of co-oxidant.[19] In 2001, Tanaka and co-workers[20] reported an efficient enantioselective Mn(salen)-catalyzed electro-epoxidation of olefins in a two-phase CH2Cl2/NaCl (aq.) electrolysis system in an undivided cell (Scheme 2a). The electro-epoxi- dation of indene 3e gave the corresponding epoxide 4e in rather low yield and enantioselectivity. Due to the high reactivity of the double bond, which enabled undesired oxidations by an electro-generated chlorine species [Cl+].
The CH2Cl2/water two-phase system played an important role in the reaction. When using water-miscible organic solvents, such as t-BuOH and MeCN, a complex mixture of unidentified products was formed in lieu of the desired epoxide, possibly a result of decomposition of Mn(salen) via oxidation at the anode. In the two-phase system, electrochemical oxidation occurs only in the aqueous phase, thereby suppressing direct electrochemical oxidation of Mn(salen) which resides in the organic phase. A plausible mechanism is presented in Scheme 2b.
Recently, Lin and co-workers[21] reported the development of highly enantioselective cyanophosphinoylations or cyanosulfinylations of alkenes using rational ligand design and optimization of the electrolysis conditions (Scheme 3a). Substantial modification of the ligand scaffold was required to break a selectivity ceiling that they encountered. Introduction of an ancillary ligand to the BOX (bisoxazolines) scaffold (specifically an ester group) was used to further stabilize the putative pentacoordinated Cu(III) complex prior to reductive elimination. This modification also served to increase the rigidity of the transition state and improve the stereochemical fidelity of the cyanation process (Scheme 3b). Moreover, Lin and co-workers hypothesized that the multidentate ligand further stabilized the Cu catalyst against cathodic demetallation and present a cationic intermediate (I, Scheme 3b) that was more susceptible to reductive elimination.

Selective oxidation of hydroxyl groups into carbonyl groups is an important organic transformation. In 2008, Onomura and co-workers[22] reported a copper-catalyzed electrochemical oxidation kinetic resolution of racemic cis-cycloalkane-1, 2-diols, aminoalcohols, and aminoaldehydes. Their efficient procedure employed copper(II) triflate and (R, R)-Ph-BOX ligand with tetraethylammonium bromide as the oxidizing mediator (Scheme 4). The method was applicable to the kinetic resolution of some piperidine-3, 4-diols (rac-10 and rac-12, Schemes 4b and 4c respectively). It bears mentioning that these substrates are not efficiently oxidized by NBS (N-Bromosuccinimide).[23]
In 1975, Shono and co-workers[25] demonstrated an an-odic oxidation of carbamates to N-carbamoyl iminium ions.[24] The resulting N-carbamoyl iminium ions can be trapped with various nucleophiles, including allyl silanes, cyanide, fluoride, furans, and isocyanides. This provided a powerful tool for the α-C—H functionalization of amines.[26] Early attempts at asymmetric Shono-type oxidative cross-couplings relied on chiral auxiliaries and afforded varying levels of stereoselectivity when using alkyl nucleophiles.[27] In 2017, Luo and coworkers[28] developed a protocol using enamine catalysis and electrochemical C—H oxidation, providing C1-alkylated tetrahydroisoquinolines (THIQs) in good-to-excellent di- astereoselectivity and enantioselectivity.
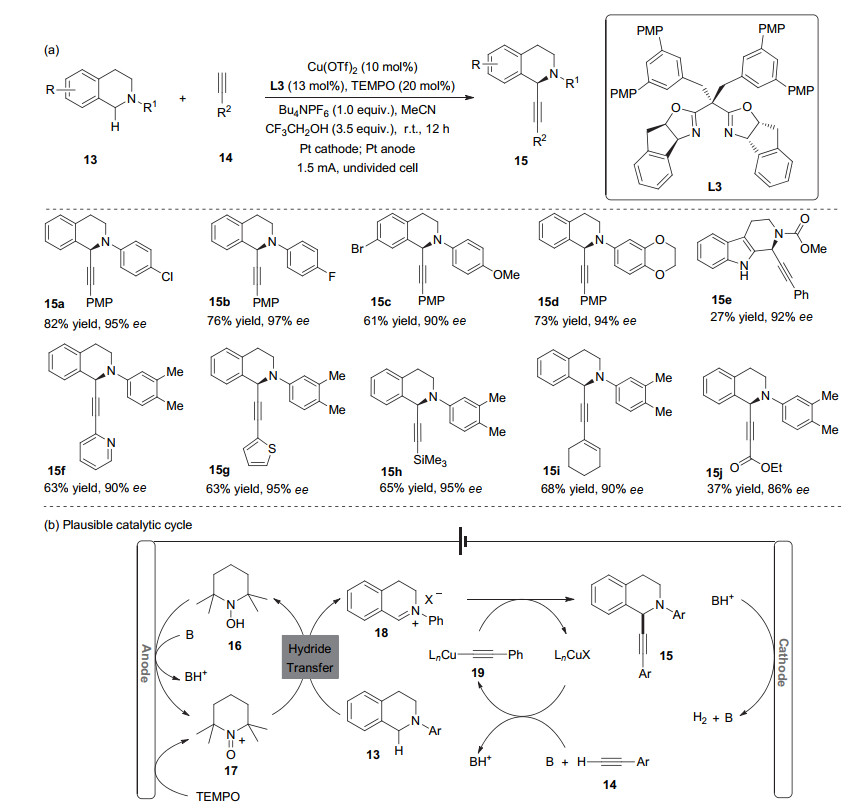
Very recently, Mei and co-workers[29] reported the first example of Cu/TEMPO (2, 2, 6, 6-tetramethylpiperidinyl- N-oxyl) co-catalyzed electrochemical enantioselective oxidative cross-coupling between cyclic tertiary amines and terminal alkynes using a novel chiral bisoxazoline ligand in an undivided cell (Scheme 5a). The reaction exhibited high enantioselectivity and broad functional group tolerance. The use of TEMPO as a co-catalytic redox mediator was crucial not only for oxidizing a tetrahydroisoquinoline to an iminium ion species, but also for dimishing the oxidation potential of the reaction. A plausible catalytic cycle is shown in Scheme 5b. First, oxoammonium species (TEMPO+) 17 is formed via anodic oxidation of TEMPO. Upon hydride transfer, [30] iminium intermediate 20 is generated from 13, with the concomitant formation of TEMPO-H 16, which can be converted into 17 by anodic oxidation. Addition of the chiral acetylide species 19 (derived from Cu(I) and alkyne 14 in the presence of base) to electrophilic 18 results in the formation of product 15. Active Cu(I) could be generated by either the reaction of Cu(II) with TEMPO-H, or cathodic reduction of Cu(II).[31] Alternatively, other cooperative modes involving both Cu(II) and TEMPO in the key oxidative iminium-formation step could not be ruled out.[32]
In 2019, Meggers and co-workers[33] reported an electricity-driven asymmetric ∆-Rh-catalyzed oxidative cross- coupling of 2-acyl imidazoles with silyl enol ethers for the generation of enantioenriched 1, 4-dicarbonyls in high yield and high enantionselectivity, including access to quaternary stereocenters (Scheme 6a). The reaction exhibited broad functional group tolerance. The chiral Lewis acid catalyst ∆-Rh was shown to be involved in both the electrochemical step and the asymmetric induction. Substrate binding to the catalyst and subsequent deprotonation raised the highest occupied molecular orbital and triggered a mild and selective anodic oxidation to provide a catalyst-bound reactive intermediate that engaged in a stereo-controlled radical C— C bond-forming reaction. A plausible mechanism is shown in (Scheme 6b). Later in 2019, Guo and co-workers[34] reported a Ni-catalyzed asymmetric electrochemical alkylation via benzylic C—H bond functionalization (Scheme 7a). The imidazole protecting group in substrate 23 proved crucial for the transformation, as there was no alkylation product with other protecting groups. The electrochemical alkylation products were then applied to the synthesis of target compound 26, which has antibacterial activity (Scheme 7b).[35] The proposed mechanism of the reaction is depicted in Scheme 7c. First, the coordination of Ni-catalyst to substrate 23 affords intermediate F, which is deprotonated to afford G. Intermediate G is then oxidized at the anode to form the radical intermediate H, which cross-coupled to electrochemically generated benzylic radical J to afford the alkylation product 25.
Very recently, Ackermann and co-workers published the first electrochemical enantioselective synthesis of axially-chiral biaryls, which used a catalytic amount of transient directing group (TDG) in Pd-catalyzed asymmetric electrochemical C—H olefinations. This protocol afforded highly enantiomerically-enriched biaryls and axially chiral C—N bond scaffolds that the authors showed could be used to expediently access to novel enantio-enriched helicenes, dicarboxylic acids, and 1, 1′-Binaphthalene-2, 2'-diyl)bis- (diphenylphosphine) (BINOLs) (Scheme 8).[36]
Catalytic carbon-carbon (C—C) bond-forming reactions that utilize carbon dioxide (CO2) to directly access carbox-ylic acids have received extensive attention in recent decades due to the attractiveness of CO2 as a C1 synthon.
In 2014, Lu and co-workers[37] reported the first investigation of asymmetric electrocarboxylation of 1-phenylethyl chloride catalyzed by an electrogenerated chiral Co(salen) complex. 2-Phenylpropionic acid was afforded in 37% yield and 83% ee (Scheme 9a). Although low yielding, this study laid the foundation for further research.
In 2018, Mei and co-workers[38] reported the first Pd-cata- lyzed regioselective carboxylation of homostyrenyl acetates utilizing electric current as the reductant, and also demonstrated the first catalytic asymmetric carboxylation of cinnamyl acetate, which provided a heretofore unavailable means to construct chiral phenyl acetic acids from homostyrenyl acetates (Scheme 9b). The proposed mechanism for this Pd-catalyzed carboxylation reaction via electrochemical reduction is shown in Scheme 9c.
In 2002 Duñach and co-workers[39] reported the electro- reductive cleavage of the allyl group of allyl 2-methyl-1- tetralone-2-carboxylate which afforded optically active 2-methyl-1-tetralone in low enantioselectivity in the presence of a chiral nickel-complex (Scheme 10). The reaction probably proceeded through the electrochemical generation of Ni(0) complex, which added oxidatively to the C—O bond of the allyl ester group of 40 to form a π-allyl-Ni(II) complex. The formation of 41 and 42 could be the result of deallylation with or without decarboxylation, and 43 was formed from an intramolecular allyl transfer reaction to the carbonyl group.
Transition metal-catalyzed reductive cross-coupling is a powerful strategy for the formation of carbon-carbon bonds. However, a superstoichiometric proportion of metal powder is often needed as a reductant for catalyst turnover. In 2019 Reisman and co-workers[40] reported the first asymmetric electrochemical reductive cross-coupling of alkenyl bromides and benzyl chlorides with a nickel catalyst and chiral ligand L9. The reaction exhibited high efficiency, high enantioselectivity, and broad functional group tolerance (Scheme 11).
Very recently, Mei and co-workers[41] reported the first example of a Ni-catalyzed enantioselective electrochemical reductive coupling of aryl bromides under very mild conditions. This process afforded axially chiral BINOL derivatives in good yield and enantiomeric excess (Scheme 12). Common metal reductants such as Mn or Zn powder resulted in significantly lower yields in the absence of electric current under otherwise identical conditions, which underscores the enhanced reactivity enabled by the combination of transition metal catalysis and electrochemistry.
This review summarized recent progress in asymmetric organotransition metal-catalyzed electrochemistry (AOM- CE). Chiral ligands combined with transition metals in the presence of electricity have been established for efficient enantioselective synthesis. Although AOMCE is readily understood, designed, and implemented, examples of AOMCE are rare, some challenges still need to be addressed. For example, AOMCE methods limited to Os, Mn, Cu, Rh, Co, Pd, or Ni catalysis, thus the design of diversified catalysts is highly desired. Furthermore, free metal ions are prone to accept electrons from cathode to yield inert zero-valent metals, which result in poor yields or no reaction at all. We hope that this review provides helpful overview of the current state of AOMCE. We anticipate that the problems mentioned above will be solved in the near future after further investigation, and novel AOMCE methods will also be developed with some regularity.
Kolbe, H. J. Prakt. Chem. 1847, 41, 137. doi: 10.1002/prac.18470410118
Baizer, M. M. J. Electrochem. Soc. 1964, 111, 215. doi: 10.1149/1.2426086
(a) Wang, F.; Stahl, S. S. Acc. Chem. Res. 2020, 53, 561.
(b) Siu, J. C.; Fu, N.; Lin, S. Acc. Chem. Res. 2020, 53, 547.
(c) Fuchigami, T.; Inagi, S. Acc. Chem. Res. 2020, 53, 322.
(d) Flexer, V.; Jourdin, L. Acc. Chem. Res. 2020, 53, 311.
(e) Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300.
(f) Robinson, S.; Sigman, M. S. Acc. Chem. Res. 2020, 53, 289.
(g) Jing, Q.; Moeller, K. D. Acc. Chem. Res. 2020, 53, 135.
(h) Leech, M. C.; Lam, K. Acc. Chem. Res. 2020, 53, 121.
(i) Yamamoto, K.; Kuriyama, M.; Onomura, O. Acc. Chem. Res. 2020, 53, 105.
(j) Ackermann, L. Acc. Chem. Res. 2020, 53, 84.
(k) Kingston, C.; Palkowitz, M. D.; Takahira, Y.; Vantourout, J. C.; Peters, B. K.; Kawamata, Y.; Baran, P. S. Acc. Chem. Soc. 2020, 53, 72.
(l) Röckl, J. L.; Pollok, D.; Franke, R.; Waldvogel, S. R. Acc. Chem. Res. 2020, 53, 45.
(m) Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339.
(n) Yuan, Y.; Lei, A. Acc. Chem. Res. 2019, 52, 3309.
(o) Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485.
(p) Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338.
(q) Ma, C.; Fang, P.; Mei, T.-S. ACS Catal. 2018, 8, 7179.
(r) Karkas, M. D. Chem. Soc. Rev. 2018, 47, 5786.
(s) Horn, E. J.; Rosen, B. R.; Baran, P. S. ACS Cent. Sci. 2016, 2, 302.
(a) Hou, Z.-W.; Xu, H.-C. Chin. J. Chem. 2020, 38, 394.
(b) Ye, Z.; Zhang, F. Chin. J. Org. Chem. 2020, 40, 241 (in Chinese).
(叶增辉, 张逢质, 有机化学, 2020, 40, 241.)
(c) Qian, X.-Y.; Xiong, P.; Xu, H.-C. Acta Chim. Sinica 2019, 77, 879 (in Chinese).
(钱向阳, 熊鹏, 徐海超, 化学学报, 2019, 77, 879.)
(d) Yang, Q.-L.; Wang, X.-Y.; Weng, X.-J.; Yang, X.; Xu, X.-T.; Tong, X.; Fang, P.; Wu, X.-Y.; Mei, T.-S. Acta Chim. Sinica 2019, 77, 866 (in Chinese).
(杨启亮, 王向阳, 翁信军, 杨祥, 徐学涛, 童晓峰, 方萍, 伍新燕, 梅天胜, 化学学报, 2019, 77, 866.)
(e) Zhang, H. Tang, R.; Shi, X.; Xie, L.; Wu, J. Chin. J. Org. Chem. 2019, 39, 1837 (in Chinese).
(张怀远, 唐蓉萍, 石星丽, 颉林, 伍家卫, 有机化学, 2019, 39, 1837.)
(f) Feng, E.-Q.; Hou, Z.-W.; Xu, H.-C. Chin. J. Org. Chem. 2019, 39, 1424 (in Chinese).
(冯恩祺, 侯中伟, 徐海超, 有机化学, 2019, 39, 1424.)
(g) Zhou, Z.; Yuan, Y.; Cao, Y.; Qiao, J.; Yao, A.; Zhao, J.; Zou, W. Chen, W.; Lei, A. Chin. J. Chem. 2019, 37, 611.
(h) Lu, F.; Yang, Z.; Wang, T.; Wang, T.; Zhang, Y.; Yuan, Y.; Lei, A. Chin. J. Chem. 2019, 37, 547.
(i) Hou, Z.-W.; Yan, H.; Song, J.; Xu, H.-C. Chin. J. Chem. 2018, 36, 909.
(j) Wu, Y.; Xi, Y.; Zhao, M.; Wang, S. Chin. J. Org. Chem. 2018, 38, 2590 (in Chinese).
(吴亚星, 席亚超, 赵明, 王思懿, 有机化学, 2018, 38, 2590.)
(a) Chang, X.; Zhang, Q.; Guo, C. Angew. Chem., Int. Ed. 2020, 59, 12612.
(b) Ghosh, M.; Shinde, V. S.; Rueping, M. Beilstein J. Org. Chem. 2019, 15, 2710.
(c) Lin, Q.; Li, L.; Luo, S. Chem. Eur. J. 2019, 25, 10033.
Seebach, D.; Oei, H. A. Angew. Chem., Int. Ed. 1975, 14, 634.
(a) Louafi, F.; Moreau, J.; Shahane, S.; Golhen, S.; Roisnel, T.; Sinbandhit, S.; Hurvois, J.-P. J. Org. Chem. 2011, 76, 9720.
(b) Feroci, M.; Inesi, A.; Orsini, M.; Palombi, L. Org. Lett. 2002, 4, 2617.
(a) Kodama, Y.; Fujiwara, A.; Kawamoto, H.; Ohta, N.; Kitani, A.; lto, S. Chem. Lett. 2001, 30, 240.
(b) Horner, L.; Degner, D. Tetrahedron Lett. 1971, 12, 1241.
(a) Shiigi, H.; Mori, H.; Tanaka, T.; Demizu, Y.; Onomura, O. Tetrahedron Lett. 2008, 49, 5247.
(b) Kuroboshi, M.; Yoshihisa, H.; Cortona, M. N.; Kawakami, Y.; Gao, Z.; Tanaka, H. Tetrahedron Lett. 2000, 41, 8131.
(c) Kashiwagi, Y.; Kurashima, F.; Kikuchi, C.; Anzai, J.; Osa, T.; Bobbitt, J. M. Chem. Commun. 1999, 1983.
(a) Kashiwagi, Y.; Kurashima, F.; Chiba, S.; Anzai, J.; Osa, T.; Bobbitt, J. M. Chem. Commun. 2003, 114.
(b) Moutet, J. C.; Duboc-Toia, C.; Ménage, S.; Tingry, S. Adv. Mater. 1998, 10, 665.
(c) Komori, T.; Nonaka, T. J. Am. Chem. Soc. 1984, 106, 2656.
(d) Firth, B. E.; Miller, L. L. J. Am. Chem. Soc. 1976, 98, 8272.
(e) Watkins, B. F.; Behling, J. R.; Kariv, E.; Miller, L. L. J. Am. Chem. Soc. 1975, 97, 3549.
Gourley, R. N.; Grimshaw, J.; Millar, P. G. Chem. Commun. 1967, 1278.
(a) Kobayashi, S.; Sugiura, M. Adv. Synth. Catal. 2006, 348, 1496.
(b) Lohray, B. B. Tetrahedron: Asymmetry 1992, 3, 1317.
(a) Sugimoto, H.; Kitayama, K.; Mori, S.; Itoh, S. J. Am. Chem. Soc. 2012, 134, 19270.
(b) Metin, O.; Alp, N. A.; Akbayrak, S.; Bicer, A.; Gultekin, M. S.; Ozkar, S.; Bozkaya, U. Green Chem. 2012, 14, 1488.
(c) Sugimoto, H.; Kitayama, K.; Mori, S.; Itoh, S. J. Am. Chem. Soc. 2012, 134, 19270.
(a) Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2024.
(b) Kolb, H. C.; VanNieuwenhze, M. S.; Sharpless, K. B. Chem. Rev. 1994, 94, 2483.
(c) Katsuki, T.; Sharpless, K. B. J. Am. Chem. Soc. 1980, 102, 5974.
Amundsen, R.; Balko, E. N. Appl. J. Electrochem. 1992, 22, 810. doi: 10.1007/BF01023722
Torii, S.; Liu, P.; Tanaka, H. Chem. Lett. 1995, 24, 319. doi: 10.1246/cl.1995.319
Torii, S.; Liu, P.; Bhuvaneswari, N.; Amatore, C.; Jutand, A. J. Org. Chem. 1996, 61, 3055. doi: 10.1021/jo952137r
Noyori, R. Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994.
Guo, P.; Wong, K.-Y. Electrochem. Commun. 1999, 1, 559. doi: 10.1016/S1388-2481(99)00110-1
Tanaka, H.; Kuroboshi, M.; Takeda, H.; Kanda, H.; Torii, S. J. Electroanal. Chem. 2001, 507, 75. doi: 10.1016/S0022-0728(01)00387-4
Fu, N.; Song, L.; Liu, J.; Shen, Y.; Siu, J. C.; Lin, S. J. Am. Chem. Soc. 2019, 141, 14480. doi: 10.1021/jacs.9b03296
Minato, D.; Arimoto, H.; Nagasue, Y.; Demizu, Y.; Onomura, O. Tetrahedron. 2008, 64, 6675. doi: 10.1016/j.tet.2008.05.015
Onomura, O.; Arimoto, H.; Matsumura, Y.; Demizu, Y. Tetrahedron Lett. 2007, 48, 8668. doi: 10.1016/j.tetlet.2007.10.014
Shono, T.; Hamaguchi, H.; Matsumura, Y. J. Am. Chem. Soc. 1975, 97, 4264. doi: 10.1021/ja00848a020
(a) Jones, A. M.; Banks, C. E. Beilstein J. Org. Chem. 2014, 10, 3056.
(b) Onomura, O. Heterocycles 2012, 85, 2111.
(c) Shono, T. Top. Curr. Chem. 1988, 148, 1.
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230. doi: 10.1021/acs.chemrev.7b00397
(a) D'Oca, M. G. M.; Pilli, R. A.; Pardini, V. L.; Curi, D.; Comni- nos, F. C. M. J. Braz. Chem. Soc. 2001, 12, 507.
(b) Sierecki, E.; Errasti, G.; Martens, T.; Royer, J. Tetrahedron 2010, 66, 10002.
(c) Lee, D.-S. Tetrahedron: Asymmetry 2009, 20, 2014.
(d) Shankaraiah, N.; Pilli, R. A.; Santos, L. S. Tetrahedron Lett. 2008, 5098.
(e) Zelgert, M.; Nieger, M.; Lennartz, M.; Steckhan, E. Tetrahedron 2002, 58, 2641.
(f) Matsumura, Y.; Kanda, Y.; Shirai, K.; Onomura, O.; Maki, T. Tetrahedron 2000, 56, 7411.
Fu, N.; Li, L.; Yang, Q.; Luo, S. Org. Lett. 2017, 19, 2122. doi: 10.1021/acs.orglett.7b00746
Gao, P.-S.; Weng, X.-J.; Wang, Z.-H.; Zheng, C.; Sun, B.; Chen, Z.-H.; You, S.-L.; Mei, T.-S. Angew. Chem., Int. Ed. 2020, 59, 15254. doi: 10.1002/anie.202005099
(a) Lennox, A. J. J.; Geos, S. L.; Webster, M. P.; Koolman, H. F.; Djuric, S. W.; Stahl, S. S. J. Am. Chem. Soc. 2018, 140, 11227.
(b) Wang, F.; Rafiee, M.; Stahl, S. S. Angew. Chem., Int. Ed. 2018, 57, 6686.
(c) Wu, Y.; Yi, H.; Lei, A. ACS Catal. 2018, 8, 1192.
(d) Li, C.; Zeng, C.-C.; Hu, L.-M.; Yang, F.-L.; Yoo, S. J.; Little, R. D. Electrochim. Acta 2013, 114, 560.
(e) Cao, Y.; Suzuki, K.; Tajima, T.; Fuchigami, T. Tetrahedron 2005, 6854.
Ryan, M. C.; Whitemire, L. D.; Mccann, S. D.; Stahl, S. S. Inorg. Chem. 2019, 58, 10194. doi: 10.1021/acs.inorgchem.9b01326
Badalyan, A.; Stahl, S. S. Nature 2016, 535, 406. doi: 10.1038/nature18008
Huang, X.; Zhang, Q.; Lin, J.; Harms, K.; Meggers, E. Nat. Catal. 2019, 2, 34. doi: 10.1038/s41929-018-0198-y
Zhang, Q.; Chang, X.; Peng, L.; Guo, C. Angew. Chem., Int. Ed. 2019, 58, 6999. doi: 10.1002/anie.201901801
Lu, P.; Jackson, J. J.; Eickhoff, J. A.; Zakarian, A. J. Am. Chem. Soc. 2015, 137, 656. doi: 10.1021/ja512213c
Dhawa, U.; Tian, C.; Wdowik, T.; Oliveira, J. C. A.; Hao, J.; Ackermann, L. Angew. Chem., Int. Ed. 2020, 59, 13451. doi: 10.1002/anie.202003826
Chen, B.-L.; Zhu, H.-W.; Xiao, Y.; Sun, Q.-L.; Wang, H.; Lu, J.-X. Electrochem. Commun. 2014, 42, 55. doi: 10.1016/j.elecom.2014.02.009
Jiao, K.-J.; Li, Z.-M.; Xu, X.-T.; Zhang, L.-P.; Li, Y.-Q.; Zhang, K.; Mei, T.-S. Org. Chem. Front. 2018, 5, 2244. doi: 10.1039/C8QO00507A
Franco, D.; Riahi, A.; Hénin, F.; Muzart, J.; Duñach, E. Eur. J. Org. Chem. 2002, 2257.
DeLano, T. J.; Reisman, S. E. ACS Catal. 2019, 9, 6751. doi: 10.1021/acscatal.9b01785
Qiu, H.; Shuai, B.; Wang, Y.-Z.; Liu, D.; Chen, Y.-G.; Gao, P.-S.; Ma, H.-X.; Chen, S.; Mei, T.-S. J. Am. Chem. Soc. 2020, 142, 9872. doi: 10.1021/jacs.9b13117

Scheme 3 Cu-catalyzed asymmetric electro-cyanophosphinoylation or -cyanosulfinylation of alkenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:










 下载:
下载:









