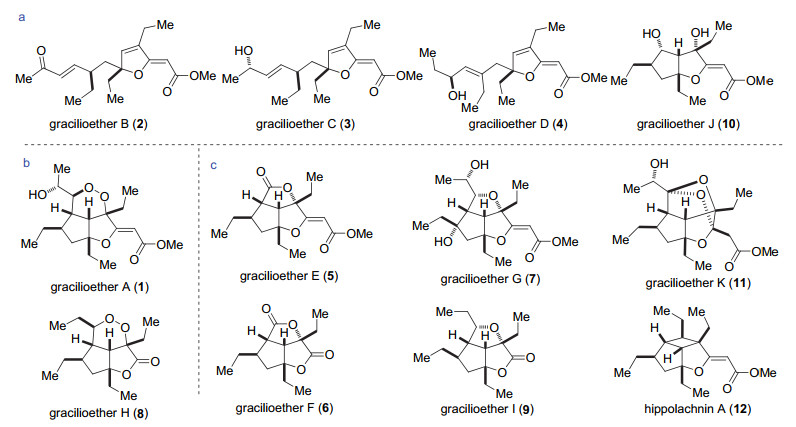
图 1.
gracilioethers A~K以及hippolachnin A的结构
Figure 1.
Structures of gracilioethers A~K and hippolachnin A

海洋是天然产物的宝库, 目前已经从海洋中分离出数以万计的天然产物, 其中很多表现出抗病毒、抗肿瘤和抗心脑血管病等活性[1]. 2009年Fusetani小组[2]从日本南部的深海海绵Agelas gracilis中分离得到三个具有抗疟疾活性(IC50 0.5~10 μg/mL)的天然产物gracilioethers A~C (1~3)(图 1), 其中gracilioether B (2)还表现出对利士曼原虫的生长抑制作用. 2012年, Zampella等[3-4]从南太平洋海绵Plakinastrella mamillaris中分离得到了gracilioethers D~K (4~11), 其中gracilioether H (9)具有较好的抗疟疾活性, gracilioethers E, G, I, K具有潜在的抗炎活性.林厚文小组[5]从南海海绵Hippospongia lachne中分离得到了hippolachnin A (12), 该天然产物具有很好的抗真菌活性, 其对三种致病真菌Cryptococcus neoformans, Trichophyton rubrum和Microsporum gypseum的最低抑菌浓度(MIC)均为0.41 μmol•L-1.
如图 1a所示, gracilioether家族包括一部分单环和双环结构的天然产物gracilioethers B, C, D, J (2, 3, 4, 10), 这几个天然产物很有可能是gracilioether家族其它天然产物的生源前体, 它们可以通过分子内环加成过程转化成家族中其它天然产物[5]. Gracilioether家族还包含具有5/5/6三环高张力核心骨架的天然产物gracilio- ethers A (1), H (8)(图 1b)和5/5/5三环核心骨架gracilio- ethers E~G, I, K (5~7, 9, 11)(图 1c).它们的核心骨架包含5~6个连续手性中心, 其中gracilioether K具有氧杂七元环结构. Hippolachnin A (12)具有碗状5/5/4三环高张力核心骨架, 与gracilioether类具有三环骨架的天然产物相比, 除具有的四元环结构不同以外, 手性中心、取代基都非常相似.如果将hippolachnin A中的四元环转化为五元内酯环, 就能够得到gracilioether类天然产物的骨架.
通过对gracilioethers与hippolachnin A的结构分析, 合成化学家采用了多种方式来构建这类天然产物骨架.出于合成发散性及hippolachnin A骨架可能转化到gracilioethers骨架两方面考虑, 如何高效合成hippolachnin A的5/5/4骨架尤为重要, 其中最具挑战性的是环丁烷结构的合成.构建环丁烷的常用方法包括光化学过程的分子间或分子内[2+2]环加成反应和路易斯酸催化的烯酮与烯烃之间的[2+2]环加成反应等[6].非共轭的烯烃直接参与光化学过程的[2+2]环加成反应可能存在激发能量高及单线态寿命短的问题.通常需要加入光敏剂等降低反应所需的激发能, 或者通过加入金属试剂与烯烃形成配合物, 使激发非共轭烯烃的最大吸收峰红移[6c].而共轭的烯烃可以通过紫外光直接激发, 经过系间窜越(ISC)过程达到三线态[6a-6b], 具有较长的激发态寿命, 有利于与另一个烯烃发生[2+2]环加成反应.在路易斯酸促进下, 烯酮参与的[2+2]环加成反应, 由于其特异的电子效应及通过对取代基的调控, 通常表现出比较好的区域选择性与立体选择性.另外, 由于四元环的环张力较大, 容易发生开环或者扩环过程, 所以完成四元环的构建后还需要考虑其稳定性问题.针对gracilioethers和hippolachnin A中环丁烷的构建和稳定性问题, 在合成研究的过程中, 合成化学家通过不断研究和探索, 提出了各种方案:比如通过对底物官能团、取代基等修饰进而改变反应底物的电性及位阻; 或者通过加入催化剂、光敏剂来调控反应底物的活性、反应过渡态和反应位点的空间相对位置.
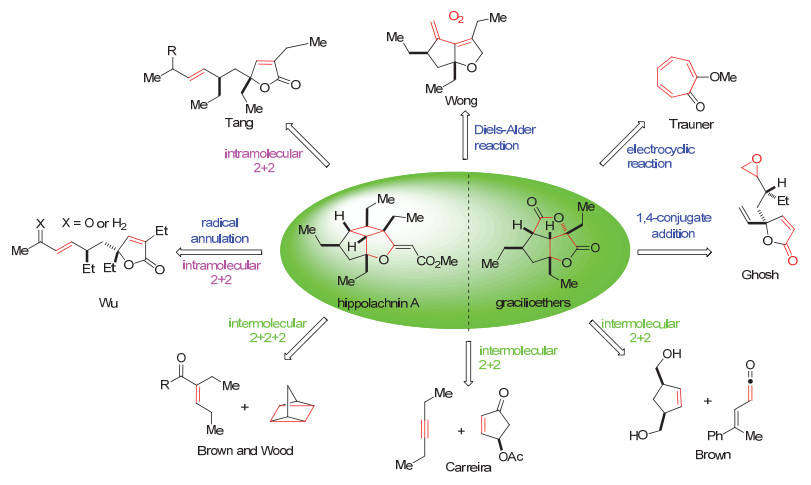
本文根据作者采用的关键步分为三类, 分别为分子间[2+2]环加成类型的合成、分子内[2+2]环加成类型的合成和其它合成策略. 2014年Brown小组[7]首次报道了利用分子间的[2+2]环加成反应作为关键步骤, 完成gracilioether F (6)的全合成.利用hippolachnin A (12)与gracilioether类具有三环骨架的天然产物骨架的相似性, Carreira小组[8-9]分别在2015年和2016年以分子间的[2+2]与分子内的ene反应作为关键步骤完成了hippolachnin A (12)和gracilioethers E (5), F (6)的全合成. 2016年, Brown小组和Wood小组[10]共同报道了以[2+ 2+2]环加成反应和C(sp3)—H键氧化为关键步骤的hippolachnin A (12)的合成. 2017年, 伍贻康小组[11]以分子内[2+2]环加成反应作为关键步骤完成了(±)-hippolachnin A (12)的全合成, 通过自由基环化反应完成了(±)-gracilioethers F (6), I (9)的全合成. 2018年唐叶峰小组[12]以光催化的分子内[2+2]环加成反应作为关键步骤完成了(+)-gracilioether A (1), (-)-gracilioethers E (5), F (6)和(+)-hippolachnin A (12)的合成. 2016年, Wong小组[13]利用单线态氧气参与的Diels-Alder环加成策略完成了(±)-gracilioether F (6)的合成. 2017年Trauner小组[14]以环庚三烯酮光照下发生的电环化反应为关键步骤合成了(±)-hippolachnin A (12). 2017年, Ghosh小组[15]通过分子内自由基1, 4-共轭加成构建骨架的策略完成了gacilioether F (6)的合成(Scheme 1).
通过分子间[2+2]环加成构建骨架, 可以构建5/4并环结构, 然后通过发生氧化环化或者其它成环反应完成实现骨架的合成.
2014年Brown小组[7]报道了gracilioether F (6)的首次全合成, 他们的策略是通过烯烃与烯酮的分子间[2+2]环加成构建5/4并环, 然后再通过C(sp3)—H键氧化构建五元内酯环, 完成gracilioether的合成.
Brown小组[7]从二醇底物13出发, 通过磺酰化和取代反应得到环戊烯14.酰氯15被路易斯酸三甲基铝活化, 然后在二异丙基乙胺作用下生成烯酮, 再与环戊烯14发生分子间[2+2]反应得到环丁酮16[16] (Scheme 2).烯酮的环加成反应本身可能有一些潜在问题, 例如烯酮自身不稳定, 有二聚问题, 烯基取代的烯酮会作为双烯体参与[4+2]反应等.研究表明, 烯酮参与的分子间[2+2]环加成反应如果是在路易斯酸或者路易斯碱催化下进行, 可以有效抑制烯酮自身的二聚问题[16f]. Brown小组目前的反应是在AlMe3, i-Pr2NEt参与下发生, 避免了烯酮的二聚. Brown小组[16a-16b, 16d]之前的研究还发现二取代的烯酮参与的反应具有较好的产率和区域选择性, 而单取代的烯酮由于活性太高而不稳定; 富电子或者缺电子且位阻较大的芳基烯酮具有较好的产率和选择性. Brown小组以酰氯15作为前体, 生成了烯基单取代的烯酮, 可能由于苯基和甲基的存在, 有一定的位阻作用, 所以不易作为双烯体发生[4+2]反应.而且苯基通过共轭作用, 更容易促进酰氯生成烯酮发生[2+2]反应, 提高了反应的整体活性.另外, 烯基取代的烯酮作为双烯体在参与[4+2]反应时, 其电性更类似于Danishefsky双烯这类富电子的双烯体[16e-16g], 需要缺电子的亲双烯体匹配, 才更容易发生[4+2]反应.而目前的亲双烯体14不是缺电子的烯烃, 并且反应是低温下进行, 这些因素可能也是[4+2]副反应不易发生的原因.
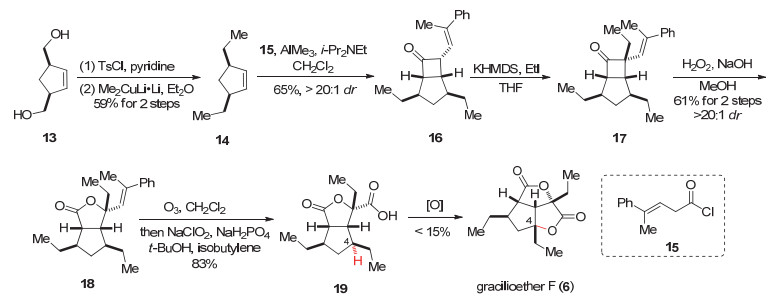
在得到环丁酮16后, 16发生α位烷基化得到17, 然后通过Baeyer-Villiger氧化得到18.化合物18发生臭氧化和Pinnick氧化得到羧酸19.接下来需要实现C(sp3)—H键氧化内酯化, 得到天然产物gracilioether F (6).从电子效应上讲, C(4)位远离拉电子基团, 其三级的C(sp3)—H键较为富电子, 在电性上是有利的.但是该位点恰好处于环系的凹面[17], 这使得其难以与外界的氧化剂作用.但基于较为有利的电子效应的考虑, 作者尝试了RuCl3, KBrO3, 吡啶, MeCN/H2O和甲基(三氟甲基)二氧杂环丙烷(TFDO), 二氯甲烷(DCM)等条件, 发现只有极少量的产物生成.基于White等[18]在羧酸导向的C—H氧化方面的工作, 作者尝试了[Fe(PDP)], H2O2, MeCN等条件, 以9%产率得到产物, 同时有48%的原料被回收.将[Fe(PDP)]换为Cu(OAc)2, 可以得到15%的天然产物, 并有51%的原料被回收(Scheme 2).
Brown小组采用了烯酮与烯烃间的[2+2]环加成反应以及对C(sp3)—H键的氧化反应作为两个关键反应, 经过8步合成了天然产物gracilioether F (6).因为最后一步氧化的效率较低, 所以对C(sp3)—H键的氧化反应需要进一步的优化. Brown小组作为第一个设计合成gracilioether F (6)的小组, 策略巧妙简洁, 为其它课题组合成gracilioether家族天然产物起到了很好的借鉴作用.
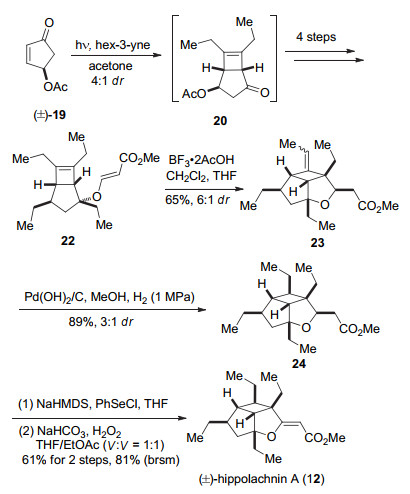
2015年, Carreira小组[8]通过分子间的[2+2]环加成反应构建了5/4并环, 然后通过分子内的ene反应完成五元内酯环的构建, 首次完成了(±)-hippolachnin A (12)的合成. 2016年Carreira小组[9]利用hippolachnin A (12)和gracilioethers E (5), F (6)骨架结构的关联性, 用类似的合成策略完成了(±)-gracilioethers E (5), F (6)的合成.
Carreira小组[8]首先以环戊烯酮19作为起始原料, 在光照条件下19与3-己炔发生分子间[2+2]反应得到环丁烯化合物20[19], 非对映选择性为4:1. 20经过4步转化生成化合物22.化合物22在BF3•2AcOH的作用下发生ene反应, 以较好的收率、区域选择性、立体选择性得到23[20].接下来在对双键还原时, 他们发现用自由基还原的方法主要得到非期望的立体选择性的产物, 而在Pd(OH)2/C, H2条件下可以以较好的选择性和产率获得24.最后通过对酯基α位硒化, 氧化消除构建双键, 得到天然产物(±)-hippolachnin A (12) (Scheme 3).
Carreira小组[9]在完成天然产物(±)-hippolachnin A (12)的基础上又完成了(±)-gracilioether F (6)的全合成.他们从化合物23出发, 对双键进行臭氧切断, 得到环丁酮化合物25和少量内酯化合物26.在间氯过氧苯甲酸(mCPBA)作用下发生Baeyer-Villiger氧化, 可以将环丁酮化合物25转化成26.然后经过硒化和氧化消除的过程构建双键得到天然产物(±)-gracilioether E (5).同时化合物21和多聚甲醛在Sc(OTf)3的催化下发生Prins环化反应构建5/5/4三环体系27, 经过臭氧切断双键和Baeyer-Villiger氧化构建内酯环29, 最后29在三氧化铬和乙酸的作用下, 进一步氧化得到天然产物(±)- gracilioether F (6) (Scheme 4).
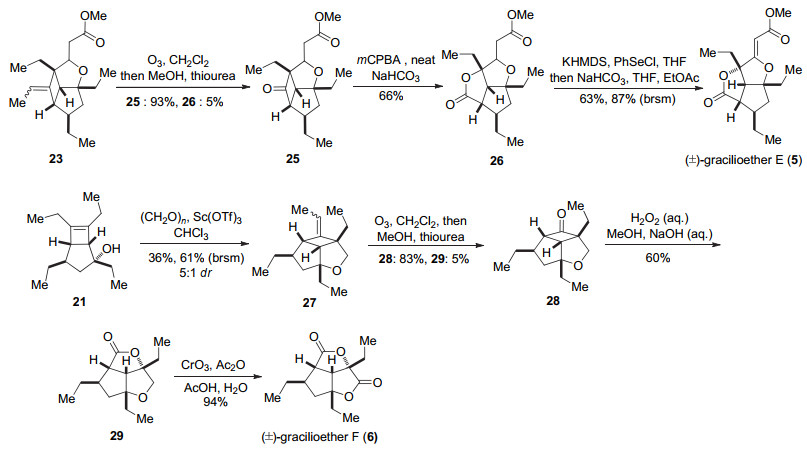
Carreira采用分子内的[2+2]环加成反应与ene反应作为关键反应, 以9步转化完成了(±)-hippolachnin A (12)的合成, 并且通过分析(±)-hippolachnin A (12)和gracilioether这类天然产物生源上的相互转化关系, 应用相似的合成策略完成了(±)-gracilioethers E (5)和F (6)的合成. Carreira通过化学合成实现了hippolachnin A (12)骨架到gracilioether F (6)骨架的转化, 为这些天然产物的生源合成假设提供了强有力的证据, 也为后续关于hippolachnin A与gracilioether家族天然产物的合成工作提供了很好的指导意义.
2016年, Brown和Wood小组[10]共同报道了hippolachnin A的合成.他们以四环庚烷和缺电子烯烃进行[2+2+2]环加成反应, 构建了[4.2.1.02, 5]桥环化合物, 之后经过开环及C(sp3)—H键氧化搭建内酯环, 完成了hippolachnin A (12)的合成.
Brown和Wood小组以四环庚烷34作为起始原料, 在光照下34和酰氯35发生[2+2+2]反应得到36[21].只有通过吸电子基团活化的烯烃才能更容易与四环庚烷发生环加成反应[21c], 鉴于对底物电性的要求, 作者采用羧酸衍生物中较为缺电子的不饱和酰氯35进行反应, 可以以较好的产率得到期望的产物.化合物36通过开环复分解反应(ROCM)打开环戊烯, 得到含两个端烯的5/4并环化合物37, 通过采用之前Brown生成内酯环的策略, 在White小组[22]发展的醋酸钯二亚砜加合物的催化下对C(4)位的C(sp3)—H键进行氧化, 以70%的收率得到化合物38, 38接着和乙酸叔丁酯缩合, 然后经过脱水、还原以及酯交换反应得到天然产物(±)-hippolachnin A (12) (Scheme 5).
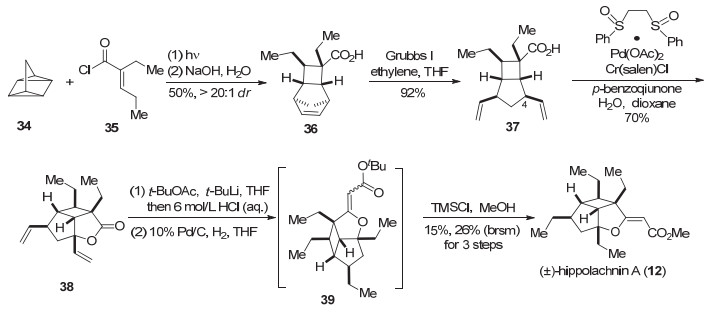
上述的合成策略较2014年Brown小组关于gracilio- ether F (6)的合成对于骨架构建效率更高, 并且解决了C(sp3)—H键氧化收率低的问题, 通过6步反应完成了(±)-hippolachnin A (12)的全合成. Brown和Wood小组通过合作, 融合了两个小组各自合成策略中的优势, 极大提高了整体合成效率, 也为各个研究小组开展科研上的合作起到了模范作用[23].
分子间的[2+2]反应可以高效地搭建5/4并环结构, 但是存在一些不足之处, 比如中间体烯酮的不稳定性. Brown小组与Wood小组在合成过程中也发现分子间[2+2+2]存在产率不高或者选择性不好的问题.从生源合成上分析, 天然产物hippolachnin A, gracilioethers A, E, F的骨架结构更可能是通过分子内的[2+2]反应构建.
2017年, 伍贻康小组[11]以相似的底物经过不同的反应构架了两类分子的骨架结构.他们以分子内[2+2]环加成反应构建了(±)-hippolachnin A (12)的骨架结构, 通过自由基环化反应构建了(±)-gracilioethers F (6), I (9)骨架结构.
他们从底物40和41出发, 通过4步转化得到γ-丁内酯42, 然后发生Julia-Lythgoe反应构建双键得到化合物43.化合物43通过Ohira小组[24]发展的方法, 在二异丁基氢化铝(DIBAL-H)还原下生成半缩醛, 然后与烯醇硅醚反应得到44, 随后在铜盐的参与下, 经过光照发生[2+2]反应构建得到三环骨架结构45.采用Carreira的方法可以得到天然产物(±)-hippolachnin A (12).或者是在光照条件下化合物43发生分子内[2+2]反应得到化合物46, 然后参考Wood小组和Brown小组的合成路线得到(±)- hippolachnin A (12) (Scheme 6).
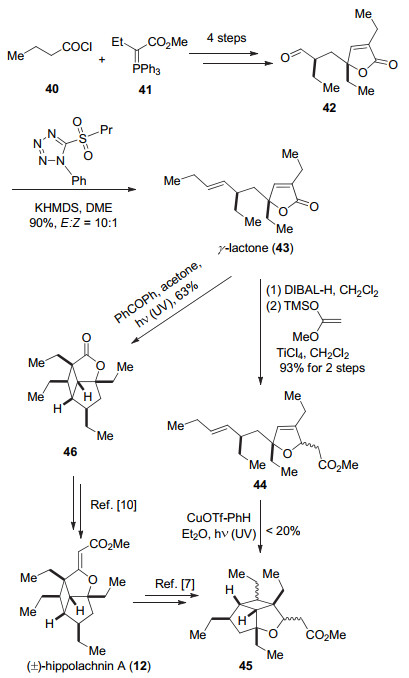
另外, 伍贻康小组通过化合物42的Horner- Wadsworth-Emmons (HWE)反应得到天然产物(±)- plakilactone (47). 47可以发生自由基1, 4-共轭加成构建5/5并环结构得到化合物48, 然后通过4步的官能团和取代基修饰得到化合物49, 再经过4步反应得到天然产物(±)-gracilioether F (6).而采用Brown小组的方法可以将49转化为天然产物(±)-gracilioether I (9). (Scheme 7).
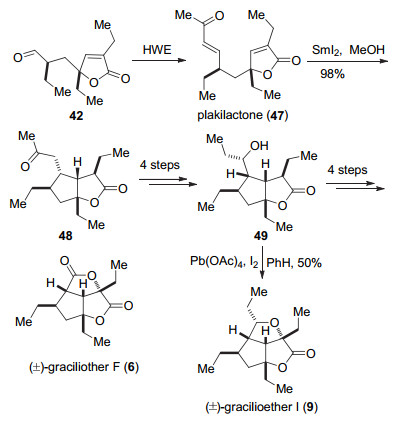
伍贻康小组通过对γ-丁内酯44的合成, 经过分子内[2+2]环加成可以高效构建了(±)-hippolachnin A (12)的三环骨架结构, 进一步证明生源合成假设.另外他们发现天然产物(±)-plakilactone (47)可以通过发生分子内的自由基加成反应得到天然产物(±)-gracilioether I (9)和(±)-gracilioether F (6)的骨架结构, 由于(±)-plakilactone (47)和gracilioethers D~K在同一种海绵中被发现[3-4], 伍贻康小组的合成策略为gracilioether这类天然产物可能的生源前体和生源转化提供了一定的证据.
唐叶峰小组[12]受生源合成启发, 以光促进的分子内[2+2]环加成反应为关键步骤, 2018年分别完成了(+)-gracilioether A (1), (-)-gracilioethers E (5), F (6)和(+)-hippolachnin A (12)的全合成.
化合物52生成烯醇硅醚后与化合物59在手性胺60的催化下, 发生不对称的Mukaiyama-Michael反应合成化合物54及其异构体化合物53.然后通过Wittig反应以70%收率及1:5的Z/E选择性得到混合物55.以二氯甲烷作为溶剂, 在三乙胺存在下用500 W的高压汞灯照射, 混合物55可以发生分子内[2+2]环加成反应, 得到单一构型的化合物56.化合物56可以在RuCl3/ NaIO4/乙腈超声作用下得到环丁酮57, 然后在间氯过氧苯甲酸和碳酸氢钠的作用下发生Baeyer-Villiger氧化将环丁酮部分氧化成内酯.遗憾的是, 反应的选择性不好, 得到了1:1的天然产物(-)-gracilioether F (6)与其异构体58.由于Brown和Carreira在相似的底物中应用该反应都有较好的选择性, 作者考虑到可能是环丁酮C(4)—C(10)键受周围羰基吸电子效应影响, 导致了反应的选择性不好.在尝试不同的氧化剂和添加剂后, 他们意外发现延长化合物56与RuCl3/NaIO4在超声作用下的时间, 两步反应可以通过一锅法完成, 并且只得到了(-)- gracilioether F (6) (Scheme 8).
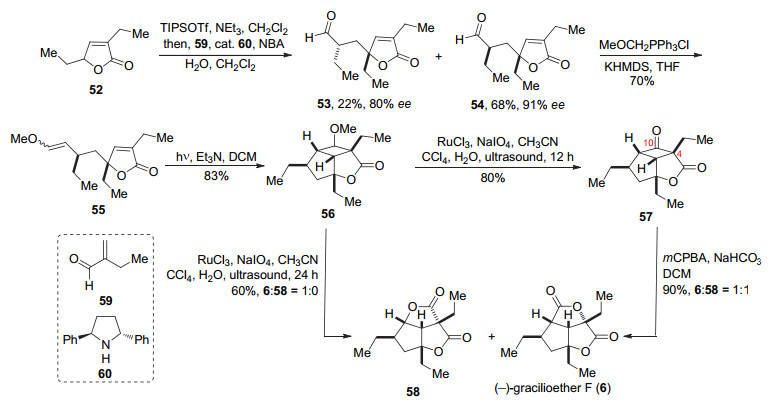
另外化合物54经过HWE反应得到化合物61, 在光照下, 发生分子内[2+2]环化反应得到62, 再经过6步转化得到化合物63, 然后在以Co(acac)2作为催化剂, 过氧叔丁醇和氧气作为氧化剂的条件下, 可以得到天然产物(+)-gracilioether A (1)和(-)-gracilioether E (5).化合物54经过5步转化可以得到64, 在汞灯的照射下发生分子内[2+2]环加成反应, 可以得到天然产物(+)- hippolachnin A (12) (Scheme 9).
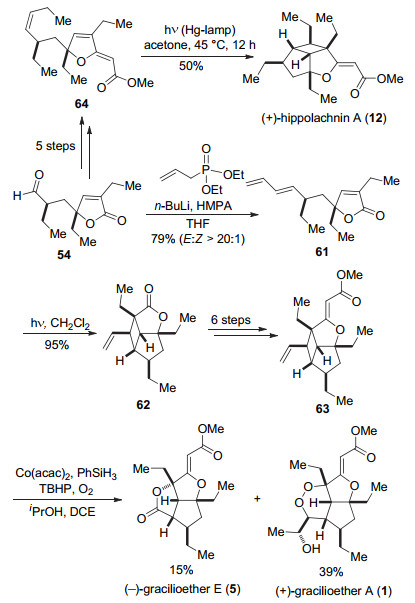
唐叶峰课题组合成了(+)-gracilioether A (1), (-)- gracilioethers E (5), F (6)和(+)-hippolachnin A (12)四个天然产物, 他们的策略是受γ-丁内酯类天然产物gracilioethers B~D (2~4)作为生源前体的生源合成假说的启发[5], 实现了这类天然产物的仿生合成.
合成化学家们主要采用了分子间和分子内两种类型的[2+2]环加成反应构建环丁烷体系, 其中Brown小组、Carreira小组以及Brown和Wood的合作研究都采用分子间途径.分子间环加成可能会存在反应条件剧烈和非对映、区域选择性差等问题. Brown小组采用路易斯酸促进下烯酮与烯烃之间的环加成反应, 因为电性匹配的原因, 反应较容易发生.并且通过底物取代基的位阻效应, 以较好的非对映选择性完成了环丁烷的构建. Carreira小组采用的炔与烯之间的环加成反应, 生成的是环丁烯体系, 可能因为反应中间体两个乙基取代基与底物中的OAc取代基相互作用不明显, 所以反应的非对映选择性不是特别好. Brown和Wood采用的四环庚烷参与的[2+2+2]环加成反应, 因为四环庚烷自身断键裂解需要的温度比较高, 导致了该反应的温度比较高.
而分子内[2+2]环加成反应, 由于反应位点在空间上比较接近, 较容易发生反应.而且由于底物本身的手性控制原因, 在区域和对映选择性方面会得到较好的控制, 这一点可以从伍贻康和唐叶峰小组的光化学过程的分子内[2+2]环加成反应的结果可以看到.但是底物为非共轭的烯烃时, 因为反应需要的能量比较高, 仍然需要金属试剂的参与, 以促进加成反应的进行.比如伍贻康小组在完成分子内[2+2]环加成过程中, 采用非共轭的烯烃时, 除了光化条件, 还需要加入铜盐, 才能促使期望的反应发生.原因可能一方面是Cu(I)与双键配位, 可以拉近反应官能团之间的空间距离.另外, 这样的络合物的形成, 能够更好地吸收光能, 诱使反应进行[24].
具有三环骨架的gracilioether家族天然产物和hippolachnin A生源合成上都可能经过了[2+2]环加成构建骨架结构, 很多课题组受生源合成启发设计了不同类型的[2+2]环加成反应策略, 完成对这类天然产物的全合成. Carreira和唐叶峰等课题组[8-9, 12]还根据这类天然产物骨架之间的联系, 实现了骨架之间的直接转化.而Wong, Trauner, Ghosh等课题组[10, 14-15]则采用了非[2+2]环加成反应构建骨架, 进而完成了天然产物的全合成.
2016年, Wong小组[13]通过单线态氧气参与的Diels- Alder环加成反应, 构建了包含过氧六元环在内5/5/6三环结构, 然后通过还原切断过氧键以及后续的转化完成了(±)-gracilioether F (6)的合成.
他们以联烯酸酯65为起始原料, 首先发生碘引发的内酯化反应得到内酯化合物66[25], 再通过5步转化引入端烯边链得到Heck反应前体67.然后发生Heck反应构建5/5并环得到化合物68.在亚甲基蓝作为光敏剂, 日光灯的照射下, 化合物68和氧气发生Diels-Alder反应得到过氧化合物69[26], 经过对双键的还原以及对过氧键还原切断得到化合物70, 最后借鉴Carreira小组的方法完成(±)-gracilioether F (6)的合成(Scheme 10).
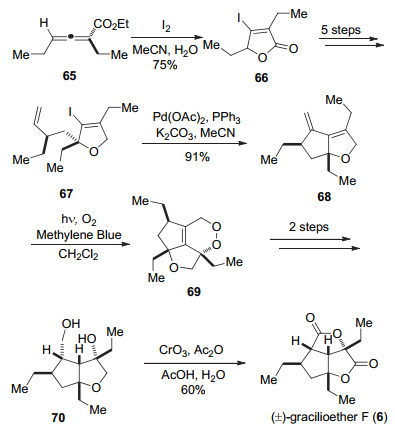
Wong小组通过11步反应完成了(±)-gracilioether F (6)的全合成.关键步骤是通过单线态氧气参与的[4+2]环加成反应构建了过氧六元环, 利用这一策略可以有效地将具有过氧环结构的gracilioether H (8)和家族内其它天然产物联系到一起, 为生源合成提供了一定支持.
通过生源合成过程来指导化学合成天然产物的策略是一种高效的途径, 同时按照可能的生源途径设计完成天然产物的合成, 也为生源合成提供证据.但是非仿生途径的化学合成有时候也比较高效, 比如Trauner小组创造性地通过非仿生过程设计并完成了hippolachnin A (12)的合成. 2017年, Trauner小组[14]通过环庚三烯酮衍生物发生电环化反应构建了5/4并环, 并且以非常好的非对映选择性实现了两个乙基的引入, 完成了对(±)-hippolachnin A (12)的全合成.
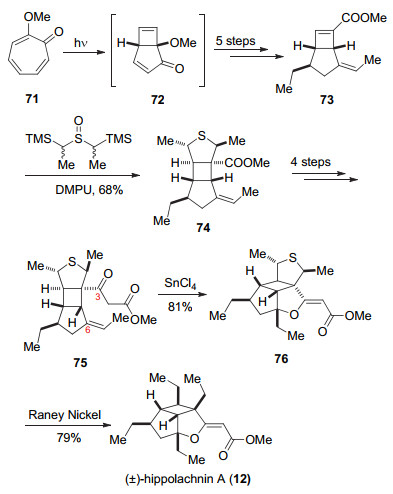
以环庚三烯酮衍生物71作为起始原料, 通过电环化反应构建5/4并环得到化合物72[27], 然后经过四步转化得到73, 接着通过与原位生成的硫代羰基叶立德发生分子间1, 3-偶极环加成反应得到化合物74.化合物74经过5步官能团转化得到化合物75, 在SnCl4作用下C(6)位生成三级碳正离子, 然后被分子内C(3)位生成的烯醇锡捕获, 发生关环得到76[28], 最后在Raney镍作用下脱硫得到天然产物(±)-hippolachnin A (12) (Scheme 11).
2017年, Ghosh小组[15]通过分子内自由基1, 4-共轭加成构建了5/5并环骨架, 然后巧妙地应用炔基加成, 完成了三级羟基的构建并且引入天然产物所需的碳原子, 并通过后续转化完成对(-)-gracilioether F (6)的合成.
Ghosh小组以化合物77作为起始原料经过还原、加成等官能团转化得到化合物78, 再经过烯烃复分解以及保护基的调整得到内酯79.在NaH的作用下发生分子内SN2反应生成环氧80.通过Cp2TiCl2和Zn作用原位生成一价Ti[29], 然后与化合物80中的环氧作用, 开环以后生成的二级自由基对内酯进行加成, 生成双环化合物81.化合物81经过4步转化可以得到α-羰基酯82, 通过炔基加成和去保护得到二醇化合物83, 经过Jones氧化、内酯化得到化合物84, 最后将炔基还原得到天然产物(-)-gracilioether F (6) (Scheme 12).
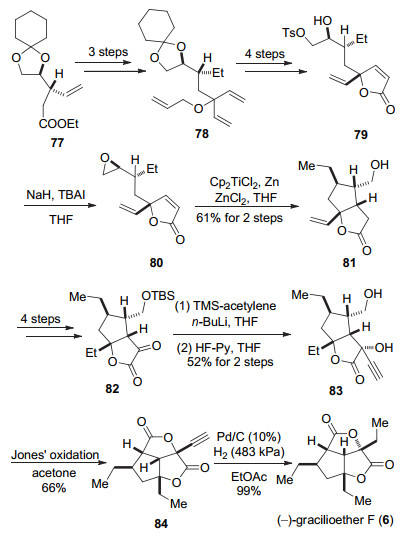
Ghosh小组通过一价钛引发的自由基反应完成了五元环的构建, 这一方法还能应用于gracilioether家族其它天然产物的合成.
除了以上总结的合成报道以外, gracilioether家族还有一些其它单环天然产物的合成研究包括gracilioethers B (2), C (3), 在这里没有总结. Brown小组最早采用[2+2]环加成反应的策略完成了gracilioether F (6)的首次全合成, Carreira小组也设计了[2+2]环加成的策略, 并且经过Baeyer-Villiger氧化实现了hippolachnin A (12)骨架与gracilioethers E (5), F (6)骨架的直接转化.相比于伍贻康和唐叶峰课题组的主要策略是仿生的分子内[2+2]环加成, Trauner等课题组采用非仿生的方法, 都十分高效地完成了对这类天然产物的全合成. Wood小组和Brown小组的相互合作, 结合各自合成路线的优点, 很大程度上提高了整体的合成效率.由于Gracilioether类天然产物和hippolachnin A的所表现出来的生物活性在药物开发领域有很好的前景, 我们相信将来随着新方法的发展或者新策略的应用, 会有更多高效实用的合成方法被报道.
Cragg, G. M.; Newman, D. J. Biochim. Biophys. Acta, Gen. Subj. 2013, 1830, 3670. doi: 10.1016/j.bbagen.2013.02.008
Ueoka, R.; Nakao, Y.; Kawatsu, S.; Yaegashi, J.; Matsumoto, Y.; Matsunaga, S.; Furihata, K.; Soest, R. W. M. V.; Fusetani, N. J. Org. Chem. 2009, 74, 4203. doi: 10.1021/jo900380f
Festa, C.; Lauro, G.; De Marino, S.; D'Auria, M. V.; Monti, M. C.; Casapullo, A.; D'Amore, C.; Renga, B.; Mencarelli, A.; Petek, S.; Bifulco, G.; Fiorucci, S.; Zampella, A. J. Med. Chem. 2012, 55, 8303. doi: 10.1021/jm300911g
(a) Festa, S.; De Marino, S.; D'Auria, M. V.; Deharo, E.; Gonzalez, G.; Deyssard, C.; Petek, S.; Bifulco, G.; Zampella, A. Tetrahedron 2012, 68, 10157.
(b) Festa, C.; D'Amore, C.; Renga, B.; Lauro, G.; De Marino, S.; D'Auria, M. V.; Bifulco, G.; Zampella, A.; Fiorucci, S. Mar. Drugs 2013, 11, 2314.
Piao, S.; Song, Y.; Jiao, W.; Yang, F.; Liu, X.; Chen, W.; Han, B.; Lin, H. Org. Lett. 2013, 15, 3526. doi: 10.1021/ol400933x
(a) Lee-Ruff, E.; Mladenova, G. Chem. Rev. 2003, 103, 1449.
(b) Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Chem. Rev. 2016, 116, 9748.
(c) Salomon, R. G. Tetrahedron 1983, 39, 485.
Rasik, C. M.; Brown, M. K. Angew. Chem., Int. Ed. 2014, 53, 14522. doi: 10.1002/anie.201408055
Ruider, S. A.; Sandmeier, T.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 2378. doi: 10.1002/anie.201410419
Ruider, S. A.; Carreira, E. M. Org. Lett. 2016, 18, 220. doi: 10.1021/acs.orglett.5b03356
McCallum, M. E.; Rasik, C. M.; Wood, J. L.; Brown, M. K. J. Am. Chem. Soc. 2016, 138, 2437. doi: 10.1021/jacs.5b13586
Xu, Z. J.; Wu, Y. Chem.-Eur. J. 2017, 23, 2026. doi: 10.1002/chem.201605776
Li, Q.; Zhao, K.; Peuronen, A.; Rissanen, K.; Enders, D.; Tang, Y. J. Am. Chem. Soc. 2018, 140, 1937. doi: 10.1021/jacs.7b12903
Shen, X.; Peng, X.; Wong, H. N. C. Org. Lett. 2016, 18, 1032. doi: 10.1021/acs.orglett.6b00161
Winter, N.; Trauner, D. J. Am. Chem. Soc. 2017, 139, 11706. doi: 10.1021/jacs.7b06815
Datta, R.; Ghosh, S. J. Org. Chem. 2017, 82, 7675. doi: 10.1021/acs.joc.7b01179
(a) Rasik, C. M.; Brown M. K. J. Am. Chem. Soc. 2013, 135, 1673.
(b) Rasik, C. M.; Brown, M. K. Synlett 2014, 760.
(c) Wang, Y.; Wei, D.; Li, Z.; Zhu, Y.; Tang, M. J. Phys. Chem. A 2014, 118, 4288.
(d) Rasik, C. M.; Hong, Y. J.; Tantillo, D. J.; Brown, M. K. Org. Lett. 2014, 16, 5168.
(e) Huston, R.; Rey, M.; Dreiding, A. S. Helv. Chim. Acta 1982, 65, 1563.
(f) Nelson, S. G.; Dura, R. D.; Peelen, T. J. Org. React. 2012, 82, 471.
(g) Loebach, J. L.; Bennett, D. M.; Danheiser, R. L. J. Org. Chem. 1998, 63, 8380.
(a) Godula, K. Science 2006, 312, 67.
(b) Gutekunst, W. R.; Baran, P. S. Chem. Soc. Rev. 2011, 40, 1976.
(c) Newhouse, T.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 3362.
(a) Chen, M. S.; White, M. C. Science 2007, 318, 783.
(b) Bigi, M. A.; Reed, S. A.; White, M. C. J. Am. Chem. Soc. 2012, 134, 9721.
(c) Shi, G.; Zhang, Y. Adv. Synth. Catal. 2014, 356, 1419.
(a) Song, L.; Yao, H.; Zhu, L.; Tong, R. Org. Lett. 2013, 15, 6.
(b) Noyori, R.; Tomino, I.; Yamada, M.; Nishizawa, M. J. Am. Chem. Soc. 1984, 106, 6717.
(a) Dias, E. L.; Brookhart, M.; White, P. S. Chem. Commun. 2001, 423.
(b) Gao, H.; Zhang, J. Chem.-Eur. J. 2012, 18, 2777.
(a) Smith, C. D. J. Am. Chem. Soc. 1966, 88, 4273.
(b) Tabushi, I.; Yamamura, K.; Yoshida, Z. J. Am. Chem. Soc. 1972, 94, 787.
(c) Petrov, V. A.; Vasil'ev, N. V. Curr. Org. Synth. 2006, 3, 215.
(a) Osberger, T. J.; White, M. C. J. Am. Chem. Soc. 2014, 136, 11176.
(b) Ammann, S. E.; Rice, G. T.; White, M. C. J. Am. Chem. Soc. 2014, 136, 10834.
(c) Malik, M.; Witkowski, G.; Jaroz, S. Org. Lett. 2014, 16, 3816.
(d) White, M. C. J. Am. Chem. Soc. 2013, 135, 14052.
Morrill, L. A.; Susick, R. B.; Chari, J. V.; Garg, N. K. J. Am. Chem. Soc. 2019, 141, 12423. doi: 10.1021/jacs.9b05588
Kiyama, M.; Isoda, Y.; Nishimoto, M.; Kobayashi, A.; Togawa, D.; Hirao, N.; Kuboki, A.; Ohira, S.; Tetrahedron Lett. 2005, 46, 7483. doi: 10.1016/j.tetlet.2005.09.011
Fu, C.; Ma, S. Eur. J. Org. Chem. 2005, 2005, 3942. doi: 10.1002/ejoc.200500296
(a) Blay, G.; Garcia, B.; Molina, E.; Pedro, J. R. Org. Lett. 2005, 7, 3291.
(b) Paquette, L. A.; Green, K. E.; Gleiter, R.; Schäfer, W.; Gallucci, J. C. J. Am. Chem. Soc. 1984, 106, 8232.
Dauben, W. G.; Koch, K.; Smith, S. L.; Chapman, O. L. J. Am. Chem. Soc. 1963, 85, 2616. doi: 10.1021/ja00900a021
Reetz, M. T.; Chatziiosifidis, I.; Schwellnus, K. Angew. Chem. 1981, 93, 716. doi: 10.1002/ange.19810930837
(a) RajanBabu, T. V.; Nugent, W. A. J. Am. Chem. Soc. 1994, 116, 986.
(b) RajanBabu, T. V.; Nugent, W. A.; Beattie, M. S. J. Am. Chem. Soc. 1990, 112, 6408.
(c) RajanBabu, T. V.; Nugent, W. A. J. Am. Chem. Soc. 1989, 111, 4525.
(d) Nugent, W. A.; RajanBabu, T. V. J. Am. Chem. Soc. 1988, 110, 8561.

图 1 gracilioethers A~K以及hippolachnin A的结构
Figure 1 Structures of gracilioethers A~K and hippolachnin A

图式 2 Brown小组关于gracilioether F (6)的全合成
Scheme 2 Total synthesis of gracilioether F (6) by Brown and co-workers

图式 3 Carreira小组关于(±)-hippolachnin A (12)的全合成
Scheme 3 Total synthesis of (±)-hippolachnin A (12) by Carreira and co-workers

图式 4 Carreira小组关于(±)-gracilioethers E (5), F (6)的全合成
Scheme 4 Total synthesis of (±)-gracilioethers E (5), F (6) by Carreira and co-workers

图式 5 Brown和Wood小组关于(±)-hippolachnin A (12)的全合成
Scheme 5 Total synthesis of (±)-hippolachnin A (12) by Brown and Wood

图式 6 伍贻康小组关于(±)-hippolachnin A (12)的全合成
Scheme 6 Total synthesis of (±)-hippolachnin A (12) by Wu and co-workers

图式 7 伍贻康小组关于(±)-gracilioethers F (6), I (9)的全合成
Scheme 7 Total synthesis of (±)-gracilioethers F (6), I (9) by Wu and co-workers

图式 8 唐叶峰小组关于(-)-gracilioether F (6)全合成
Scheme 8 Total synthesis of (-)-gracilioether F (6) by Tang and co-workers

图式 9 唐叶峰小组关于(+)-gracilioether A (1), (-)-gracilio- ether E (5)和(+)-hippolachnin A (12)全合成
Scheme 9 Total synthesis of (+)-gracilioether A (1), (-)- gracilioether E (5) and (+)-hippolachnin A (12) by Tang and co-workers

图式 10 Wong小组关于(±)-gracilioether F (6)的全合成
Scheme 10 Total synthesis of (±)-gracilioether F (6) by Wong and co-workers

图式 11 Trauner小组关于(±)-hippolachnin A (12)的全合成
Scheme 11 Total synthesis of (±)-hippolachnin A (12) by Trauner and co-workers

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

