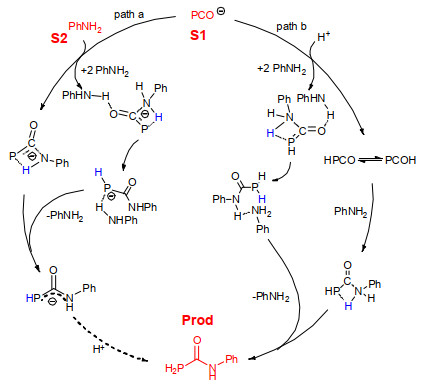
Scheme 1.
Plausible mechanisms

Phosphorus plays a key role in all life processes. The phosphorus chemistry has been made great development and many fields, especially largely important catalysis reactions were discovered at the turn of the 20th century.[1] Among the numerous phosphorus-containing compounds primary phosphines[2] are especially interest one due to their wide variety of applications as starting materials for asymmetric catalysis, macrocyclic synthesis, [3] medicinal chemistry, [4] and polymer science.[5] More than, phosphinocarboxamides were applied as ligands in both small- and large-scale catalytic processes due to their unique structural versatility.[6]
NaPCO was first reported by Becker[7] and has been investigated by several groups recently.[8] The unique reactivity of PCO- anion toward organic substrates leading to the generation of some very unusual compounds including inorganic and organic phosphorus-containing compounds.
Recently, Goicoechea et al.[8d, 9] reported the novel preparation of phosphinecarboxamide and N-alkyl phosphine-carboxamides by the addition of primary amines (NH4+, RNH2) into the 2-phosphaethynolate anion (PCO-) under acidic conditions, such as in the presence of pyridiniumtriflate (Py-HOTf), EtNH3Cl, HCl or HOTf, which are new members of the air-stable primary phosphines. These pioneered works approached phosphinecarboxamide derivatives, which are the heavier analog of urea. However, the substrate scope is limited (only N-alkyl substituted phosphinecarboxamides were prepared). Additionally, some newly chemistry of PCO- acts as a "P-" transfer reagent with the release of CO in a variety of reactions has been discovered.[10] Bertrand, [11] Grutzmacher[12] and Zhao et al, [13] developed serious synthetic methodologies using phosphaketenes as building blocks for novel phosphinidenes and phosphorus heterocycles. Inspired by these pioneered works, we have carried out the reasonably synthesized strategies of heaver analog of N-aryl/alkyl-ureas and that the reaction occurred under grinding, mild and acid-free conditions at room temperature.[14]
Although the mechanism of the reaction of 2-phosphae-thynolate with aryl/alkyl primary amines was investigated and the possible synthetic mechanism was proposed, [14] why the reaction possessed rapid reaction rate, higher yield, and the mild and acid-free conditions was not clear. Therefore, to understand the title reaction deeply, we further perform a detailed theoretical analysis on its mechanism and which can further give a clue to synthesize the versatile functional phosphinecarboxamides.
All the structures were fully optimized at the B3LYP[15]/ BSI level [BSI: 6-31G(d, p)] because this computational method has been successfully applied in the studies.[16] All the calculations were performed with the GAUSSIAN09 program suite.[17] Unscaled harmonic vibrational frequency calculations were used to characterize all of the stationary points as either minima or transition states. Intrinsic reaction coordinates (IRC)[18] were employed to verify the connection of the transition states to two relevant minima. At B3LYP/BSI optimized geometries, the free energy results were refined by calculating the single point energy at the B3LYP/BSII and M062X/BSII levels (BSII: 6-311++G(d, p)) with solvation effect. The solvent, water and acetic acid are simulated with the solvation model based on density (SMD)[19] continuum model at the B3LYP/BSII level. The natural bond orbital (NBO) analysis was also carried out using the NBO 5.0 procedure.[20] The Cartesian coordinates and the varied energies for all optimized structures along with the IRC cures for all translation states are listed in the supporting information.
From the experiment, we examined the detailed reaction mechanisms depicted in Scheme 1. In the paper, the Xx-m-n/Xx-m-TSn denotes the nth intermediate n/transition state TSn on the pathway Xx-m based on the number of assisted/catalyzed molecules (H2O or AcOH) m (X=a, b; x=1~4, m=0~4).
The first event is to investigate why the mixture of PCO- (S1) and PhNH2 (S2) cannot give the product (Prod) under proton source-free conditions in the experiment without the aid of H2O. The calculated geometries and energy profiles are shown in Figures 1 and 2, respectively.
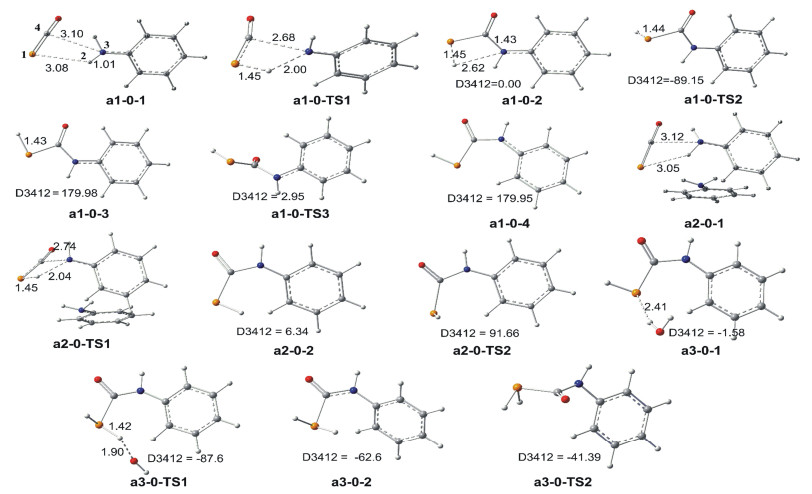
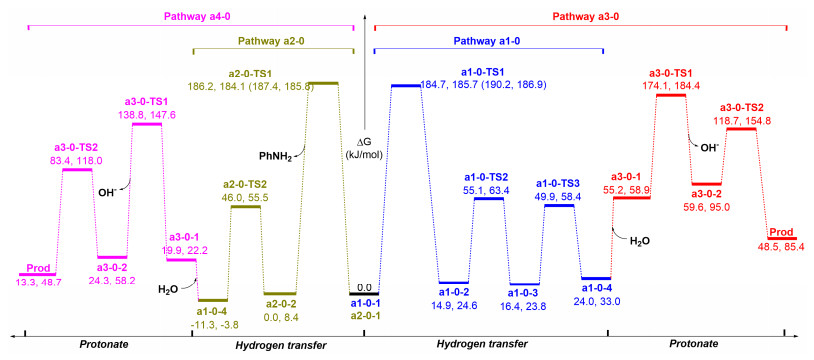
As figures showing, pathway a includes two subpathways of a1-0 and a2-0. In pathway a1-0, the structure of pre-complex a1-0-1 is like experimentally proposed. The distance between P and H atoms of NH2 is 0.308 nm and which is comparable to the sum of the van der Waals radii for one H atom and one P atom (0.31 nm). This case demonstrates that the H…P hydrogen bond which is the driving force of complexation of S1 and S2, is weak. Subsequently, the reaction undergoes a1-0-TS1 (H transferring and N—C bond-forming)→a1-0-2→a1-0-TS2 (P—H bond rotation) process with high activation barriers 184.6 and 185.9 kJ/mol in H2O and AcOH phase, respectively. And, the system is under proton source-free conditions, therefore the pathway a1-0 is terminated at the a1-0-4 and Prod is not given via path a1-0.
As above discussed, two factors prevent path a1-0 from undergoing, one is a higher activation barrier and another is a lack of the proton-donor in the system. Based on this case, we consider another reaction pathway a2-0 in which the ratio of the substrate molecule S1 with S2 is one to two. This performance aims to investigate whether the substrate molecule S2 can act as the proton-donor as well as aid system to decrease the activation barrier. The calculated geometries and energy profiles are shown in Figures 1 and 2, respectively. The activation barrier for this path is 186.3 and 184.2 kJ/mol H2O and AcOH phase, respectively. And, the calculated results show that the PhNH2 cannot afford proton in the system.
Therefore, the reaction between S1 and S2 under proton source-free case without the aid of H2O or AcOH is unfavored and the Prod is not given in the experiment.
Another mechanistic possibility arises because the H- shift process commonly occurs with aid (catalysis) of other functional groups or molecules, such as catalyst, [16e] hydroxyl, [18b] NH3[16k] and H2O.[16k, 21] Inspired by these ideas, a potential mechanistic scenario (H2O-aided hydrogen shift pathway a1-m) is considered. Figures 3 and 4 showed the calculated geometries and energy profile, respectively.
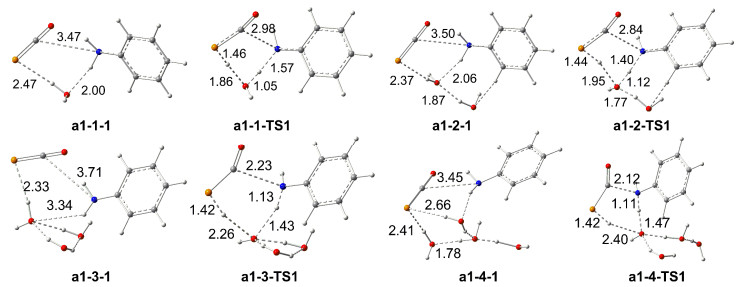
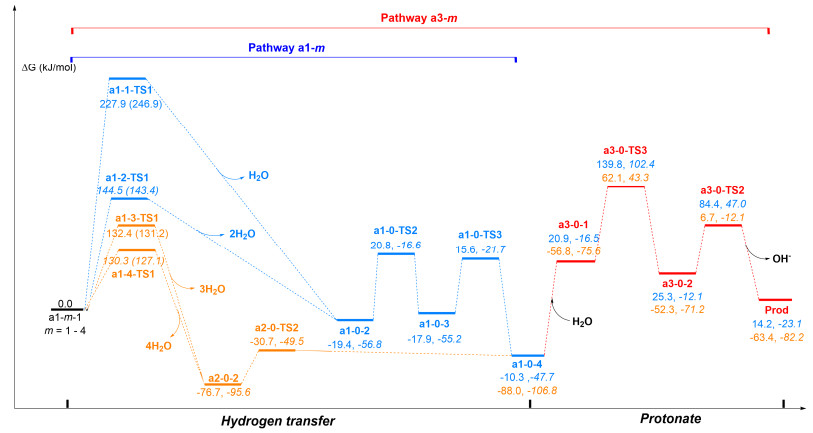
As for H2O aided process, a set of pathways, (1) one H2O (pathway a1-1), (2) two H2O (pathway a1-2), (3) three H2O (pathway a1-3), or (4) four H2O molecules (pathway a1-4) are considered.
For pathway a1-1, the H of NH2 shift with one water molecule aided initialed from six-member ring complex a1-1-1. In a1-1-1, one H of NH2 is attached by O atom of H2O and the P atom interacts with the H atom of H2O. Triggered by interactions of LP(P)→LP*(H) (11.3 kJ/mol) and LP(O)→BD*(H—N) (52.3 kJ/mol) in a1-1-1, the H2O as the medium of the H-exchange spurring the H-shift in a1-1-TS1.
From Figure 4, one can find that the assistance of a single water molecule in the proton transfer in pathway a1-1 (a1-1-TS1: 227.7 kJ/mol) increases the barrier heights by 43.1 kJ/mol compared to a1-0 (a1-0-TS1: 184.6 kJ/mol) in the water phase. Notable the activated barrier gradually reduces from two to four H2O molecules involved with 144.4 (pathway a1-2), 132.3 (pathway a1-3) and 130.2 kJ/mol (pathway a1-4) in the reaction, by amounts 40.2, 52.3 and 54.4 kJ/mol contrast with that of pathway a1-0 in the water phase (184.6 kJ/mol). Figure 4 also lists the barrier heights for a1-1-TS1 (247.0 kJ/mol), a1-2-TS1 (143.6 kJ/mol), a1-3-TS1 (131.4 kJ/mol) and a1-4-TS1 (127.2 kJ/mol) at M062X/BSII//B3LYP/BSI level, and which are reasonable ones at B3LYP/BSII//B3LYP/BSI level. Based on this instance, although increasing the number of H2O molecules involved in the catalysis (to five or more molecules) is predicted to further decrease the energy barrier, their increased extent is smaller and smaller. With the aid of H2O molecule from one to four, although it can decrease the activated energy of the reaction gradually, the energy barrier of the reaction a1-4 is yet very high (above 125.6 kJ/mol) and therefore the detailed processes of path a1-m in Figure 4 are not discussed.
In this section, the essential cases in the calculation are to investigate why the Prod can be higher yielded in the experiment in presence of proton donors, such as H2O and AcOH, comparing to under proton source-free conditions. The optimized structures and potential energy surface are presented in Figures 5 and 6, respectively.
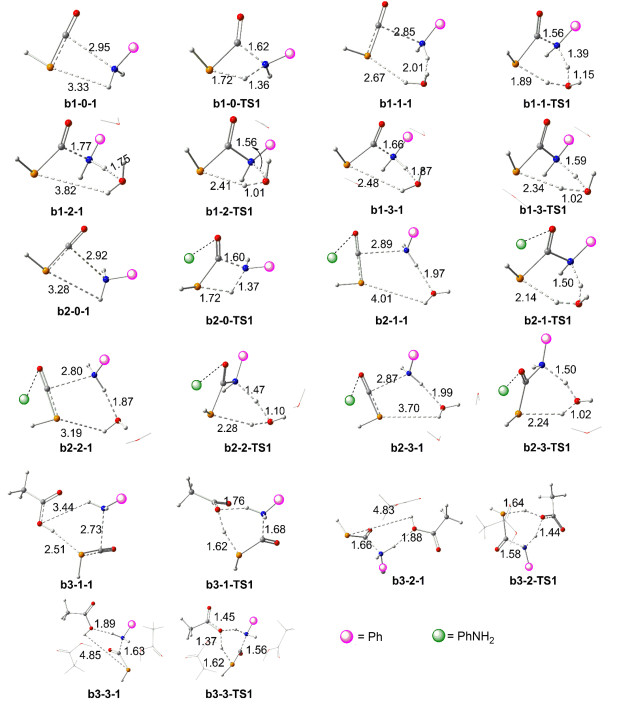
The anion PCO- can firstly accept the proton in this case and the reaction is initialed from the neutral HPCO and S2.
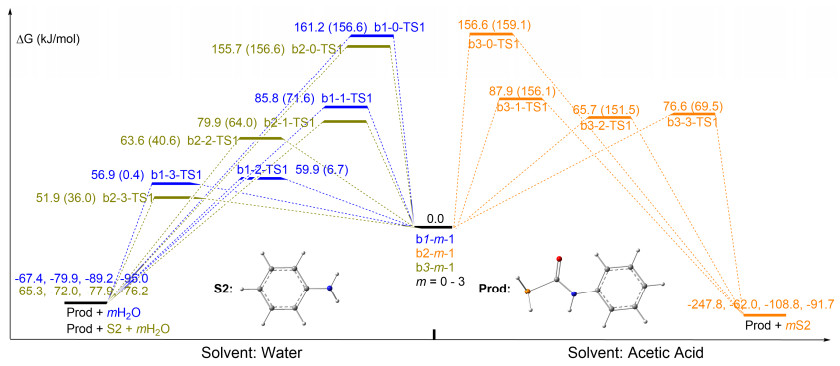
Non-aided mechanism of H2O or AcOH: The first series pathways are the H2O/AcOH non-assisted in the system (pathways b1-0, b2-0 and b3-0). As Figures 5 and 6 illustrate, pathways b1-0, b2-0 and b3-0 are similar and succinct, and they only include three steps. The first step is the complexation preliminary intermediate b1-0-1/b2-0-1/b3-0-1 of HPCO and PhNH2, the second is the H-transfer- ring transition state b1-0-TS1/b2-0-TS1/b3-0-TS1 and the final is the Prod produced. For these pathways, the H- final is the Prod produced. For these pathways, the H- transferring and the N—C bond formation are concerted in b1-0-TS1/b2-0-TS1/b3-0-TS1. It is unfortunately pointed out, the activated energy is higher 161.2/155.7/156.6 kJ/mol and the pathway b1-0/b2-0/b3-0 is unfavored, and which is inconsistent with the high yield in the experiment. When a comparison between Entries 7 and 11 in the experiment was made, [14] the difference between them is H2O involving in Entry 7 and which leads to that giving higher yield by 43% than that of Entry 11. Therefore, it is unambiguous that the H2O in reaction acts as a crucial role.
Aided mechanism of H2O or AcOH: The calculations illustrated that the reaction undergoing is strongly assisted by H2O, actively participating as a catalyst in the course of the H-shifting rearrangement. The geometries and energy profile of the H-shifting assisted by one H2O (pathway b1-1, b2-1), two H2O (pathways b1-2 and b2-2) or three H2O (pathways b1-3 and b2-3) molecules, along with the reactant complexes. The transition structures were displayed in Figures 5 and 6. Along the pathway b1-1, the initial six-member ring reactant complex b1-1-1 proceeds across a barrier of 85.8 kJ/mol through transition state b1-1-TS1, giving the Prod. The H2O molecule assists the H shift, dramatically lowering the activation barrier by 75.3 kJ/mol with respect to that of the non-catalytic reaction pathway b1-0 (161.2 kJ/mol). Along the pathway b1-2, the initial HPCO…(H2O)2…H2NPh reactant complex b1-2-1 proceeds across a barrier of 59.9 kJ/mol through b1-2- TS1, giving the Prod. The H2O molecule further assists the H shift, lowering the activation barrier by 26.0 kJ/mol with respect to that of catalytic reaction by one H2O molecule. Three H2O molecules involved in pathway b1-3 and the reaction is initiated from complex b1-3-1, undergoing a transition state with a 56.9 kJ/mol energy barrier to give Prod. Therefore, the H2O molecule in the system acts as the role of the catalyst which can decrease the activation barrier by ca. 104.6 kJ/mol and the pathways b1-2/b1-3 are favoured in path b1. It is noteworthy that Prod is stabilized by 95.0 kJ/mol regarding the precursor b1-3-1 (Figure 6).
The second series pathways with aid of H2O is b2-m, in which one HPCO and two PhNH2 molecules are involved. It can be seen from Figure 6 that the activated energies are also gradually decreased with the number of H2O molecules increased. The activated energies of paths b2-1 (79.9 kJ/mol), b2-2 (63.6 kJ/mol) and b2-3 (51.9 kJ/mol) are respectively similar to that of pathways b1-1 (85.5 kJ/mol), b1-2 (60.7 kJ/mol) and b1-3 (56.9 kJ/mol). Therefore, pathways b1-m and b2-m are no distinct difference and they are all favored in dynamics for the reaction.
Inspiring by H2O assist in Entry 7, we want to know whether the H-shift in Entry 11[14] can be aided by AcOH (pathway b3-m). Figures 5 and 6 are clearly shown that AcOH can also assist H-shift from N to P as well as the activated energies are 156.6 kJ/mol→87.9 kJ/mol→65.7 kJ/mol→76.6 kJ/mol with the number of AcOH molecules increased from zero (pathway b3-0) to three (pathway b3-3), which illustrates that the reaction through Entry 11 can also give Prod.
It can be concluded that pathways b1 and b2 with H2O molecules aided are both occurred in Entry 7, while Entry 11 is only involved b3-m with AcOH molecules assisted. And, pathways b1-m and b2-m have lower activated energy than path b3-m. Therefore, the Entry 7 system is a three-channel reaction, and which leads to a higher yield than that of Entry 11 in the experiment.[14]
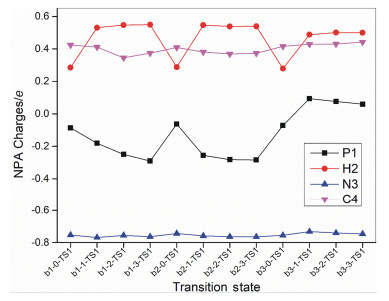
To further elucidate the mechanistic difference among the pathways b1-m, b2-m and b3-m, the NPA (Natural Population Analysis) and NBO analysis were employed.[22] As Figure 7 shown, the NPA charges for N3 (ca. -0.8 e) and C4 (ca. 0.4 e) remained almost constant in all transi- tion states and with the solvents H2O/AcOH number increasing, the NPA charges on P1 (more negative) and H2 (more positive) atoms in pathways b1-m and b2-m changed regularly. On the contrary, in b3-m pathway, the NPA charges on P1 atom changed from negative to positive.
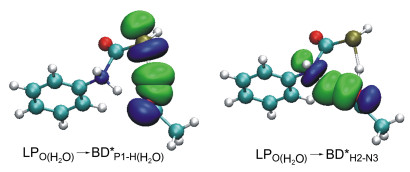
For pathway b1-m, the electrons transfer from LPN3 to BDP*1-C4 in transition states with the secondary stabilization energy Eij(2) increasing gradually: 60.7 kJ/mol (b1-1-TS1)→117.2 kJ/mol (b1-2-TS1)→149.4 kJ/mol (b1-3-TS1). In pathway b2-m, excepting b2-1-TS1 (LPN3→BDP*1-C4, 110.0 kJ/mol), orbitals LPN3 interacts with BDC*4-O (Eij(2): ca. 79.5 kJ/mol) for both b2-2-TS1 and b2-3-TS1. The electron donors are the orbital LPO- (H2O) and the electron acceptors are BDP*1-H(H2O) and BDH*2-N3 in transition states of b3-m pathway (Figure 8), which is different from those in pathways b1-m and b2-m. The Eij(2) values of LPO(H2O)→BDP*1-H(H2O) are decreased gradually whereas those of LPO(H2O)→BDH*2-N3 are increased in the order: b3-1-TS1→b3-2-TS1→b3-3- TS1.
In this case, the anion PCO- can firstly complex with the substrate S2, and the pathway corresponding to a3/a4.
For pathways a3-0 and a4-0 (Figures 1 and 2) without the aid of H2O molecules, although the intermediate a1-0-4 can be protonated and the final Prod can be given seemingly in the procedure, the precursor processes of a3-0 and a4-0 are the same with the pathways a1-0 and a2-0, respectively and their activation barriers are ca. 167.4 kJ/mol. Therefore, pathways a3-0 and a4-0 without H2O assisted are disfavored.
For the pathway a3-m with aid of varies number H2O molecules m (m=1~4), the a3-m are also similar to a1-m with the same number of H2O molecules and they have the same H-transfer processes. The energy barriers of them are all above 30 kJ/mol (Figure 4), which results in the later protonation that cannot perform in the subsequent procedure.
The reaction mechanism of 2-phosphaethynolate anion and primary amines for phosphinecarboxamides synthesis was studied by using DFT method in B3LYP/6-31G(d, p) and M062X/6-311++G**(PCM)//B3LYP/6-31G(d, p) levels. Calculated investigating shows that the anion PCO- is protonated firstly and the reaction is initiated from the complexation of HPCO with substrate PhNH2. Attributed to aid/catalysis with various sizes (from one to four) of solvents H2O and AcOH cluster, the activation free energies barrier can be dramatically decreased and the reaction has its own merits, such as rapid reaction rate, higher yield, and the mild reaction condition.
Supporting Information The Cartesian coordinates for the calculated stationary structures, and the energies for the transition and ground states obtained from the DFT calculations are given.. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(a) Solařová, H.; Císařová, I.; Štěpnička, P. Organometallics 2014, 33, 4131.
(b) Gómez Arrayás, R.; Adrio, J.; Carretero, J. C. Angew. Chem., Int. Ed. 2006, 45, 7674.
(a) Hiney, R. M.; Ficks, A.; Müller-Bunz, H.; Gilheany, D. G.; Higham, L. J. Organometallic Chemistry, the Royal Society of Chemistry, London, 2011, Vol. 37, p. 27.
(b) Li, X.; Robinson, K. D.; Gaspar, P. P. J. Org. Chem. 1996, 61, 7702.
(c) Chatterjee, S.; George, M. D.; Salem, G.; Willis, A. C. J. Chem. Soc., Dalton Trans. 2001, 1890.
(d) Herrbach, A.; Marinetti, A.; Baudoin, O.; Guénard, D.; Guéritte, F. J. Org. Chem. 2003, 68, 4897.
(e) Hoge, G.; Samas, B. Tetrahedron: Asymmetry 2004, 15, 2155.
(f) Clark, T.; Landis, C. Tetrahedron: Asymmetry 2004, 15, 2123.
Kyba, E. P.; Liu, S. T. Inorg. Chem. 1985, 24, 1613. doi: 10.1021/ic00205a005
Katti, K. V.; Gali, H.; Smith, C. J.; Berning, D. E. Acc. Chem. Res. 1999, 32, 9. doi: 10.1021/ar9800082
(a) Hooper, T. N.; Huertos, M. A.; Jurca, T.; Pike, S. D.; Weller, A. S.; Manners, I. Inorg. Chem. 2014, 53, 3716.
(b) Dorn, H.; Singh, R. A.; Massey, J. A.; Nelson, J. M.; Jaska, C. A.; Lough, A. J.; Manners, I. J. Am. Chem. Soc. 2000, 122, 6669.
(c) Dorn, H.; Singh, R. A.; Massey, J. A.; Lough, A. J.; Manners, I. Angew. Chem., Int. Ed. 1999, 38, 3321.
(d) Dorn, H.; Singh, R. A.; Massey, J. A.; Lough, A. J.; Manners, I. Angew. Chem. 1999, 111, 3540.
(a) Duckmanton, P. A.; Blake, A. J.; Love, J. B. Inorg. Chem. 2005, 44, 7708.
(b) Meeuwissen, J.; Detz, R.; Sandee, A. J.; de Bruin, B.; Siegler, M. A.; Spek, A. L.; Reek, J. N. H. Eur. J. Inorg. Chem. 2010, 2010, 2992.
(c) Škoch, K.; Císařová, I.; Štěpnička, P. Organometallics 2016, 35, 3378.
(a) Becker, G.; Heckmann, G.; Hübler, K.; Schwarz, W. Z. Anorg. Allg. Chem. 1995, 621, 34.
(b) Becker, G.; Schwarz, W.; Seidler, N.; Westerhausen, M. Z. Anorg. Allg. Chem. 1992, 612, 72.
(a) Puschmann, F. F.; Stein, D.; Heift, D.; Hendriksen, C.; Gal, Z. A.; Grützmacher, H.-F.; Grützmacher, H. Angew. Chem., Int. Ed. 2011, 50, 8420.
(b) Jupp, A. R.; Goicoechea, J. M. Angew. Chem., Int. Ed. 2013, 52, 10064.
(c) Li, Z.; Chen, X.; Benkö, Z.; Liu, L.; Ruiz, D. A.; Peltier, J. L.; Bertrand, G.; Su, C.-Y.; Grützmacher, H. Angew. Chem., Int. Ed. 2016, 55, 6018.
(d) Jupp, A. R.; Goicoechea, J. M. J. Am. Chem. Soc. 2013, 135, 19131.
(a) Jupp, A. R.; Trott, G.; Payen de la Garanderie, É.; Holl, J. D. G.; Carmichael, D.; Goicoechea, J. M. Chem.-Eur. J. 2015, 21, 8015.
(b) Robinson, T. P.; Goicoechea, J. M. Chem.-Eur. J. 2015, 21, 5727.
(a) Magnall, R.; Balázs, G.; Lu, E.; Tuna, F.; Wooles, A. J.; Scheer, M.; Liddle, S. T. Angew. Chem., Int. Ed. 2019, 58, 10215.
(b) Goicoechea, J. M.; Grützmacher, H. Angew. Chem., Int. Ed. 2018, 57, 16968.
(a) Hansmann, M. M.; Bertrand, G. J. Am. Chem. Soc. 2016, 138, 15885.
(b) Liu, L.; Ruiz, D. A.; Munz, D.; Bertrand, G. Chem 2016, 1, 147.
Liu, L.; Ruiz, D. A.; Dahcheh, F.; Bertrand, G.; Suter, R.; Tondreau, A. M.; Grützmacher, H. Chem. Sci. 2016, 7, 2335. doi: 10.1039/C5SC04504E
Wu, Y.; Liu, L.; Su, J.; Zhu, J.; Ji, Z.; Zhao, Y. Organometallics 2016, 35, 1593. doi: 10.1021/acs.organomet.6b00187
Wu, Y.-H.; Li, Z.-F.; Wang, W.-P.; Wang, X.-C.; Quan, Z.-J. Eur. J. Org. Chem. 2017, 2017, 5546. doi: 10.1002/ejoc.201700867
(a) Becke, A. D. Phys. Rev. A 1988, 38, 3098.
(b) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
(c) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
(a) Cui, C.-X.; Chen, H.; Li, S.-J.; Zhang, T.; Qu, L.-B.; Lan, Y. Coord. Chem. Rev. 2020, 412, 213251.
(b) Faza, O. N.; López, C. S.; Álvarez, R.; de Lera, A. R. J. Am. Chem. Soc. 2006, 128, 2434.
(c) Shi, F.-Q.; Li, X.; Xia, Y.; Zhang, L.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 15503.
(d) Yu, Z.-X.; Wender, P. A.; Houk, K. N. J. Am. Chem. Soc. 2004, 126, 9154.
(e) Li, Z.-F.; Fan, Y.; DeYonker, N. J.; Zhang, X.; Su, C.-Y.; Xu, H.; Xu, X.; Zhao, C. J. Org. Chem. 2012, 77, 6076.
(f) Li, Z.-F.; Yang, X.-P.; Hui-Xue, L.; Guo, Z. Organometallics 2014, 33, 5101.
(g) Zhou, T.; Xia, Y. Organometallics 2014, 33, 4230.
(h) Wang, Y.; Liao, W.; Huang, G.; Xia, Y.; Yu, Z.-X. J. Org. Chem. 2014, 79, 5684.
(i) Cohen, A. J.; Mori-Sánchez, P.; Yang, W. Chem. Rev. 2012, 112, 289.
(j) Hou, C.; Jiang, J.; Zhang, S.; Wang, G.; Zhang, Z.; Ke, Z.; Zhao, C. ACS Catal. 2014, 4, 2990.
(k) Tsipis, C. A.; Karipidis, P. A. J. Am. Chem. Soc. 2003, 125, 2307.
Frisch, M. J.; Trucks, G. W.; Schlegel, G. W.; Scuseria, G. W. Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013.
(a) Fukui, K. Acc. Chem. Res. 1981, 14, 363.
(b) Fukui, K. J. Phys. Chem. 1970, 74, 4161.
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 4538. doi: 10.1021/jp809094y
Glendening, E. D.; Badenhoop, J. K.; Reed, A. E.; Carpenter, J. E.; Bohmann, J. A.; Morales, C. M.; Weinhold, F. NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001.
Karton, A.; O'Reilly, R. J.; Radom, L. J. Phys. Chem. A 2012, 116, 4211. doi: 10.1021/jp301499y
(a) Ess, D. H.; Houk, K. N. J. Am. Chem. Soc. 2008, 130, 10187.
(b) Bickelhaupt, F. M.; Houk, K. N. Angew. Chem., Int. Ed. 2017, 56, 10070.
(c) Lv, X.; Zhang, X.; Sa, R.; Huang, F.; Lu, G. Org. Chem. Front. 2019, 6, 3629.
(d) Ogunlana, A. A.; Bao, X. Chem. Commun. 2019, 55, 11127.

Figure 1 Schematic diagrams of the optimized geometries for the pathways a1-0, a2-0, and a3-0 at B3LYP/BSI level (bond length: 0.1 nm, angle: °)

Figure 2 Free energy profiles for pathways a1-0, a2-0, a3-0, and a4-0 at B3LYP/BSII//B3LYP/BSI and M062X/BSII///B3LYP/BSI (in parentheses) levels (The parameters from left to right are in water and acetic acid phase, respectively)

Figure 3 Schematic diagrams of the partly optimized geometries for the pathways a1-m at B3LYP/BSI level (m=1~4, bond length: 0.1 nm)

Figure 4 Free energy profiles for pathways a1-m at B3LYP/BSII//B3LYP/BSI and M062X/BSII///B3LYP/BSI (in parentheses) levels (m=1~4, the parameters are in the water phase)

Figure 5 Schematic diagrams of the optimized geometries for the pathways bx-m at B3LYP/BSI level (x=1~3, m=0~3, bond length: 0.1 nm)

Figure 6 Free energy profiles for pathways bx-m (x=1~3, m=0~3) at B3LYP/BSII//B3LYP6/BSI and M062X/BSII//B3LYP6/BSI (in parentheses) levels

Figure 7 NPA charges for P1, H2, N3 and C4 atoms in transition states for pathways b1-m, b2-m and b3-m

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
