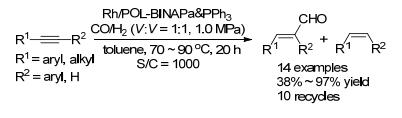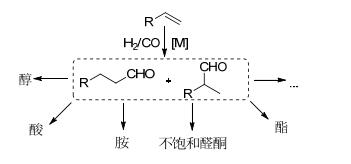
图式 1.
氢甲酰化产物醛及衍生物
Scheme 1.
Hydroformylation of olefins to aldehydes and their derivatives

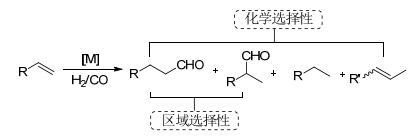
氢甲酰化反应也称之为“Oxo合成”, 是指在催化剂的作用于下, 烯烃与H2/CO反应得到醛的过程[1], 1938年由德国科学家Roellen首次发现[2].经过几十年的发展, 氢甲酰化反应作为工业中最重要的均相反应之一, 已经是商业上合成醛主要的方法, 每年生成的醛及其衍生物达一千万多吨[3].醛作为重要的化工原料, 不仅可以用作香料的添加剂, 而且经氢化、氧化、羟醛缩合、还原胺化等反应制备醇、酸、α, β-不饱和醛酮和胺等化合物(Scheme 1).另外, 氢甲酰化反应是一类100%原子经济性制备醛的过程.因此, 研究氢甲酰化反应具有重要的理论意义和应用价值.
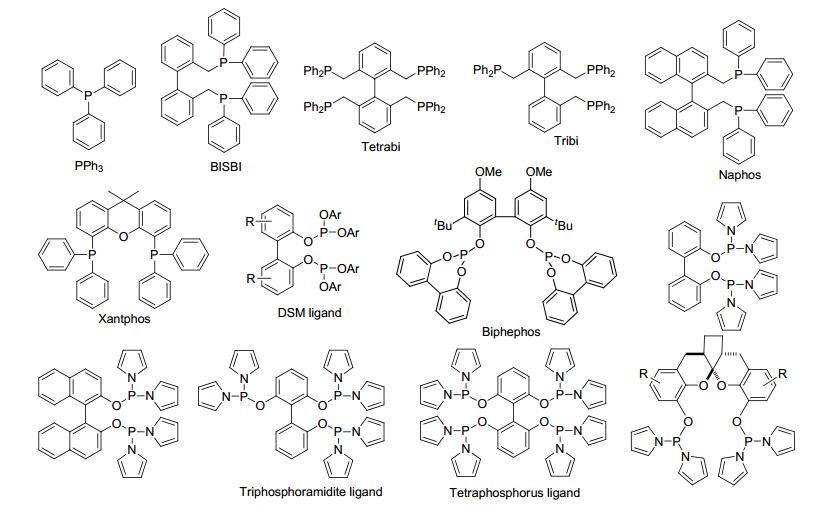
在取代烯烃的氢甲酰化反应过程中, 不仅存在着反应活性问题, 而且还有区域选择性和化学选择性问题(Scheme 2).醛基可以生成在双键的末端和内端, 分别得到直链醛和支链醛.目前, 直链醛是工业上的主要需求, 如直链醛衍生的直链醇广泛应用于洗涤剂和增塑剂的原材料.另外, 在反应中还伴随着氢化产物和异构化烯烃的生成.催化剂对氢甲酰化反应的活性和选择性起着决定性作用.因此, 发展高活性和高选择性的催化剂具有重要的意义.钴是最早应用于工业氢甲酰化反应的催化剂.由于存在着催化活性低、高温高压及化学选择性差等问题, 工业上钴催化剂逐渐被铑催化剂代替.在铑催化的氢甲酰化反应中, 配体对反应的活性、区域选择性和化学选择性具有重要的决定性作用.一系列的膦配体被用于均相铑催化的烯烃氢甲酰化反应, 代表性的配体有: PPh3、BISBI[4a-4b]、Tetrabi[4c-4d]、Tribi[4e]、Naphos[5]、Xantphos[6]、亚磷酸酯配体(DSM ligand[7], Biphephos[8])和吡咯基亚磷酰胺配体配体[9]等(Scheme 3).
与均相反应相比, 非均相催化体系具有产物与催化剂分离容易, 催化剂可回收循环利用等优点, 更具有广阔的工业应用前景.在非均相氢甲酰化反应中, 首先需要解决几个主要问题: (1)达到与均相体系相当的或更高的催化剂活性; (2)反应的选择性, 包括直链醛和支链醛的选择性及异构化产物和氢化产物; (3)催化剂的流失, 包括配体和铑金属的流失; (4)催化剂的循环次数及使用寿命.
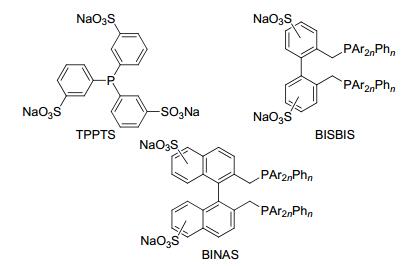
实现均相氢甲酰化反应的非均相化, 主要包括液/液两相和液/固两相催化体系[10].液/液两相催化体系分为水/有机两相[11]、氟两相[12]、离子液体两相[13]、超临界流体[14]等.但用于工业化只有水/有机两相催化体系, 代表性的为RCH/RP工艺[15].水/有机两相是指以水为反应溶剂, 使用水溶性的磺酸钠膦配体与铑络合催化烯烃的氢甲酰化反应; 反应之后产物醛和水溶液为两相, 且催化剂溶于水相, 所以容易实现产物和催化剂的分离及回收.目前用于水/有机两相的膦配体包括TPPTS (trisulfonated triphenylphosphane)[16]、BISBIS (sulfonated 2, 2'-bis(diphenylphosphinomethyl)-l, l'-biphenyl)[17]和BINAS (bis[disulphonatophenylphosphinomethyltetrasul-phonatobinaphthene])[18] (Scheme 4).由于烯烃在水相中的溶解性问题, 该工艺主要用于短链C3和C4烯烃的氢甲酰化反应.
液/固两相体系是指将催化剂负载于固体载体上, 产物醛为液相, 反应完成后容易实现产物与催化剂的分离.固载载体分为无机载体和聚合物载体两大类.无机载体主要包括分子筛[19]、碳材料[20]和金属氧化物[21]等.通过利用无机材料的多孔结构, 包覆、封装或共价键结合固载催化剂.该类固载催化剂可以有效解决催化剂的活性, 但并不能得到理想的区域选择性.氢甲酰化反应的催化剂中, 配体对铑金属的反应活性尤其是选择性起到了主要的决定性作用.由于无机载体本身的局限性, 使得其与有机膦配体之间难以构建稳定的键合结构, 导致无法满足固载催化剂中铑周围的配体浓度, 而且在反应中容易发生膦配体的流失.低的配体浓度及配体流失将会引起反应选择性的降低和铑金属的流失, 进而影响催化剂的使用寿命.另外, 使用无机载体难以实现配体修饰的多样化.
以有机聚合物为载体, 将膦配体通过共价键固定在聚合物链中, 进一步通过膦配体与铑金属的配位实现催化剂的负载.聚合物固载催化剂可以实现负载膦配体的多样化, 有效提高催化剂性能和稳定性.因此, 本文概述了近年来有机聚合物负载催化剂在氢甲酰化反应中应用研究进展, 探讨了固载催化剂的合成、结构与氢甲酰化反应活性、选择性和稳定性之间的关系.
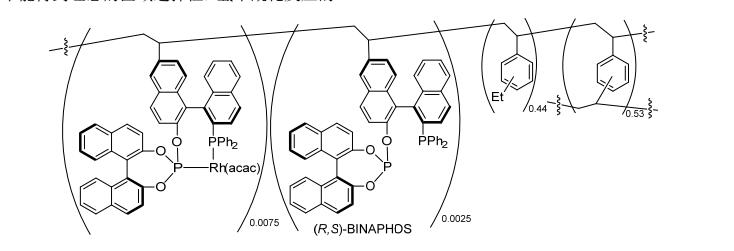
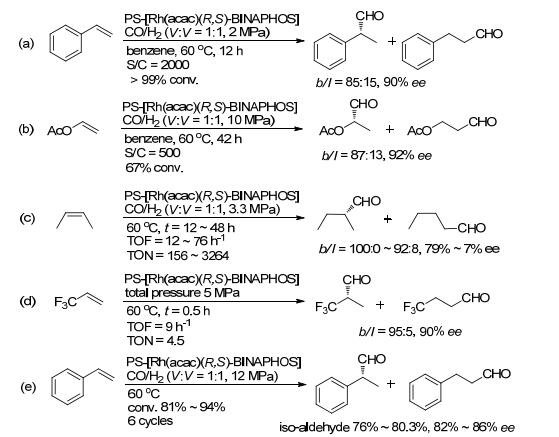
传统的有机聚合物主要包括聚苯乙烯、聚甘油和壳聚糖等.经聚合反应和缩合反应等, 以C—C、C—O、C—N等共价键等将配体固定在聚合物链中.在1998年, Nozaki课题组[22]用乙烯基修饰的(R, S)-BINAPHOS ((R)-(2-(diphenylphosphino)-1, 1'-binaphthalen-2'-yl)-((S)-l, l'-binaphthalen-2, 2'-yl)phosphite)先与Rh(acac)(CO)2络合, 再将其与商业化的二乙烯基苯聚合可制备得到PS-[Rh(acac)(R, S)-BINAPHOS] (Scheme 5).该方法制备的催化剂在苯乙烯和醋酸乙烯酯的不对称氢甲酰化反应中均可以取得与均相体系相当的催化效果(Schemes 6a, 6b).另外, 该类催化剂不会在反应中发生溶胀, 容易实现催化剂的分离. 2003, Nozaki课题组[23]使用固定床反应器探索了PS-[Rh(acac)(R, S)-BINA-PHOS]催化剂在氢甲酰化反应中的使用时间和循环次数.当以液化的顺式-二丁烯为反应底物和反应溶剂时, 反应48 h后, 产物醛的ee值由最初的79% (12 h)降至7% (Scheme 6c).当以三氟甲基乙烯为反应底物时, 产物的对映选择性可以保持, 但催化剂的活性明显降低(Scheme 6d).为了提高催化剂的活性, 当采用超临界CO2作为反应介质和高的合成气压力(12.1 MPa)条件下, 催化剂能够循环使用6次, 且保持好的催化活性和选择性(Scheme 6e).
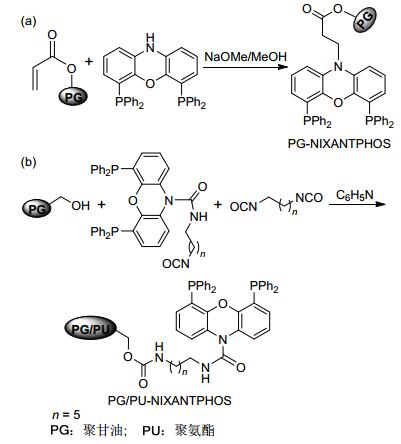
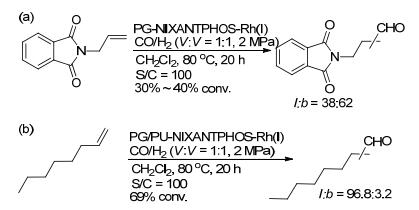
树枝状聚甘油是一类含有大量羟基的超支化聚合物, 通过共价键的形式将催化剂固定在其分子内部或周围, 来提高催化剂稳定性、分散性和催化效率[24]. 2006年, Haag和Eilbracht课题组[25]分别通过迈克尔加成反应和异氰酸酯的亲核加成反应, 将修饰的NIXANT- PHOS [4, 6-bis(diphenylphosphino)-10H-phenoxazine]固定在树状的聚甘油链中, 进一步通过与铑催化前体络合分别得到两类聚合物负载催化剂PG-NIXAN-TPHOS- Rh(I)和PG/PU-NIXANTPHOS-Rh(I) (Scheme 7).在PG-NIXANTPHOS-Rh(I)催化的N-烯丙基邻苯二甲酰亚胺的氢甲酰化反应中, 仅以30%~40%的转化率和低的区域选择性(l:b=38:62)得到相应脂肪醛(Scheme 8a).在PG/PU-NIXANTPHOS-Rh(I)催化的1-辛烯的氢甲酰化反应中, 以69%的转化率和高的区域选择性(l:b=96.8:3.2)得到相应的醛产物(Scheme 8b).但这两类负载催化剂的催化性能均低于相应的均相催化体系.
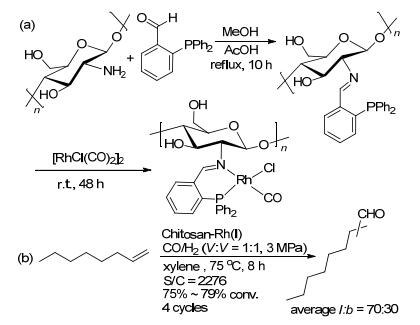
壳聚糖是甲壳素脱乙酰化的产物, 具有无毒无害易降解的特点.在壳聚糖的分子链中含有大量的氨基和羟基, 易于通过化学修饰来负载催化剂[26]. 2012年, Smith课题组[27]以壳聚糖和2-苯基膦苯甲醛为原料, 经缩合反应得到含有Schiff碱的壳聚糖.通过壳聚糖中C=N和PPh2单元与铑的配位实现催化剂的固载(Scheme 9a).该类催化剂在1-辛烯的氢甲酰化反应中, 能够循环使用4次, 以75%~79%的转化率和中等的区域选择性(l: b=70:30)得到相应的醛(Scheme 9b).在滤液中, 经ICP-MS可以检测到0.02%的铑的流失.
从以上研究结果可以发现, 聚合物可以实现催化剂修饰的的多样性, 但传统的有机聚合物的比表面积比较低, 导致有些固载催化剂难以达到相应的均相催化剂活性.另外, 由于配体是固定在其它的有机聚合物链中, 并不能控制配体的分布及铑金属周围的配体浓度, 因而反应的选择性也会受到一定的影响, 同时导致铑的流失, 进而影响催化剂的循环.
近年来, 多孔有机聚合物(POPs)的发展成为一个新的研究热点[28].该类材料可以应用于气体吸收、分子分离和电化学等方向, 同时也为负载催化剂提供了一类新的聚合物载体.尤其是以有机配体单体聚合构建的多孔有机聚合物(POLs)作为负载体具有以下优势: (1)相对比传统的聚合物载体, 多孔配体聚合物具有可更高的比表面积, 可以有效分散金属催化剂, 防止催化剂的聚集.另外, 由于该类聚合物为交联产物, 所以不溶于大多数溶剂, 防止溶解而导致的催化剂损失. (2)该类聚合物具有多孔结构.有利于反应物与催化活性位点的接触及产物的扩散.同时, 根据反应需要, 可以调节孔径的大小. (3)该类聚合物负载金属催化剂时, 主要通过聚合物中的配体单元与金属配位螯合, 这样容易形成单原子催化中心, 使得催化剂具有更高的催化活性. (4)在负载的金属中心周围有很多的配体单元, 高浓度的配体可以稳定催化剂, 防止金属的流失, 使催化剂具有更多的循环次数和使用时间.基于以上的特点, 该类多孔聚合物负载铑催化剂在氢甲酰化的反应中表现出优异的催化性能.
均聚物负载催化剂是指由一种单体聚合而成的聚合物负载的催化剂.构建多孔结构聚合物往往需要反应单体包含两个或两个以上的聚合反应位点.因此, 从合成的角度出发, 就需要对膦配体进行进一步的修饰.结合目前的研究工作, 聚合单元以乙烯基基团为主.在膦配体结构中构建乙烯基单元主要包括三种方法: (1)直接使用乙烯基的原料; (2)使用含有羰基的原料, 通过Wittig反应合成乙烯基; (3)通过在配体骨架上引入卤素, 通过偶联反应合成乙烯基.
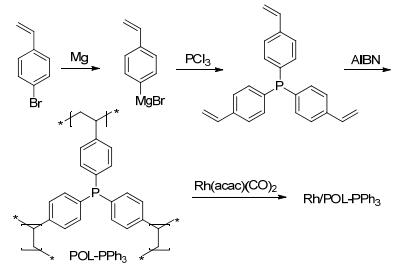
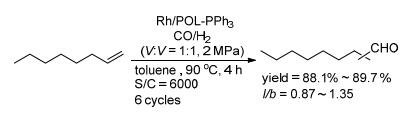
2014年, 丁云杰课题组和刘景月课题组[29]报道了Rh(CO)2(acac)/POL-PPh3催化剂的合成, 并将其应用于1-辛烯的氢甲酰化反应.以对溴苯乙烯为起始原料制备格氏试剂, 再与PCl3反应得到三乙烯基三苯基膦; 在AIBN的催化下, 乙烯基三苯基膦单体经过聚合反应得到POL-PPh3 (Scheme 10).经结构表征, 聚合物POL-PPh3的BET (Brunauer、Emmett和Teller)比表面积和孔容分别可达到1086 m2• g-1和1.70 cm3•g-1; 孔径主要分布在0.7、1.5和3~70 nm, 说明该类材料含有多级孔状结构(表 1, Entry 1);分解温度可以高达440 ℃, 甚至高于全氟磺酸树脂Nafion NR50的分解温度(330 ℃), 表明POL-PPh3有好的热稳定性.负载Rh以后的催化剂仍能保持高的比表面积和孔容(表 1, Entry 2).在1/6000的催化剂用量下, Rh/POL-PPh3催化的1-辛烯的氢甲酰化反应中, 能以88.1%~89.7%的收率和0.87~1.35的正异比得到目标醛; 催化剂循环使用6次, 且保持稳定的转化率、收率和选择性; 与均相的Rh/PPh3催化效果相比, Rh/POL-PPh3能得到更高的区域选择性(Scheme 11).
 下载:
导出CSV
下载:
导出CSV
| Entry | Sample | BET surface area/(m2•g-1) |
Pore volume/ (cm3•g-1) |
Pore size distribution/ nm |
Decomposition temperature/℃ |
Reference |
| 1 | POL-PPh3 | 1086 | 1.70 | 0.7, 1.5, 3~70 | 440 | [29] |
| 2 | Rh/POL-PPh3 | 1032 | 1.69 | — | — | [29] |
| 3 | Phosphite-POP | 643 | 0.43 | 0.5–1.4, 2~10 | — | [31] |
| 4 | Rh/phosphite-POP | 616 | 0.43 | — | — | [31] |
| 5 | POL-dppe | 943 | 1.51 | 0.7−1.5, 2.8~150 | >400 | [33] |
| 6 | Rh/POL-dppe | 856 | 1.41 | — | — | [33] |
| 7 | POL-dppm | 892 | 1.07 | — | — | [33] |
| 8 | POL-dppb | 846 | 0.81 | — | — | [33] |
| 9 | POL-Xantphos & PPh3 | 1022 | 1.24 | 0.6–1, 1.2~2, 2~10 | >450 | [34] |
| 10 | Rh/POL-Xantphos & PPh3 | 883.9 | 1.16 | — | - | [34] |
| 11 | CPOL-BP & PPh3 | 1088.0 | 2.07 | 0.70, 0.84, 1.38, 2.18 | >430 | [35] |
| 12 | Rh/CPOL-BP & PPh3 | 985.3 | 1.94 | — | — | [35] |
| 13 | Rh/CPOL-BP & Ph | 1116 | 1.66 | — | — | [36] |
| 14 | CPOL-BP & P(OPh)3 | 635 | 0.72 | 0.7, 0.85, 1.38, 1.89, 2~10 | — | [37] |
| 15 | Rh/CPOL-BP & P(OPh)3 | 556 | 0.68 | — | — | [37] |
| 16 | Rh/CPOL-BPa & PPh3 | 423.6 | 0.41 | 1.2~2.0, 2.3~25 | 370 | [38] |
| 17 | Rh/POL-BINAPa & PPh3 | 492.2 | 0.41 | — | 456 | [40] |
| 18 | PSA-PPh3 | 547 | 0.56 | — | 400 | [41] |
| 19 | PSA-Xantphos | 290 | 0.69 | — | — | [41] |
2015年, 丁云杰课题组和刘景月课题组[30]研究了Rh/POL-PPh3催化剂在固定床反应器中的氢甲酰化催化性能.当乙烯为反应底物时, 能以高的转化率(96.2%)和优秀的催化活性(TOF=4530 h-1)得到丙醛产物; 催化剂可以使用1000多个小时, 催化活性几乎没有降低且在产物中未检测到铑的流失.当长链的十二烯用于反应时, 经过504 h的反应, 转化率仍然保持在86.6%, 且直链醛与支链醛比值可以达到6.4.该结果进一步证明了Rh/POL-PPh3催化性能和稳定性.高角环形暗场扫描透射电子显微镜(HAADF-STEM)显示, Rh/POL-PPh3催化剂中Rh以单原子形态存在.使用后的催化剂也没有出现铑金属的烧结现象.拓展X射线吸收精细结构谱图(EXAFS)分析表明, 在使用后的催化剂中含有Rh—P和Rh—C的键, 并没有发现Rh—Rh键, 进一步说明了铑的单原子形态, 同时证明聚合物中Rh与PPh3配位非常稳定.
亚磷酸酯配体对水比较敏感, 容易发生配体分解, 导致催化剂的活性和选择性的降低. 2016年, 肖丰收课题组和马胜前课题组[31]发展的亚磷酸酯均聚物, 在实现催化剂固载化的同时, 解决了亚磷酸酯对水的敏感问题, 代表性的聚合物为Phosphite-POP.以邻叔丁基苯酚为底物, 经过六步反应得到乙烯基亚磷酸酯单体; 在偶氮二异丁腈(AIBN)的引发下, 单体经自由基反应得到Phosphite-POP (Scheme 12).聚合物Phosphite-POP的BET比表面积和孔容分别为643 m2•g-1和0.43 cm3•g-1; 孔径主要分布在0.5~1.4和2~10 nm, 表明聚合物由微孔和介孔构成(表 1, Entry 3).更重要的是, Phosphite-POP表现出优异的超疏水性能; 该疏水性能能够有效阻止水分子进入聚合物的孔隙, 使得Phosphite-POP具有很好的水稳定性.该聚合物负载Rh(CO)2(acac)前体得到Rh/phos- phite-POP催化剂.在具有挑战性的2-辛烯的异构化/氢甲酰化反应中, Rh/phosphite-POP表现出优秀的催化活性、化学选择性和循序性, 但反应的区域选择性偏低(Scheme 12).
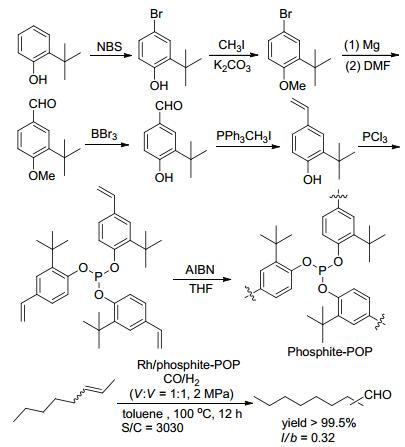
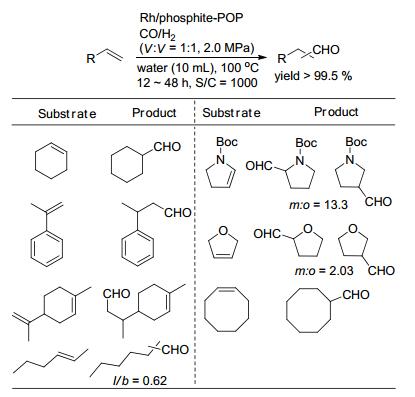
之后, 肖丰收课题组又考察了Rh/phosphite-POP催化剂中P/Rh物质的量比(P:Rh=2, 4, 9, 18, 36)对2-辛烯的异构化/氢甲酰化反应的催化活性、选择性及催化剂的循环性的影响[32].在P/Rh比为9时, 催化剂表现出最高的反应活性, 降低和升高P/Rh比都会引起催化活性的降低; P/Rh比的改变并没有引起反应的化学选择性和区域选择性的变化.当固载催化剂的P/Rh比大于9时, 催化剂循环十次仍然保持反应的活性和选择性, 经ICP-OES检测后, 未发现铑物种的流失; 当P/Rh比小于9时(P:Rh=2, 4), 五次循环之后, 铑的流失量分别达到16.9%和6.8%, 且醛的收率逐渐降低.由此可见, 膦配体的浓度对催化剂的活性和铑的流失具有重要的决定作用.高浓度的膦配体可以稳定催化剂, 且自由的膦配体可以配位流失的铑物种, 从而减少铑流失和保持催化剂的活性.另外, 以水作为反应溶剂, Rh/phosphite- POP催化不同种类烯烃的氢甲酰化反应, 并以优秀的收率得到目标醛(Scheme 13).
与单膦配体相比, 双膦配体与铑配体形成的络合物具有更稳定的构型和空间立体效应, 用于催化烯烃氢甲酰化反应时往往可以取得更高的选择性. 2015年, 肖丰收课题组[33]发展了一系列双齿膦配体聚合物负载铑催化剂, 并将其用于烯烃的氢甲酰化反应研究.用4-乙烯苯基格氏试剂与不同的烷基膦氯和芳基膦氯反应, 分别得到不同的乙烯基双膦配体单体dppm [bis-(diphen-ylphosphino)methane]、dppe [1, 2-bis(diphenylphosphi-no)ethane]和dppb [1, 4-bis(diphenylphosphino)butane].在AIBN引发剂的作用下, 得到POL-dppm、POL-dppe和POL-dppb聚合物; 进一步各聚合物与Rh(CO)2(acac)络合, 得到相应的负载催化剂(Scheme 14).
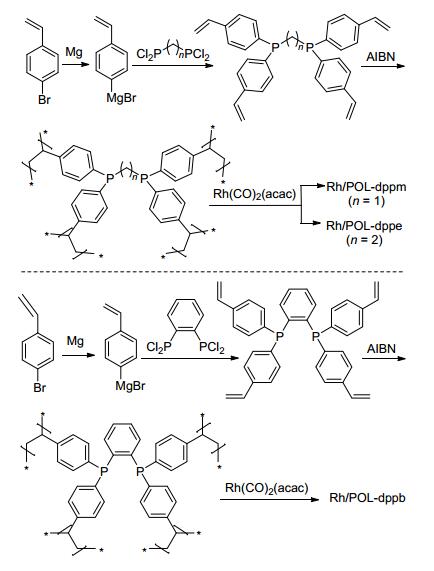
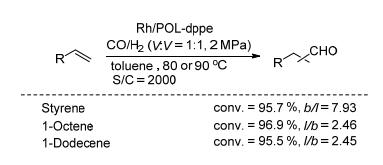
相比较Rh/POL-dppm和Rh/POL-dppb, 催化剂Rh/ POL-dppe在烯烃的氢甲酰化反应中表现出更优秀的催化性能. POL-dppe主要由微孔和介孔组成, 且具有好的热稳定性(表 1, Entry 5);该聚合物在反应溶剂中会发生溶胀, 可以使负载的Rh金属更好地分散在反应溶液中, 提高催化效率.当以苯乙烯为反应底物时, 能以优秀的转化率和高的区域选择性得到支链醛.在催化剂的循环测试中, Rh/POL-dppe循环使用五次, 均能保持稳定的催化活性和选择性.更重要的是, 该催化剂非常容易与产物分离, 滤液经ICP-OES检测未发现铑的流失(<10 ppb).另外, 在以长链的1-辛烯和1-十二烯的氢甲酰化的反应中, 能以高的转化率和中等的区域选择性得到直链产物(Scheme 15).在相同的反应条件下, 与均相的Rh/dppe催化体系相比, Rh/POL-dppe表现出更优秀的催化性能.
从以上的进展可以发现, 虽然一系列高效的多孔聚合物负载的铑催化剂用于烯烃的氢甲酰化反应, 如Rh/POL-dppe、Rh/POL-PPh3和Rh/Phosphite-POP, 但是低的区域选择性仍然是需要解决的问题.通过在乙烯基三苯基膦的聚合物中嵌入大位阻的配体, 如双齿膦配体、亚磷酸酯配体和亚磷酰胺配体等, 有助于明显提高催化剂在氢甲酰化反应催化剂的活性和选择性.在该类膦配体共聚物中, 三苯基膦单元常用来构建稳定的多孔结构; 嵌入的膦配体单元用来提高催化剂的活性和选择性.另外共聚物中的双膦配体和三苯基膦可以与铑同时配位形成新的活性物种, 该协同作用有利于提高催化剂的活性和选择性.
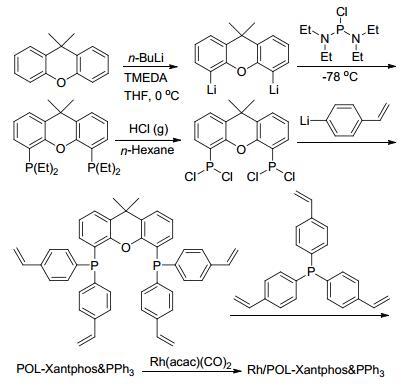
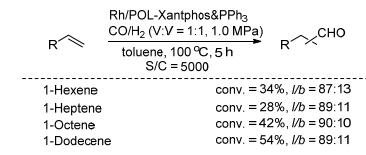
双膦配体Xantphos在均相的烯烃氢甲酰反应中有着重要的应用. 2016年, 丁云杰和严丽课题组[34]合成了乙烯基的Xantphos单体, 并将其与乙烯基PPh3聚合得到共聚物POL-Xantphos & PPh3 (Scheme 16). Rh/POL- Xantphos & PPh3催化C(6)~C(12)的末端烯烃的氢甲酰化反应中, 能以高的催化活性(TOF: 300~500 h-1)、化学选择性(87%~91%)和区域区域选择性得到直链醛(Scheme 17).在1-辛烯的反应中, 该催化剂可以循环使用5次, 并能保持的催化活性和选择性.更重要的是, 结合高角环形暗场扫描透射电子显微镜(HAADF- STEM)和拓展X射线吸收精细结构谱图(EXAFS)分析表明, 铑在固载催化剂中以单原子形态分散.进一步综合DFT (Discrete fourier transform)计算和实验数据对比, 表明在反应中铑同时与两种膦配体配位, 形成了三膦配位的活性中间体.该催化物种有助于催化剂的稳定性和选择性.
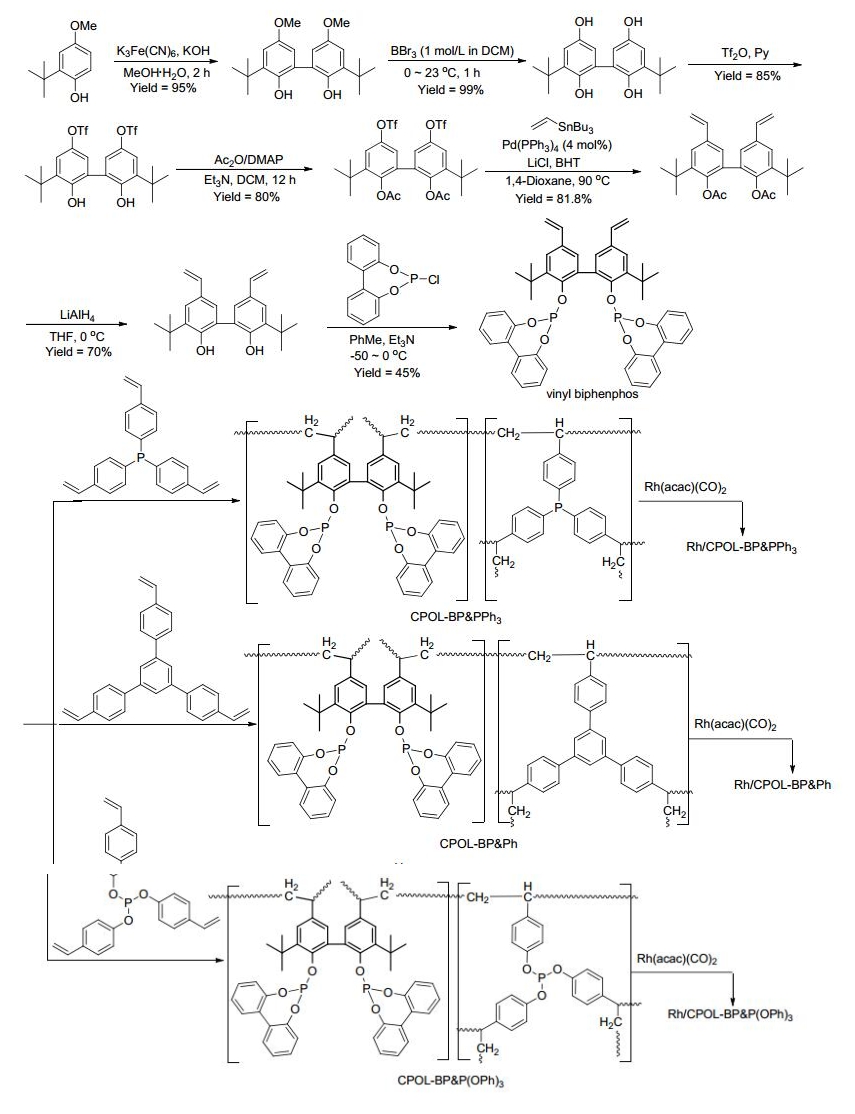
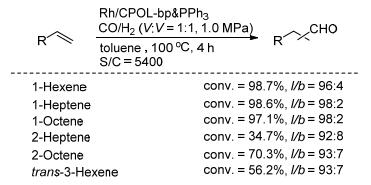
相比较膦配体、亚磷酸酯磷配体和亚磷酰胺磷配体具有强的π酸性和大的空间效应, 在烯烃的氢甲酰化反应中, 可以得到更高的催化活性和区域选择性. 2016年, 丁云杰和严丽课题组[35]以2-叔丁基-4-甲氧基苯酚为原料, 经七步反应得到乙烯基Biphenphos单体; 该单体与三乙烯基PPh3共聚得到聚合物CPOL-BP & PPh3; 共聚物进一步与Rh(acac)(CO)2反应得到催化剂Rh/CPOL- BP & PPh3 (Scheme 18).经表征, CPOL-BP & PPh3具有高的比表面积(643 m2•g-1)、孔容(2.07 cm3•g-1)和好的热稳定性(430 ℃, 表 1, Entry 11). Rh/CPOL-bp & PPh3非均相催化剂在末端烯烃氢甲酰化反应中表现出优秀的性能, 以高的催化活性和优秀的区域选择性(l/b=96:4~98:2)得到相应的直链醛(Scheme 19); 催化性能甚至优于Rh/乙烯基Biphenphos催化体系.另外, 该催化剂可以循环使用六次, 且能保持稳定的催化活性和选择性.值得注意的是, Rh/CPOL-BP & PPh3还能催化内烯烃的氢甲酰化反应, 以高的区域选择性(l/b=92:8~93:7)得到直链醛产物(Scheme 19).之后, 丁云杰课题组将Rh/CPOL-BP & PPh3应用于固定床反应器, 研究了其在丁烯的氢甲酰化反应中的催化性能[36].在Rh/CPOL- BP & PPh3摧毁的1-丁烯的反应中, 以优秀的催化活性(TOF=11200 h-1)和优秀的区域选择性(l:b=62.2)得到直链的戊醛.该催化剂在100到300 h的连续反应中, 能保持高的催化活性(TOF=5400 h−1)和区域选择性(l: b>61).为了研究催化剂单元结构对催化性能的影响, 还合成了Rh/CPOL-BP & Ph催化剂(Scheme 18).在Rh/CPOL-BP & Ph催化的1-己烯的氢甲酰化中, 反应时间为100~300 h内, TOF值为2500 h−1且l:b值从45降至18.通过实验对比及催化剂结构表征, Rh/CPOL- BP & PPh3优秀的催化性能与两种不同膦配体(Bi-phen- phos和PPh3)和铑金属的协同作用有关.
2018年, 丁云杰和严丽课题组[37]将乙烯基Biphen- phos和乙烯基的三苯基亚磷酸酯配体聚合得到CPOL- BP & P(OPh)3 (Scheme 18), 并研究了该类聚合物负载铑催化剂在氢甲酰化反应中的应用.在1-丁烯为底物反应中, 该催化剂能以高的活性(TOF=2490.3 h-1)和区域选择性(l:b=40.0)得到直链戊醛.作者进一步通过在线FT-IR考察了反应中催化剂物种的变化.经实验发现, 在聚合物负载催化反应过程中, 铑金属更倾向同时与不同类型的膦(Biphenphos和P(OPh)3)配位形成新的Rh-H物种(Scheme 20).当Rh/CPOL-BP & P(OPh)3应用于固定床反应器测试时, 反应36 h后, TOF值从4626降至2806 h-1; 但反应24 h后, l:b值从18.6升高到49.4.进一步证明反应中逐渐形成的Rh-H催化物种对区域选择性的提高有重要的决定作用.
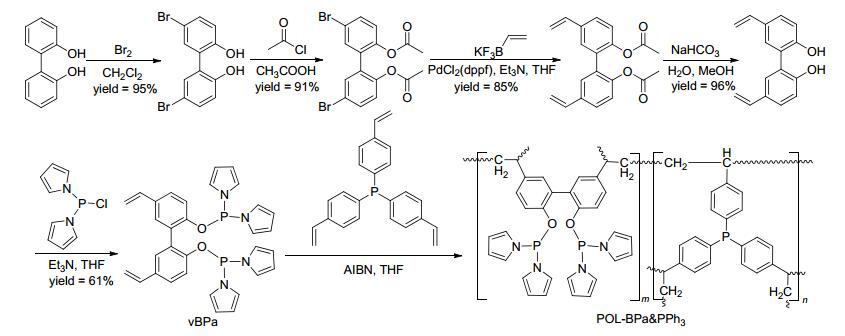
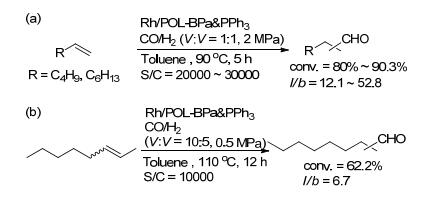
我们课题组[38]发展了一类双齿亚磷酰胺配体与PPh3聚合物, 将其负载铑金属后得到Rh/POL-BPa & PPh3.以2, 2'-二羟基联苯为原料, 经溴化反应、羟基保护、Suzuki偶联等五步反应得到乙烯基双齿亚磷酰胺配体单体(Scheme 21). Rh/POL-BPa & PPh3具有高的表面积和热稳定性(表 1, Entry 16).该催化剂在末端烯烃的氢甲酰化反应中表现出优秀的活性、区域选择性(l/b=12.1~52.8)和循环性(Scheme 22a).在1-己烯为底物的反应中, TON最高可达45.3×104; 且催化剂可以循环使用10次, 并能保持反应活性和区域选择性.通过实验对比, Rh/POL-BPa & PPh3的催化性能要优于Rh/POL-PPh3催化剂, 说明聚合物中双齿亚磷酰胺配体单元对催化剂的性能提高有着重要的影响.另外, 该类催化剂在内烯烃的异构化/氢甲酰化反应中也表现出优秀的催化性能(Scheme 22b).
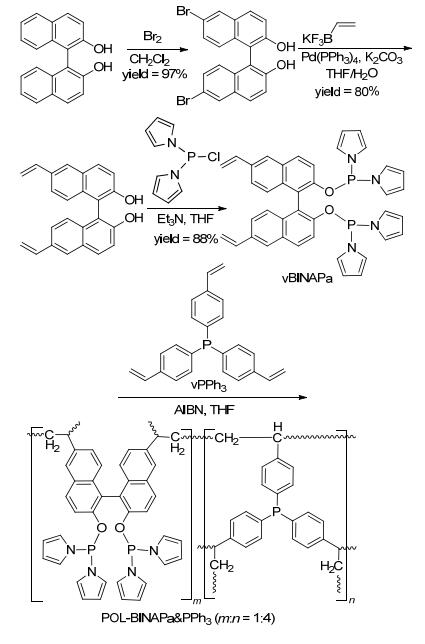
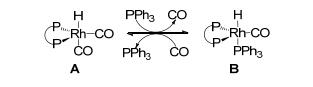
基于以上的研究基础, 我们进一步将该类催化剂用于非均相的炔烃氢甲酰化反应研究.与烯烃相比, 炔烃氢甲酰化反应表现出低的催化活性、难以控制的化学选择性和区域选择性[39].首先合成了以联萘为骨架的乙烯基亚磷酸酰胺单体, 并与乙烯基三苯基膦聚合得到POL-BINAPa & PPh3 (Scheme 23)[40]; 该聚合物与Rh- (acac)(CO)2配位络合得到Rh/POL-BINAPa & PPh3催化剂. Rh/POL-BINAPa & PPh3在炔烃的氢甲酰化反应中, 该类催化剂表现出优秀的催化活性、立体选择性和化学选择性(Scheme 24).在二苯乙炔为底物的氢甲酰化反应中, 催化剂用量最低可降至五万分之一.产物不饱和醛的E/Z构型比值高达40:1.该催化体系对各种官能团具有兼容性, 如甲基、甲氧基、醛基、溴基.另外, 杂芳环底物也能顺利转化得到相应的不饱和醛.该类催化剂也能高效地催化烷基炔, 以高的收率得到烷基不饱和醛. Rh/POL-BINAPa & PPh3在苯乙炔的循环催化测试中可重复使用十次, 且没有检测到铑的损失.值得注意的是, Rh/POL-BINAPa & PPh3的催化活性要高于均相Rh/BINAPa/PPh3体系.我们推测在反应中双齿亚磷酸酰胺配体和三苯基膦与铑配位, 得到三膦配位的Rh-H活性中间体B.与中间体A相比, 中间体B铑周围的空间位阻较大, 使得其在炔烃的氢甲酰化反应中表现出低的催化活性(Scheme 25).在聚合物中, 由于配体单体固定在聚合链中, 降低了两种配体之间与铑的协同效应, 因此负载后的催化剂具有更高的催化活性.
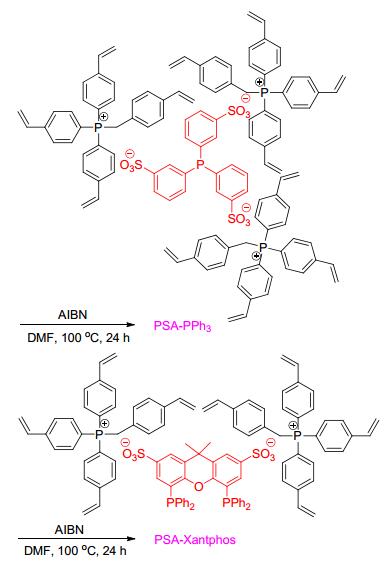
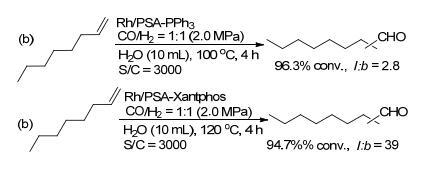
在氢甲酰化反应中, 以水作为反应溶剂, 采用水溶性的离子型膦配体, 可以实现催化剂和产物分离, 以及催化剂的循环使用. Ruhrchemie/Rhône-Poulenc作为代表型工艺, 水溶性TPPTS配体用于丙烯的氢甲酰化反应.但对于水溶性差的长链烯烃, 该类工艺表现出低的反应性能.肖丰收课题组和马胜前课题组[41]发展的离子型多空聚合物, 可以有效地解决该类问题.首先将阳离子的4-乙烯基苄基乙烯基三苯基膦与阴离子的磺酸基膦配体结合; 进一步在AIBN的催化下聚合乙烯基官能团得到离子型多孔有机膦配体聚合物.通过嵌入不同的阴离子配体可以分别得到PSA-PPh3和PSA-Xantphos, 再负载铑金属后得到相应的催化剂(Scheme 26)[41].该类催化剂具有两亲性, 不仅具有亲水性, 而且与烯烃也具有很好的相容性.以水为溶剂, Rh/PSA-PPh3和Rh/PSA- Xantphos催化1-辛烯的氢甲酰化反应, 分别以96.3%和94.7%的转化率及2.8和39的l/b值得到相应的醛产物(Scheme 27).而以Rh/TPPTS作为催化剂的反应, 仅得到29.7%转化率和2.9正异比.该工作为水相催化烯烃的氢甲酰化反应的催化剂设计开辟了新的方向.
从以上的结果可以发现, 经过近二十年的研究, 聚合物负载催化剂的发展及其应用已经取得很大的进展.特别是近几年, 由有机配体单体构建的多孔聚合物出现, 为固载催化剂的发展提供了新的契机.该方法不仅可以把单齿和双齿膦配体嵌入到聚合物链中, 而且可以把不同类型的配体修饰到多孔聚合物中, 包括膦配体、亚磷酸酯配体、亚磷酰胺配体和离子型膦配体.发展的多孔聚合物负载催化剂具有高的比表面积和好的稳定性; 在氢甲酰化反应的应用中, 该类多相催化剂可以取得与均相体系一样甚至更加优异催化效果.更重要的是, 由配体构建的聚合物结构可以有效避免铑的流失, 使催化剂具有好的循环性.基于实验结果和材料的优秀性能, 都为多孔聚合物负载催化剂的工业化奠定了基础.与无机物固载催化剂相比, 多孔有机聚合物负载催化剂在活性、选择性和稳定性方面都表现出更优的性能.因此, 多孔有机聚合物锚定铑的多相催化剂可能成为非均相氢甲酰化反应催化剂的重要研究方向.
尽管多孔聚合物负载催化剂的研究工作取得了一定得进展, 但仍存在许多亟待解决的问题: (1)进一步探索聚合物中配体和铑的配位关系及催化剂活性中间体, 为提高催化剂性能和设计新型催化剂提供理论基础; (2)多孔聚合物的孔径主要分布在微孔和介孔范畴.孔径的大小与催化活性、选择性及反应底物的影响关系需要进一步探索; (3)非均相氢甲酰化催化剂的研究主要集中于非手性催化剂, 手性聚合物负载催化剂发展和应用仍是一个具有挑战和潜力的研究方向[42]; (4)目前探索的底物相对比较狭窄, 因此有必要进一步拓展烯烃底物的范围.
另外, 目前聚合物负载催化剂在实验室取得了一定的成功, 但该类催化剂在工业化过程中仍然有很多问题需要解决: (1)氢甲酰化反应是一个放热反应, 但聚合物载体的热传导性较差, 反应体系内部温度过高将会影响催化剂的活性和选择性, 甚至导致催化剂的钝化或失活; (2)相比较无机载体, 聚合物载体的机械强度较差, 这样会增加该类催化剂在使用中的损耗; (3)聚合物负载催化剂的合成步骤较多, 势必会导致“三废”问题的产生.另外, 合成过程中对无水无氧条件要求严格.这些都会影响到催化剂的大量生产.
因此, 在铑催化的非均相的氢甲酰化反应研究中, 新型高效多孔聚合物负载催化剂的研究不仅是一个具有挑战性和发展潜力的方向, 而且具有巨大的应用前景.
(a) van Leeuwen, P. W. N. M.; Claver, C. Rhodium catalyzed hydroformylation, Kluwer Academic Publishers, Dordrecht, 2000.
(b) Haumann, M.; Riisager, A. Chem. Rev. 2008, 108, 1474.
(c) Hebrard, F.; Kalck, P. Chem. Rev. 2009, 109, 4272.
(d) Franke, R.; Selent, D.; Borner, A. Chem. Rev. 2012, 112, 5675.
(e) Pospech, J.; Fleischer, I.; Franke, R.; Buchholz, S.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 2852.
Roelen, O. U. S. 2327066, 1943[Chem. Abstr. 1944, 38, 363].
Börner, A.; Franke, R. Hydroformylation: Funda Mentals, Processes, and Applications in Organic Synthesis, Wiley-VCH, Weinheim, 2016.
(a) Casey, C. P.; Paulsen, E. L.; Beuttenmueller, E. W.; Proft, B. R.; Petrovich, L. M.; Matter, B. A.; Powell, D. R. J. Am. Chem. Soc. 1997, 119, 11817.
(b) Herrmann, W. A.; Kohlpaintner, C. W.; Herdtweck, E.; Kiprof, P. Inorg. Chem. 1991, 30, 4271.
(c) Yu, S.; Zhang, X.; Yan, Y.; Cai, C.; Dai, L.; Zhang, X. Chem. Eur. J. 2010, 16, 4938.
(d) Yan, Y.; Zhang, X.; Zhang, X. Adv. Synth. Catal. 2007, 349, 1582.
(e) Chen, C.; Li, P.; Hu, Z.; Wang, H.; Zhu, H.; Hu, X.; Wang, Y.; Lv, H.; Zhang, X. Org. Chem. Front. 2014, 1, 947.
Klein, H.; Jackstell, R.; Wiese, K.-D.; Borgmann, C.; Beller, M. Angew. Chem., Int. Ed. 2001, 40, 3408.
(a) Carbó, J. J.; Maseras, F.; Bo. C.; van Leeuwen, P. W. N. M. J. Am. Chem. Soc. 2001, 123, 7630.
(b) Kranenburg, M.; van der Burgt, Y. E. M.; Kamer, P. C. J.; van Leeuwen, P. W. N. M.; Goubitz, K.; Fraanje, J. Organometallics 1995, 14, 3081.
(c) Van der Veen, L. A.; Boele, M. D. K.; Bregman, F. R.; Kamer, P. C. J.; van Leeuwen, P. W. N. M.; Goubitz, K.; Fraanje, J.; Schenk, H.; Bo, C. J. Am. Chem. Soc. 1998, 120, 11616.
(d) van der Veen, L. A.; Kamer, P. C. J.; van Leeu wen, P. W. N. M. Angew. Chem., Int. Ed. 1999, 38, 336.
Burke, P. M.; Garner, J. M.; Kreutzer, K. A.; Teunis sen, A. J. J. M.; Snijder, C. S.; Hansen, C. B. WO 97/33854, 1997.
(a) Cunny, G. D.; Buchwald, S. L. J. Am. Chem. Soc. 1993, 115, 2066.
(b) Behr, A.; Obst, D.; Schulte, C. J. Mol. Catal. A-Chem. 2003, 206, 179.
(a) van der Slot, S. C.; Duran, J.; Luten, J.; Kamer, P. C. J.; van Leeuwen, P. W. N. M. Organometallics 2002, 21 3873.
(b) Yan, Y.; Zhang, X.; Zhang, X. J. Am. Chem. Soc. 2006, 128, 16058.
(c) Yu, S.; Chie, Y.; Guan, Z.; Zou, Y.; Li, W.; Zhang, X. Org. Lett. 2009, 11, 241.
(d) Jia, X.; Wang, Z.; Xia, C.; Ding, K. Chem. Eur. J. 2012, 18, 15288.
(e) Ren, X.; Zheng, Z.; Zhang, L.; Wang, Z.; Xia, C.; Ding, K. Angew. Chem., Int. Ed. 2017, 56, 310.
(f) Jia, X.; Ren, X.; Wang, Z.; Xia, C.; Ding, K. Chin. J. Org. Chem. 2019, 39, 207(in Chinese)
(贾肖飞, 任新意, 王正, 夏春谷, 丁奎岭, 有机化学, 2019, 39, 207.)
(g) Chen, C.; Qiao, Y.; Geng, H.; Zhang, X. Org. Lett. 2013, 15, 1048.
(a) Li, C; Wang, W; Yan, L.; Ding, Y. Front. Chem. Sci. Eng. 2018, 12, 113.
(b) Zhang, J.; Sun, P.; Zhao, Z. L.; Li, F. W. Chin. Sci. Bull. 2019, 64, 3173.
(a) Arhancet, J. P.; Davis, M. E.; Merola, J. S.; Hanson, B. E. Nature 1989, 339, 454.
(b) Chaudhari, R. V.; Bhanage, B. M.; Deshpande, R. M. Nature 1995, 373, 501.
(c) Sharma, S. K.; Jasra, R. V. Catal. Today 2015, 247, 70.
(d) Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, Compt. Rend. Chim. 2011, 14, 149.
(e) Paganelli, S.; Piccolo, O.; Pontini, P.; Tassini, R.; Rathod, V. D. Catal. Today 2015, 247, 64.
(a) Horváth, I. T.; Kiss, G.; Cook, R. A., Bond, J. E.; Stevens, P. A.; Rábai, J.; Mozeleski, E. J. J. Am. Chem. Soc. 1998, 120, 3133.
(b) Cornils, B. Angew. Chem., Int. Ed. 1997, 36, 2057.
(c) Chen, W. P.; Xu, L. J.; Xiao, J. L. Chem. Commun. 2000, 10, 839.
(d) Horvath, I. T.; Rabai, J. Science 1994, 266, 72.
(a) Mehnert, C. P.; Cook, R. A.; Dispenziere, N. C.; Afeworki, M. J. Am. Chem. Soc. 2002, 124, 12932.
(b) Riisager, A.; Fehrmann, R.; Flicker, S.; van Hal, R.; Haumann, M.; Wasserscheid, P. Angew. Chem., Int. Ed. 2005, 44, 815.
(c) Jin, X.; Feng, J.; Ma, Q.; Song, H.; Liu, Q.; Xu, B.; Zhang, M.; Li, S.; Yu, S. Green Chem. 2019, 21, 3267.
(d) Walter, S.; Spohr, H.; Franke, R.; Hieringer, W.; Wasserscheid, P.; Haumann, M. ACS Catal. 2017, 7, 1035
(a) David, J.; Cole-Hamilton, O. J. Science 2003, 299, 1702.
(b) Jessop, P. G.; Hsion, Y.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1996, 118, 344.
(c) Kainz, S., Koch, D.; Baumann, W.; Leitner, W. Angew. Chem., Int. Ed. 1997, 36, 1628.
(d) Koeken, A. C. J.; Smeets, N. M. B. Catal. Sci. Technol. 2013, 3, 1036.
(e) Estorach, C. T.; Orejon, A.; Masdeu-Bulto, A. M. Green Chem. 2008, 10, 545.
Gärtner, L.; Cornils, B.; Lappe, P. (to Ruhrchemie AG) EP 0107006, 1983[Chem. Abstr. 1984, 101, 55331].
(a) Kuntz, E. G. CHEMTECH 1987, 17, 570.
(b) Cornils, B.; Kuntz, E. G. J. Organomet. Chem.1995, 502, 177.
Herrmann, W. A.; Kohlpaintner, C. W.; Bahrmann, H.; Konkol, W. J. Mol. Catal. 1992, 73, 191.
Bahmann, H.; Bergrath, K.; Kleiner, H.-J.; Lappe, P.; Naumann, C.; Peters, D.; Regnat, D. J. Organomet. Chem. 1996, 520, 97.
(a) Vunain, E.; Ncube, P.; Jalama, K.; Meijboom, R. J. Porous Mater. 2018, 25, 303.
(b) Malihan, L. B.; Nisola, G. M.; Mittal, N.; Lee, S.-P.; Seo, J. G.; Kim, H.; Chung, W. J. RSC Adv. 2016, 6, 33901.
(c) Sudheesh, N.; Parmar, J. N.; Shukla, R. S. Appl. Catal. A Gen. 2012, 415, 124.
(d) Yan, L.; Ding, Y. J.; Lin, L. W.; Zhu, H. J.; Yin, H. M.; Li, X. M.; Lu, Y. J. Mol. Catal. A-Chem. 2009, 300, 116.
(a) Wolf, P.; Logemann, M.; Schorner, M.; Keller, L.; Haumann, M.; Wessling, M. RSC Adv. 2019, 9, 27732.
(b) Weiss, A.; Munoz, M.; Haas, A.; Rietzler, F.; Steinruck, H.-P.; Haumann, M.; Wasserscheid, P.; Etzold, B. J. ACS Catal. 2016, 6, 2280.
(c) Weiβ A.; Giese, M.; Lijewski, M.; Franke, R.; Wasserscheid, P.; Haumann, M. Catal. Sci. Technol. 2017, 7, 5562.
(a) Chuai, H. Y.; Su, P.; Liu, H.; Zhu, B.; Zhang, S.; Huang, W. Catalysts 2019, 9, 194.
(b) Liu, J.; Yan, L.; Ding, Y.; Jiang, M.; Dong, W.; Song, X.; Liu, T.; Zhu, H. Appl. Catal. A Gen. 2015, 492, 127.
(a) Nozaki, K.; Itoi, Y.; Shibahara, F.; Shirakawa, E.; Ohta, T.; Takaya, H.; Hiyama, T. J. Am. Chem. Soc. 1998, 120, 4051.
(b) Nozaki, K.; Shibahara, F.; Itoi, Y.; Shirakawa, E.; Ohta, T.; Takaya, H.; Hiyama, T. Bull. Chem. Soc. Jpn. 1999, 72, 1911.
(a) Shibahara, F.; Nozaki, K.; Hiyama, T. J. Am. Chem. Soc. 2003, 125, 8555.
(b) Nozaki, K.; Shibahara, F.; Hiyama, T. Chem. Lett. 2000, 694.
(a) Stiriba, S. E.; Slagt, M. Q.; Kautz, H.; Klein Gebbink, R. J. M.; Thomann, R.; Frey, H.; van Koten, G. Chem. Eur. J. 2004, 10, 1267.
(b) Kumar, K. R.; Kizhakkedathu, J. N.; Brooks, D. E. Macromol. Chem. Phys. 2004, 205, 567.
(c) Wilms, D.; Stiriba, S. E.; Frey, H. Acc. Chem. Res. 2010, 43, 129.
(d) Slagt, M. Q.; Stiriba, S.-E.; Kautz, H.; Klein Gebbink, R. J. M.; Frey, H.; van Koten, G. Organometallics 2004, 23, 1525.
Ricken, S.; Osinski, P. W.; Eilbracht, P.; Haag, R. J. Mol. Catal. A-Chem. 2006, 257, 78.
(a) Wang, H.; Sun, W.; Xia, C. J. Mol. Catal. A: Chem. 2003, 206, 199.
(b) Makhubela, B. C. E.; Jardine, A.; Smith, G. S. Appl. Catal. A Gen. 2011, 393, 231.
(c) Hertrich, M. F.; Scharnagl, F. K.; Pews-Davtyan, A.; Kreyenschulte, C. R.; Lund, H.; Bartling, S.; Jackstell, R.; Beller, M. Chem.-Eur. J. 2019, 25, 5534.
(d) Molnar, A. Coord. Chem. Rev. 2019, 388, 126.
(e) Antony, R.; Arun, T.; Manickam, S. T. D. Int. J. Biol. Macromol. 2019, 129, 615.
Makhubela, B. C. E.; Jardine, A.; Smith, G. S. Green Chem. 2012, 14, 338.
(a) Shifrina, Z. B.; Matveeva, V. G.; Bronstein, L. M. Chem. Rev. 2020, 120, 1350.
(b) Kramer, S.; Bennedsen, N. R.; Kegnæs, S. ACS Catal. 2018, 8, 6961.
(c) Sun, Q.; Dai, Z.; Meng, X.; Xiao, F.-S. Chem. Soc. Rev. 2015, 44, 6018.
(d) Kaur, P.; Hupp, J. T.; Nguyen, S. B. T. ACS Catal. 2011, 1, 819.
Sun, Q.; Jiang, M.; Shen, Z.; Jin, Y.; Pan, S.; Wang, L.; Meng, X.; Chen, W.; Ding, Y.; Li, J.; Xiao, F.-S. Chem. Commun. 2014, 50, 11844.
Jiang, M.; Yan, L.; Ding, Y.; Sun, Q.; Liu, J.; Zhu, H.; Lin, R.; Xiao, F.; Jiang, Z.; Liu, J. J. Mol. Catal. A: Chem. 2015, 404, 211.
Sun, Q.; Aguila, B.; Verma, G.; Liu, X.; Dai, Z.; Deng, F.; Meng, X.; Xiao, F.-S.; Ma, S. Chem 2016, 1, 628.
Tang, Y.; Dong, K.; Wang, S.; Sun, Q.; Meng, X.; Xiao, F.-S. Mol. Catal. 2019, 474, 110408.
Sun, Q.; Dai, Z.; Liu, X.; Sheng, N.; Deng, F.; Meng, X.; Xiao, F. S. J. Am. Chem. Soc. 2015, 137, 5204.
Li, C.; Sun, K.; Wang, W.; Yan, L.; Sun, X.; Wang, Y.; Xiong, K.; Zhan, Z.; Jiang, Z.; Ding, Y. J. Catal. 2017, 353, 123.
Li, C.; Xiong, K.; Yan, L.; Jiang, M.; Song, X.; Wang, T.; Chen, X.; Zhan, Z.; Ding, Y. Catal. Sci. Technol. 2016, 6, 2143.
Wang, Y.; Yan, L.; Li, C.; Jiang, M.; Wang, W.; Ding, Y. Appl. Catal. A Gen. 2018, 551, 98.
Wang, Y.; Yan, L.; Li, C.; Jiang, M.; Zhao, Z.; Hou, G.; Ding, Y. J. Catal. 2018, 368, 197.
Jia, X.; Liang, Z.; Chen, J.; Lv, J.; Zhang, K.; Gao, M.; Zong, L.; Xie, C. Org. Lett. 2019, 21, 2147.
(a) Johnson, J. R.; Cuny, G. D.; Buchwald, S. L. Angew. Chem., Int. Ed. Engl. 1995, 34, 1760.
(b) Agabekov, V.; Seiche, W.; Breit, B. Chem. Sci. 2013, 4, 2418.
(c) Fang, X.; Zhang, M.; Jackstell, R.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 4645.
(d) Zhang, Z.; Wang, Q.; Chen, C.; Han, Z.; Dong, X.; Zhang, X. Org. Lett. 2016, 18, 3290.
Liang, Z.; Chen, J.; Chen, X.; Zhang, K.; Lv, J.; Zhao, H.; Zhang, G.; Xie, C.; Zong, L.; Jia, X. Chem. Commun. 2019, 55, 13721.
Dong, K.; Sun, Q.; Tang, Y.; Shan, C.; Aguila, B.; Wang, S.; Meng, X.; Ma, S.; Xiao, F.-S. Nat. Commun. 2019, 10, 3059.
Wang, T.; Wang, W.; Lyu, Y.; Xiong, K.; Li, C.; Zhang, H.; Zhan, Z.; Jiang, Z.; Ding, Y. Chin. J. Catal. 2017, 38, 691.

图式 1 氢甲酰化产物醛及衍生物
Scheme 1 Hydroformylation of olefins to aldehydes and their derivatives

图式 4 用于水/有机两相的氢甲酰化反应的膦配体
Scheme 4 Phosphine ligands used for aqueous two-phase hydroformylation

图式 5 聚苯乙烯负载的(R, S)-BINAPHOS-Rh(I)
Scheme 5 Polystyrene-supported (R, S)-BINAPHOS-Rh(I)

图式 6 PS-[Rh(acac)(R, S)-BINAPHOS]催化的烯烃的不对称氢甲酰化反应
Scheme 6 PS-[Rh(acac)(R, S)-BINAPHOS] catalyzed asymmetric hydroformylation of olefins

图式 7 PG-NIXANTPHOS和PG/PU-NIXANTPHOS的合成
Scheme 7 Synthesis of PG-NIXANTPHOS and PG/PU-NIX- ANTPHOS

图式 8 PG-NIXANTPHOS-Rh(I)和PG/PU-NIXANTPHO S- Rh(I)催化的烯烃氢甲酰化反应
Scheme 8 PG-NIXANTPHOS-Rh(I) and PG/PU-NIXANT PHOS-Rh(I) catalyzed hydroformylation of olefins

图式 9 壳聚糖负载铑催化剂的制备及应用
Scheme 9 Synthesis and application of chitosan-supported Rh catalyst

图式 11 Rh/POL-PPh3催化的烯烃氢甲酰化反应
Scheme 11 Rh/POL-PPh3 catalyzed hydroformylation of olefins

图式 13 Rh/phosphite-POP催化的烯烃氢甲酰化反应
Scheme 13 Rh/phosphite-POP catalyzed hydroformylation of olefins

图式 14 Rh/POL-dppm、Rh/POL-dppe和Rh/POL-dppb的合成
Scheme 14 Synthesis of Rh/POL-dppm, Rh/POL-dppe and Rh/ POL-dppb

图式 15 Rh/POL-dppe催化的烯烃氢甲酰化反应
Scheme 15 Rh/POL-dppe catalyzed hydroformylation of olefins

图式 17 Rh/POL-Xantphos & PPh3催化的烯烃氢甲酰化反应
Scheme 17 Rh/POL-Xantphos & PPh3 catalyzed hydroformylation of olefins

图式 18 Rh/CPOL-BP & PPh3、Rh/CPOL-BP & Ph和Rh/CPOL-BP & P(OPh)3的合成
Scheme 18 Synthesis of Rh/CPOL-BP & PPh3, Rh/CPOL-BP & Ph and Rh/CPOL-BP & P(OPh)3

图式 19 Rh/CPOL-BP & PPh3催化的烯烃氢甲酰化反应
Scheme 19 Rh/CPOL-BP & PPh3 catalyzed hydroformylation of olefins

图式 20 Rh/CPOL-BP & P(OPh)3催化的氢甲酰化反应中形成的Rh-H物种
Scheme 20 Rh-H species in the Rh/CPOL-BP & PPh3 catalyzed hydroformylation of olefins

图式 22 Rh/CPOL-BPa & PPh3催化的烯烃氢甲酰化反应
Scheme 22 Rh/CPOL-BPa & PPh3 catalyzed hydroformylation of olefins

图式 27 Rh/PSA-PPh3和Rh/PSA-Xantphos催化的1-辛烯氢甲酰化反应
Scheme 27 Rh/PSA-PPh3 and Rh/PSA-Xantphos catalyzed hydroformylation of 1-octene
表 1 多孔聚合物和及其负载催化剂的结构参数
Table 1. Textural parameters of porous organic polymers and their supported catalysts
| Entry | Sample | BET surface area/(m2•g-1) |
Pore volume/ (cm3•g-1) |
Pore size distribution/ nm |
Decomposition temperature/℃ |
Reference |
| 1 | POL-PPh3 | 1086 | 1.70 | 0.7, 1.5, 3~70 | 440 | [29] |
| 2 | Rh/POL-PPh3 | 1032 | 1.69 | — | — | [29] |
| 3 | Phosphite-POP | 643 | 0.43 | 0.5–1.4, 2~10 | — | [31] |
| 4 | Rh/phosphite-POP | 616 | 0.43 | — | — | [31] |
| 5 | POL-dppe | 943 | 1.51 | 0.7−1.5, 2.8~150 | >400 | [33] |
| 6 | Rh/POL-dppe | 856 | 1.41 | — | — | [33] |
| 7 | POL-dppm | 892 | 1.07 | — | — | [33] |
| 8 | POL-dppb | 846 | 0.81 | — | — | [33] |
| 9 | POL-Xantphos & PPh3 | 1022 | 1.24 | 0.6–1, 1.2~2, 2~10 | >450 | [34] |
| 10 | Rh/POL-Xantphos & PPh3 | 883.9 | 1.16 | — | - | [34] |
| 11 | CPOL-BP & PPh3 | 1088.0 | 2.07 | 0.70, 0.84, 1.38, 2.18 | >430 | [35] |
| 12 | Rh/CPOL-BP & PPh3 | 985.3 | 1.94 | — | — | [35] |
| 13 | Rh/CPOL-BP & Ph | 1116 | 1.66 | — | — | [36] |
| 14 | CPOL-BP & P(OPh)3 | 635 | 0.72 | 0.7, 0.85, 1.38, 1.89, 2~10 | — | [37] |
| 15 | Rh/CPOL-BP & P(OPh)3 | 556 | 0.68 | — | — | [37] |
| 16 | Rh/CPOL-BPa & PPh3 | 423.6 | 0.41 | 1.2~2.0, 2.3~25 | 370 | [38] |
| 17 | Rh/POL-BINAPa & PPh3 | 492.2 | 0.41 | — | 456 | [40] |
| 18 | PSA-PPh3 | 547 | 0.56 | — | 400 | [41] |
| 19 | PSA-Xantphos | 290 | 0.69 | — | — | [41] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们