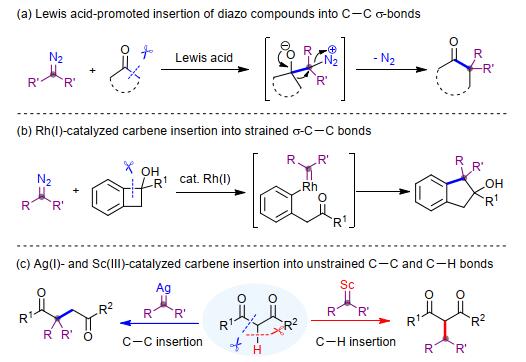
图式 1.
“一碳”插入策略构筑全碳季碳中心
Scheme 1.
Formation of all-carbon quaternary center through "one-carbon" insertion

季碳中心具有刚性和结构多样性, 是许多天然产物、药物和生物活性分子中的关键结构单元[1]. 2011年, 处于美国销售额前200名的处方药中, 有12%具有季碳中心, 但这些药物全都不是化学合成的, 而是来自天然产物的衍生化[1a].因此, 发展高效、可靠的方法来构筑季碳中心具有重要的科学意义和工业应用价值. “一碳”合成子对C—C键的选择性插入是直接构建季碳中心的理想方法, 其具有显著的原子和步骤经济性[2].该方法需要选择性地断裂惰性C—C键, 并在拥挤的碳中心同时生成两根新的C—C键, 因而极具挑战性[3].其中, 最经典的方法是重氮化合物与羰基化合物的增碳反应:重氮碳原子亲核进攻酮羰基, 随后经过1, 2-重排, 在羰基α-位生成一个季碳中心(Scheme 1a)[4].该反应通常使用路易斯酸作为催化剂, 并且底物范围往往仅限于环酮类化合物, 这是因为环酮不仅具有相对较小的空间位阻, 而且释放的环张力可驱动该反应顺利进行[3a]. 2014年, 王剑波课题组[5]以芳基重氮乙酸乙酯作为卡宾前体, 在铑的催化下, 实现了金属卡宾对苯并环丁醇C—C键的插入反应(Scheme 1b).这是首次将金属卡宾插入C—C单键中, 进而在卡宾碳上构建季碳中心, 为“一碳”插入策略开辟了新路径.最近, 我们课题组[6]在银的催化下, 实现了金属卡宾对1, 3-二羰基化合物C—C键的插入反应, 高效合成了具有重要合成用途的α-位含全碳季碳中心的1, 4-二羰基化合物, 首次将金属卡宾对C—C键的插入反应扩展到了非张力体系中, 从而打破了对环张力的依赖.更重要的是, 使用Sc(OTf)3作为催化剂时, 重氮化合物与1, 3-二羰基化合物会以完全不同的化学选择性方式发生C—H键的插入反应[7], 得到含叔碳中心的1, 3-二羰基化合物(Scheme 1c).这一反应充分展示了过渡金属催化剂在调控重氮化合物活化模式方面所具有的巨大潜力, 但是重氮化合物的具体活化模式和化学选择性产生的根本原因尚不清楚.
从完全相同的底物出发, 通过精妙的催化剂调控, 选择性地合成了结构多样、构造复杂的化合物.通过研究其结构和功能之间的关系, 可以大大加速新药研发的进程.另外, 研究不同催化体系下的反应机制, 揭示反应选择性产生的原因, 对新反应、新理论的发现以及反应的工业化应用尤为重要[8].因此, 我们最近使用密度泛函理论(DFT)计算方法, 进一步研究了银和钪催化重氮化合物与1, 3-二羰基化合物化学选择性地发生C—C键和C—H键插入的反应机理, 揭示了化学选择性产生的原因.
在我们的论文准备时期, 白若鹏等[9]基于同样的反应体系, 发表了一篇机理研究报道, 他们认为产生选择性的原因在于两种烯醇金属中间体上α-碳的亲核性和布朗斯特酸性之间的差异.而我们的计算结果表明, 银和钪金属中心之间配位数的差异可能是两种金属催化该反应产生化学选择性的根本原因.
我们采用高斯09程序包进行密度泛函理论(DFT)计算[10].使用B3LYP的泛函方法在气相中对所有构型进行了几何优化[11], 同时使用了混合基组, 即对C, H, N, O, S使用6-31G(d, p)基组, 对Ag和Sc使用SDD基组[12].在相同的计算水平上计算了振动频率, 以确保每个优化的结构或者过渡态结构都具有能量最小值, 并考虑了所有吉布斯自由能的热力学校正.数值积分计算中使用的是Gaussian 09默认积分精度10-10.对所有过渡态进行了内禀反应坐标(IRC)计算[13], 以确定处于反应势能面一阶鞍点位置的过渡态与反应物中间体或者产物能在势能面上正确连接.在最近的研究报道中, 由Truhlar及其同事提出的M06泛函方法[14]已经被广泛用来计算处理关于过渡金属体系的非共价相互作用[15], 包括银催化的反应体系[16].因此基于气相优化的几何结构, 我们采用溶剂化模型SMD[17], 在M06/6-311+G(d, p)-SDD(Ag/Sc)水平上进行了溶剂化效应的单点能计算, 溶剂为二氯甲烷(DCM)或二氯乙烷(DCE), 在标准压力下以及分别在313.5和353.5 K温度下对银体系和钪体系进行的频率计算.采用CYL View展示所有过渡态和中间体的立体结构[18].文中讨论的数值均为溶剂化效应下的热力学校正的自由能.
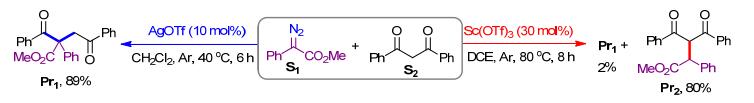
以苯基重氮乙酸甲酯(S1)和二苯甲酰基甲烷(S2)为底物模型, 分别探究了在AgOTf和Sc(OTf)3的催化下生成二烷基产物Pr1和单烷基产物Pr2的不同反应机制, 比较了金属卡宾的性质和生成路径以及银卡宾和钪卡宾与1, 3-二羰基化合物进行C—C键和C—H键插入反应的不同路径(Scheme 2).
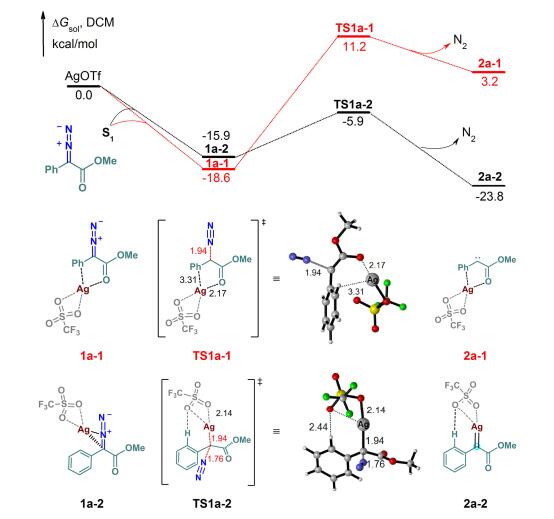
首先研究了AgOTf作为催化剂时, 重氮化合物S1与1, 3-二羰基化合物S2发生的C—C键和C—H键插入反应的不同路径.选择单独自由的AgOTf作为参考零点.计算结果表明, 该反应优先发生以下过程:首先, AgOTf催化剂促进重氮化合物脱去氮气生成银卡宾中间体, 随后银卡宾中间体与1, 3-二羰基化合物经历亲电加成、分子内环化、选择性开环和烯醇异构过程, 最终得到了C—C键插入产物Pr1.
首先, AgOTf和重氮化合物配位复合, 由于配位点的不同, 可以形成2种不同的复合物:重氮化合物上的羰基氧原子和苯环分别与银中心配位形成1a-1, 重氮化合物中重氮基团与银中心配位形成1a-2.复合物1a-1和1a-2可以分别经过TS1a-1和TS1a-2生成卡宾中间体2a-1和2a-2(图 1).虽然1a-1 (18.6 kcal/mol)比1a-2 (15.9 kcal/mol)更稳定, 前者的相对自由能比后者低了2.7 kcal/mol, 但是TS1a-1的相对自由能比TS1a-2高了17.1 kcal/mol, 且生成2a-1的过程相对于1a-1需要吸收21.8 kcal/mol的热量, 所以该反应过程在热力学上是不利的.同时卡宾化合物2a-2比2a-1更稳定, 因此, 从动力学和热力学上排除了经过过渡态TS1a-1发生脱氮气生成银卡宾的可能性.此外, 单线态的银卡宾2a-2比三线态的自由能低了23.7 kcal/mol, 其Ag—C键的距离为0.206 nm, 这个距离类似于银与N-杂环卡宾配体之间的距离, 反映了银卡宾的亲电特性[19], 这与文献上报道的银卡宾是Fisher类型的卡宾相一致[20].
探究了银卡宾中间体2a-2和1, 3-二羰基化合物S2之间的反应情况, 如图 2所示.首先, 银卡宾2a-2和1, 3-二羰基化合物的烯醇异构体S2-1形成复合物3a.银卡宾3a的卡宾碳亲电进攻S2-1的双键, 得到双性离子中间体4a.该分子内亲电加成过程需要克服的过渡态TS2a能垒仅为0.4 kcal/mol, 相对于复合物3a整体放热12.1 kcal/mol.从中间体4a, 我们分别探究了两种不同的反应路径:中间体4a上带负电荷的卡宾碳C(1)可能进攻邻近的羰基碳C(2), 得到环丙烷中间体5a, 也有可能直接发生质子化, 脱去催化剂AgOTf得到C—H插入产物Pr2.
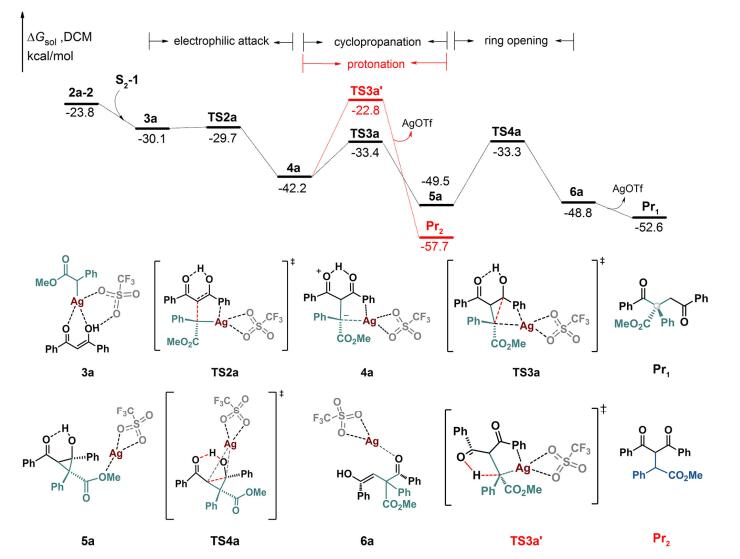
计算结果表明, 从4a经过能垒为8.8 kcal/mol的三元环过渡态TS3a, 得到环化中间体5a, 之后经过过渡态TS4a (∆G=16.2 kcal/mol)发生开环作用, 得到烯醇中间体6a, 最终经过烯醇互变异构生成α-位含全碳季碳中心的1, 4-二羰基化合物Pr1.另外, 从中间体4a, 也可以经过五元环过渡态TS3a'直接发生质子化过程, 能垒为19.4 kcal/mol, 得到C—H插入产物Pr2.环化过渡态TS3a的相对自由能比质子化过渡态TS3a'低了10.6 kcal/mol, 这是银催化反应的选择性决定步骤.
接着对Sc(OTf)3催化重氮化合物S1与1, 3-二羰基化合物S2的反应路径进行了探究.首先, 催化剂Sc(OTf)3与重氮化合物反应, 脱去氮气生成钪卡宾.随后, 分别计算了钪卡宾与1, 3-二羰基化合物的烯醇中间体S2-1反应的两条路径: (1)经过亲电加成、分子内环化、开环和氢迁移过程生成C—C插入产物Pr1; (2)经过亲电加成, 随后直接质子化得到C—H插入产物Pr2.计算结果表明, 后者反应路径更优, 计算结果与实验结果一致.
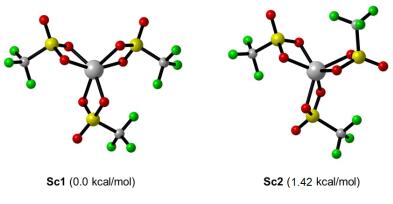
首先, 通过计算模拟了两种不同的Sc(OTf)3结构模型(Sc1和Sc2), Sc1和Sc2的相对自由能相差1.42 kcal/mol, Sc1的结构更稳定(图 3).因此, 在本工作中, 仅使用Sc1的催化剂模型进行反应机理探究.
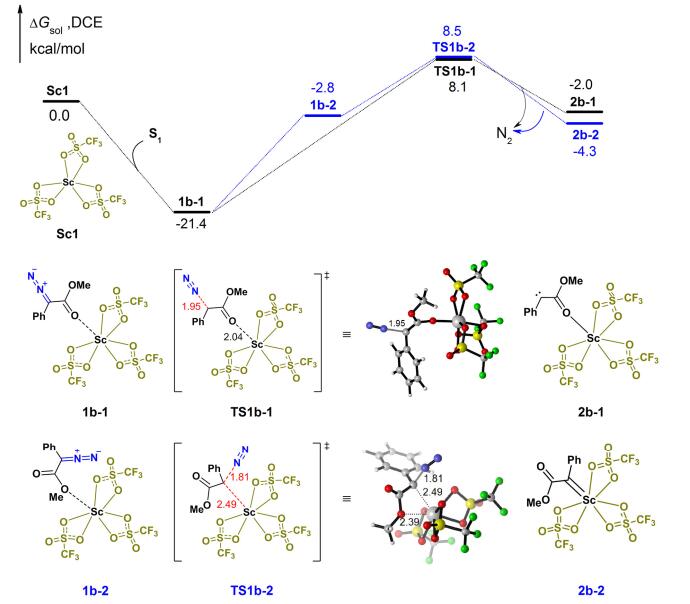
随后, 探究了催化剂Sc1催化重氮化合物的分解过程(图 4).首先Sc1和重氮化合物S1配位复合, 底物上羰基氧和钪中心配位, 得到稳定的复合物1b-1, 放热21.4 kcal/mol.随后我们考虑了两种不同的反应路径, 一种是由稳定的复合物1b-1直接发生脱氮气过程, 经过过渡态TS1b-1 (8.1 kcal/mol), 需要跨越29.5 kcal/mol的能垒, 发生脱氮气过程生成中间体2b-1 (-2.0 kcal/mol), 但由于2b-1相对于1b-1的能量高了19.4 kcal/mol, 此过程是可逆的; 另外, 我们还考虑了先由1b-1经过配体旋转, 底物和钪中心的配位点从羰基氧迁移到甲氧基的氧上, 得到复合物1b-2 (-2.8 kcal/mol), 需要吸热18.6 kcal/mol.随后钪中心配位迁移到临近的卡宾碳位点, 促进脱氮气过程的发生, 经过过渡态TS1b-2 (8.5 kcal/mol), 能垒为11.3 kcal/mol, 发生脱氮气过程生成了钪卡宾中间体2b-2 (-4.3 kcal/mol), 此过程全局所需跨越能垒为29.9 kcal/mol. TS1b-1比TS1b-2仅低了0.4 kcal/mol, 两条反应路径是竞争反应路径, TS1b-1为较优过渡态, 是该反应的决速步骤, 此计算结果与实验观测的反应温度一致.
随后, 生成的钪自由卡宾2b-1或者钪卡宾2b-2和烯醇中间体S2-1的反应过程如图 5所示, 首先形成复合物3b (-13.1 kcal/mol)或者4b (-20.9 kcal/mol), 3b可以通过配体旋转异构得到更稳定的4b, 释放出卡宾碳位点.中间体4b的卡宾碳对S2-1上的双键进行亲电加成, 经过能垒为4.1 kcal/mol的过渡态TS2b, 得到烯醇中间体5b.接下来, 类似于银催化路径, 我们对两种不同的反应路径进行了探究:中间体5b上带负电荷的卡宾碳C(1)可能进攻邻近的羟基碳C(2), 得到环丙烷中间体6b, 也可能直接发生质子化, 得到C—H插入产物Pr2.
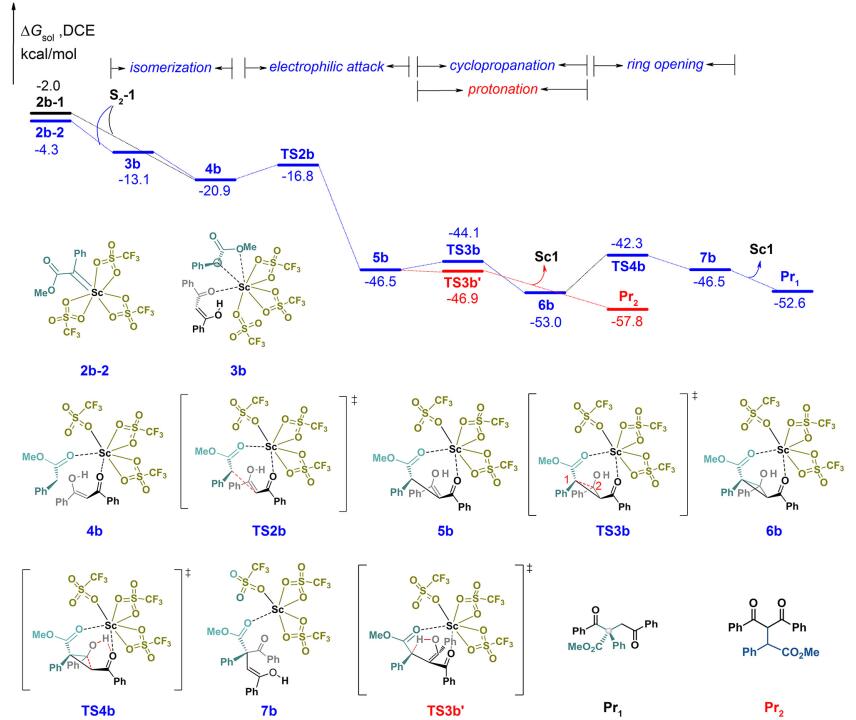
首先, 我们探究了中间体5b的环化过程: 5b经过能垒仅为2.4 kcal/mol的三元环过渡态TS3b, 得到环化中间体6b, 其相对于2b-2放热48.7 kcal/mol. 6b上的羟基氢迁移到邻近的羰基氧上, 从而促使邻近的三元环发生开环反应, 此过程过渡态TS4b的反应能垒为10.7 kcal/mol, 得到烯醇中间体7b.最终烯醇互变异构生成C—C插入产物Pr1.接着, 我们探究了中间体5b的质子化过程: 5b经过无能垒的五元环过渡态TS3b', 氢质子从羟基迁移到卡宾碳上, 得到C—H插入产物Pr2.通过对比, 氢质子迁移过渡态TS3b'比环化过渡态TS3b的相对自由能低了2.8 kcal/mol, 因此, 钪催化更容易生成C—H插入产物Pr2.此外, 从复合物3b, 我们还考虑了1, 3-二羰基化合物异构配体先发生脱质子化, 随后钪卡宾对其阴离子亲电进攻的过程, 但由于该反应路径相对自由能较高所以排除.为了进一步分析产生选择性的原因, 对银和钪催化的关键选择性决定步骤过渡态及其前驱体进行结构分析(图 6).银作为催化剂时, 在烯醇中间体4a中, 由于Ag(I)离子4d10的核外电子构型, 呈现出低配位的特点, 1, 3-二羰基异构配体上羰基氧和银中心不存在配位作用, 而存在其自身的分子内强氢键作用(0.139 nm)稳定羟基氢.虽然随后的氢迁移五元环过渡态TS3a'具有较低的张力, 但是TS3a' (-22.8 kcal/mol)的相对自由能比环化过程的三元环过渡态TS3a (-33.4 kcal/mol)高了10.6 kcal/mol, 这是因为在发生氢迁移过程TS3a'时, 需要额外克服前驱体4a分子内羟基氢和羰基氧之间强的氢键相互作用能.钪作为催化剂时, 在烯醇中间体5b中, 由于Sc(III)离子3d0的核外电子构型, 呈现高配位的特点, 5b中的1, 3-二羰基异构配体上羰基氧保持和钪中心的配位, 羟基氢和羰基氧之间仅存在较弱的氢键相互作用(0.242 nm), 以及羟基氢和三氟磺酸根离子之间存在较弱的氢键相互作用(0.182 nm).因此具有较小张力的氢迁移五元环过渡态TS3b'比环化三元环过渡态TS3b低了2.8 kcal/mol, 因此主要生成C—H插入产物, 计算结果和实验的现象一致.
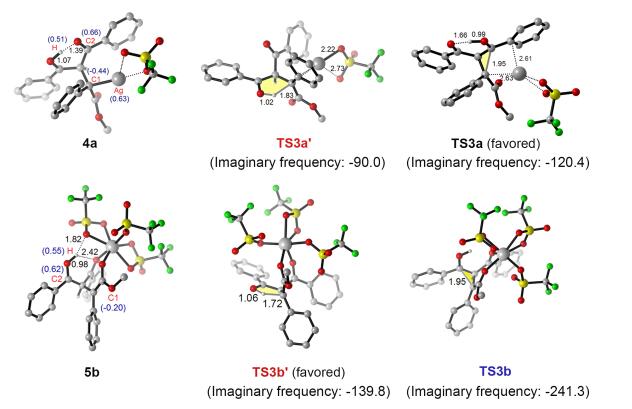
NPA charges are given in parentheses, distances are given in 0.1 nm
采用DFT理论计算方法分别研究了AgOTf和Sc(OTf)3催化重氮化合物与1, 3-二羰基化合物C—C键和C—H键插入的反应机理, 详细讨论了化学选择性的产生原因.银和钪盐催化重氮化合物脱氮气生成了相应的金属卡宾, 而具有低配位的银卡宾和高配位的钪卡宾与1, 3-二羰基化合物之间存在不同的反应机制.银卡宾的卡宾碳对1, 3-二羰基化合物的烯醇异构体的亲电进攻, 得到关键的双性离子中间体.之后可能发生分子内环化、选择性开环、烯醇异构得到C—C键插入产物; 也可能直接发生质子化过程得到C—H键插入产物.选择性决定步骤发生在分子内环化过程, 其比质子化过程的相对自由能低了10.6 kcal/mol.银催化C—C插入反应的决速步骤发生在选择性开环过程, 其全局反应能垒为16.2 kcal/mol.钪催化重氮化合物脱氮气生成钪自由卡宾或者钪卡宾.两种卡宾都能和1, 3-二羰基化合物异构体复合配位得到相同的稳定复合物中间体, 之后发生亲电加成得到关键的烯醇异构体, 随后可能发生环化、开环、异构化得到最终的C—C插入产物Pr1; 也可能直接发生氢迁移得到C—H插入产物Pr2.氢迁移过程比环化过程的相对自由能低了2.8 kcal/mol, 是该反应的选择性决定步骤.钪催化C—H插入反应的决速步骤发生在钪卡宾生成的过程, 反应能垒为29.5 kca/mol.
通过分析两种催化剂催化产生选择性关键步骤的过渡态和前驱体, 发现选择性产生的原因是关键过渡态的环张力以及银和钪中心配位数差异引起的前驱体中氢键相互作用的不同.低配位的银催化反应路径中, 氢迁移过程前驱体中存在羟基氢和羰基氧之间强的氢键, 不利于五元环氢迁移过渡态的发生, 反而环张力较大的三元环环化过渡态的相对能量更低, 得到C—C插入产物.高配位的钪催化路径中, 氢迁移过程前驱体中氢键较弱, 因此更容易发生张力较小的五元环氢迁移过渡态, 得到C—H插入产物.本文中的计算结果与实验结果一致, 并进一步解释了实验结果, 为过渡金属催化的卡宾转移反应提供了新视点.
辅助材料(Supporting Information) 所有化合物的分子坐标以及计算得到的能量.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Quasdorf, K. W.; Overman, L. E. Nature 2014, 516, 181.
(b) Long, R.; Huang, J.; Gong, J. X.; Yang, Z. Nat. Prod. Rep. 2015, 32, 1584.
(c) Zeng, X.; Cao, Z.; Wang, Y.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330.
(d) Ling, T.; Rivas, F. Tetrahedron 2016, 72, 6729.
(e) Shimizu, M. Angew. Chem., Int. Ed. 2011, 50, 5998.
(f) Wang, B.; Tu, Y. Acc. Chem. Res. 2011, 44, 1207.
Christoffers, J.; Baro, A. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis, Wiley-VCH, Weinheim, 2005.
(a) Fumagalli, G.; Stanton, S.; Bower, J. F. Chem. Rev. 2017, 117, 9404.
(b) Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613.
(a) Candeias, N. R.; Paterna, R.; Gois, M. P. Chem. Rev. 2016, 116, 2937.
(b) Guttenberger, N.; Breinbauer, R. Tetrahedron 2017, 73, 6815.
(c) Li, W.; Liu, X.; Tan, F.; Hao, X.; Zheng, J.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2013, 52, 10883.
(d) Li, W.; Liu, X.; Hao, Y.; Cai, Y.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2012, 51, 8644.
(e) Moebius, D. C.; Kingsbury, J. S. J. Am. Chem. Soc. 2009, 131, 878.
(f) Hashimoto, T.; Naganawa, Y.; Maruoka, K. J. Am. Chem. Soc. 2008, 130, 2434.
(a) Xia, Y.; Liu, Z. X.; Liu, Z.; Ge, R.; Ye, F.; Hossain, M.; Zhang, Y.; Wang, J. J. Am. Chem. Soc. 2014, 136, 3013.
(b) Wang, Y.; Wang, Y.; Zhang, W. J.; Zhu, Y.; Wei, D.; Tang, M. Org. Biomol. Chem. 2015, 13, 6587.
(a) Liu, Z.; Sivaguru, P.; Zanoni, G.; Anderson, E. A.; Bi, X. Angew. Chem., Int. Ed. 2018, 57, 8927.
(b) Liu, Z.; Zhang, X.; Virelli, M.; Zanoni, G.; Anderson, E. A.; Bi, X. iScience 2018, 8, 54.
(a) Xi, Y.; Su, Y.; Yu, Z.; Dong, B.; McClain, E. J.; Lan, Y.; Shi, X. Angew. Chem., Int. Ed. 2014, 53, 9817.
(b) Xu, K.; Shen, C.; Sheng, W.; Shan, S.; Jia, Y. Chin. J. Org. Chem. 2015, 35, 633(in Chinese).
(许恺, 沈冲, 盛卫坚, 单尚, 贾义霞, 有机化学, 2015, 35, 633.)
(a) Zhan, G.; Du, W.; Chen, Y. Chem. Soc. Rev. 2017, 46, 1675.
(b) Mahatthananchai, J.; Dumas, A. M.; Bode, J. W. Angew. Chem., Int. Ed. 2012, 51, 10954.
(c) Schreiber, S. L. Science 2000, 287, 1964.
Liu, F.; Zhu, L.; Zhang, T.; Zhong, K.; Xiong, Q.; Shen, B.; Liu, S.; Lan, Y.; Bai, R. ACS Catal. 2020, 10, 1256. doi: 10.1021/acscatal.9b02040
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision A.02, Gaussian Inc., Wallingford, CT, 2009.
(a) Becke, A. D. Phys. Rev. A 1988, 38, 3098.
(b) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
(c) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785.
(d) Qi, X.; Zhang, H.; Shao, A.; Zhu, L.; Xu, T.; Gao, M.; Liu, C.; Lan, Y. ACS Catal. 2015, 5, 6640.
(e) Xiao, P.; Yuan, H.; Liu, J.; Zheng, Y.; Bi, X.; Zhang, J. ACS Catal. 2015, 5, 6177.
(a) Schwerdtfeger, P.; Dolg, M.; Schwarz, W. H. E.; Bowmaker, G. A.; Boyd, P. D. W. J. Chem. Phys. 1989, 91, 1762.
(b) Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. J. Chem. Phys. 1987, 86, 866.
(a) Fukui, K. J. Phys. Chem. 1970, 74, 4161.
(b) Fukui, K. Acc. Chem. Res. 1981, 14, 363.
Peverati, R.; Truhlar, D. G. Phys. Chem. Chem. Phys. 2012, 14, 16187. doi: 10.1039/c2cp42576a
(a) Ji, C. L.; Hong, X. J. Am. Chem. Soc. 2017, 139, 15522.
(b) Yang, Y. F.; Cheng, G. J.; Liu, P.; Leow, D.; Sun, T. Y.; Chen, P.; Zhang, X.; Yu, J. Q.; Wu, Y. D.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 344.
(c) Xu, X.; Liu, P.; Shu, X. Z.; Tang, W.; Houk, K. N. J. Am. Chem. Soc. 2013, 135, 9271.
(d) Komagawa, S.; Wang, C.; Morokuma, K.; Saito, S.; Uchiyama, M. J. Am. Chem. Soc. 2013, 135, 14508.
(e) Liu, P.; Xu, X.; Dong, X.; Keitz, B. K.; Herbert, M. B.; Grubbs, R. H.; Houk, K. N. J. Am. Chem. Soc. 2012, 134, 1464.
(a) Liu, F.; Zhu, L.; Zhang, T.; Zhong, K.; Xiong, Q.; Shen, B.; Liu, S.; Lan, Y.; Bai, R. ACS Catal. 2020, 10, 1256.
(b) Xiao, P.; Yuan, H.; Liu, J.; Zheng, Y.; Bi, X.; Zhang, J. ACS Catal. 2015, 5, 6177.
(c) Qi, X.; Zhang, H.; Shao, A.; Zhu, L.; Xu, T.; Gao, M.; Liu, C.; Lan, Y. ACS Catal. 2015, 5, 6640.
(d) Daru, J.; Benda, Z.; Póti, Á.; Novák, Z.; Stirling, A. Chem.-Eur. J. 2014, 20, 15395.
(e) Zhou, B.; Yan, T.; Xue, X.-S.; Cheng, J.-P. Org. Lett. 2016, 18, 6128.
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. J. Phys. Chem. B 2009, 113, 6378. doi: 10.1021/jp810292n
Legault, C. Y. CYLview, version 1.0b, 2009 http://www.cylview.org/.
(a) Hu, X.; Tang, Y.; Gantzel, P.; Meyer, K. Organometallics 2003, 22, 612.
(b) Liu, Q.-X.; Yao, Z.-Q.; Zhao, X.-J.; Chen, A.-H.; Yang, X.-Q.; Liu, S.-W.; Wang, X.-G. Organometallics 2011, 30, 3732.
Zhang, J.; Shan, C.; Zhang, T.; Song, J.; Liu, T.; Lan, Y. Coord. Chem. Rev. 2019, 382, 69. doi: 10.1016/j.ccr.2018.12.009

图式 1 “一碳”插入策略构筑全碳季碳中心
Scheme 1 Formation of all-carbon quaternary center through "one-carbon" insertion

图式 2 银和钪催化的重氮化合物与1, 3-二羰基化合物的C—C键和C—H键插入反应
Scheme 2 Silver- and scandium-catalyzed insertion of diazo compound into C—C and C—H bonds of 1, 3-dicarbonyl compounds

图 1 银卡宾中间体形成路径的能量示意图
Figure 1 Energy profiles for the formation of silver carbene intermediates Distances are given in 0.1 nm

图 2 银卡宾和1, 3-二羰基化合物C—C和C—H键插入反应的能垒图
Figure 2 Energy profiles for insertion of silver carbene into C—C and C—H bonds of 1, 3-dicarbonyl compounds

图 4 钪卡宾中间体形成路径的能量示意图
Figure 4 Free energy profiles for the formation of scandium carbene intermediates Distances are given in 0.1 nm

图 5 钪卡宾和1, 3-二羰基化合物C—C和C—H键插入反应的能垒图
Figure 5 Free energy profiles for insertion of scandium carbene into C—C and C—H bonds of 1, 3-dicarbonyl compounds

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

