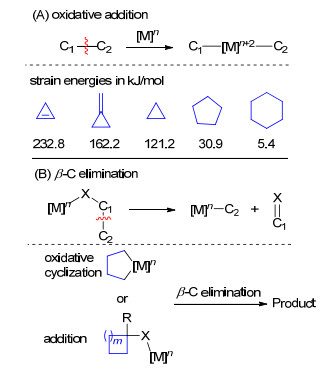
图式 1.
(A) 氧化加成或(B)β-C消除实现C—C键断裂
Scheme 1.
C—C cleavage by (A) C—C oxidative addition or (B) β-carbon elimination

C—C键广泛存在于化学化工产品中, 发展将C—C键断裂后直接官能团化的催化体系, 为医药或农药分子的合成提供简单直接的方法, 是有机化学家们长期追求的目标之一[1].然而该领域一直存在很大挑战, 主要有以下原因: (1) C—C键比较惰性, 键能较高, 高效的催化剂还不多; (2)很多分子的C—C键被C—H键或者C—X (X为杂原子)键包围, 高选择性地实现C—C键断裂的策略还比较少[1, 2].
近些年, 很多课题组通过使用过渡金属催化剂成功实现小分子化合物的C—C键断裂, 并将其应用到复杂分子的合成中[1~3].这些成功的例子主要是通过过渡金属参与的氧化加成或者β-C消除过程.当通过氧化加成过程时, 由于C—C键断裂在热力学上是不利的(C—C键的键能约为376.2 kJ/mol, 而生成的C—M键的键能约为125.4 kJ/mol), 这一过程会生成更加活泼的C—M键(M为过渡金属), 人们通常使用环张力比较大的三元环或者四元环底物, 利用这些底物释放环张力促进反应的进行.近些年人们也发展了使用含有导向基底物(或瞬态导向基策略)实现非张力环底物的C—C键断裂反应(Scheme 1, A); 而β-C消除过程主要通过环金属化或者加成后β-C消除实现C—C键断裂, 这一过程可以应用在一些环张力较小、活性较低的底物上(Scheme 1, B)[1].
已有成功的例子主要是使用贵金属催化剂如铑、钯、铱等, 而廉价过渡金属催化剂如铁、钴、镍等的报道则相对较少[4], 其中镍金属催化剂表现出独特的催化活性以及选择性.镍金属不仅价格低廉, 更重要的是镍自身具有更小的原子半径及更强的亲核性等独特的性质.在过去几十年里, 关于镍催化C—C键活化的报道不断涌现.本文主要根据环张力的大小进行划分, 就镍催化C—C键活化的反应体系展开讨论, 重点介绍近十年的研究进展, 最后介绍近些年镍催化不对称C—C键活化反应.
三元环底物具有较大的环张力, 在过渡金属催化的C—C键活化领域应用非常多, 被成功地应用到复杂分子的合成中.在镍催化的三元环的开环反应中, 主要包括环丙烯酮、亚甲基环丙烷、乙烯基环丙烷以及带有导向基的三元环等底物.
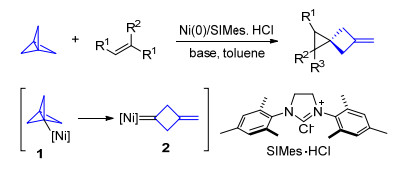
[1.1.1]螺桨烷分子的三个三元环共用一个C—C σ键, 具有非常大的环张力, 很容易发生开环反应.它可以通过自由基的加成或者接受亲核试剂的进攻发生C—C键断裂反应, 也可以在过渡金属作用下发生两次C—C键断裂生成金属卡宾物种. 2019年, Bedford和Aggarwal课题组[5]报道了镍催化[1.1.1]螺桨烷与烯烃的加成反应, 使用SIMes卡宾为配体, 在零价镍作用下, [1.1.1]螺桨烷发生两次C—C键断裂生成镍卡宾中间体2, 然后与烯烃进行金属参与的[2+2]环加成反应, 最后得到螺[2, 3]己烷产物(Scheme 2).
环丙烯酮是一类比较特殊的三元环底物, 它同时具有羰基和环内双键, 不仅具有较大的环张力, 还具有芳香性.由于具有较大环张力, 可以与低氧化态的金属发生氧化加成实现C—C键断裂; 另外羰基也可以接受亲核试剂的进攻, 随后发生β-C消除实现C—C键断裂[6].
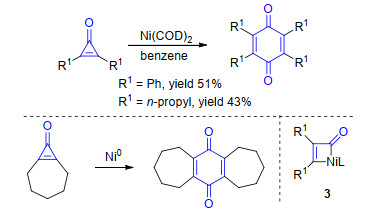
环丙烯酮参与的镍催化C—C键断裂反应可以追溯到1972年Noyori小组[7]报道的Ni(COD)2催化环丙烯酮的二聚反应.环丙烯酮在镍催化剂作用下发生自身偶联反应, 以中等的收率得到对称的1, 4-苯醌产物.作者对反应机理进行了初步的研究, 发现使用并环底物时, 反应依然可以顺利地进行.基于此实验结果作者认为反应可能经过镍金属参与的氧化C—C键断裂生成四元环镍中间体3的过程, 而不是环丙烯酮双键自身的[2+2]环加成过程(Scheme 3).
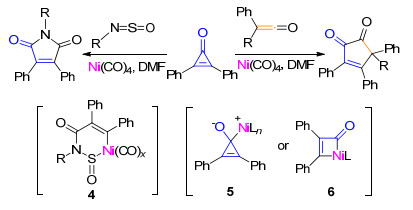
1976年, Baba等[8a]报道了Ni(CO)4催化环丙烯酮与亚磺酰亚胺的[3+2]环加成反应.在镍催化剂作用下, 环丙烯酮开环后与亚磺酰亚胺的N=S双键进行加成得到中间体4, 4进一步脱除SO, 最后还原消除得到3, 4-二苯基-吡咯啉-2, 5-二酮产物(Scheme 4).同年该课题组[8b]也报道了环丙烯酮与联烯酮的[3+2]环加成反应(Scheme 4).作者发现溶剂对反应的收率以及区域选择性都有很大的影响, 当使用N, N-二甲基甲酰胺(DMF)为溶剂时, 能够以85%的收率得到1, 2-二酮产物.作者认为当使用极性溶剂如DMF时, 体系中主要存在离子型中间体5而不是四元环镍中间体6.
2018年, 赵东兵课题组[9]报道了镍催化苯并硅杂环丁烷与环丙烯酮的[4+3]环加成反应.作者使用具有一定环张力的苯并硅杂环丁烷, 在镍催化剂作用下, 能够高选择性地发生C(sp2)—Si键的断裂, 再与环丙烯酮进行加成反应得到苯并硅杂环庚烯酮产物(Eq. 1).
|
|
(1) |
2019年, 何振宇课题组[10]报道了镍催化二取代环丙烯与炔烃的[3+2]环加成反应.通过NHC-Ni-H对环丙烯和炔烃串联加成, 最后异构化得到环戊二烯产物, 在该反应中催化剂NHC-NiII的结构对反应收率和区域选择性有非常大的影响(Eq. 2).
|
|
(2) |
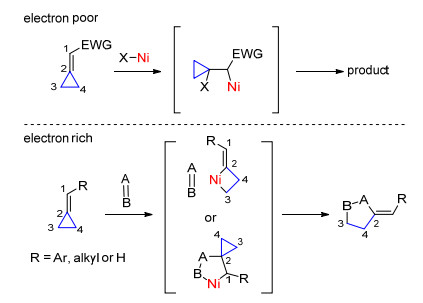
亚甲基环丙烷相比于环丙烯酮, 双键处于三元环外, 环张力较小.自1970年Noyori课题组[11]报道了镍催化亚甲基环丙烷的开环反应以来, 该类化合物参与的C—C键活化反应就受到了广泛关注.根据亚甲基环丙烷双键上取代基的电性不同可以分为缺电的亚甲基环丙烷和富电的亚甲基环丙烷.不同电性的亚甲基环丙烷参与的镍催化C—C键断裂路径可能有所不同:当使用缺电子性的亚甲基环丙烷时, 其双键比较容易接受亲核试剂的进攻, 经过β-C消除过程实现三元环开环; 当使用富电性的亚甲基环丙烷时, 容易发生氧化C—C键断裂过程得到四元环镍中间体, 进而与不饱和键进行加成反应, 或者发生环金属化后再经β-C消除过程实现C—C键断裂(Scheme 5).
2004年, Saito课题组[12a, 12b]报道了镍催化酯基取代的亚甲基环丙烷与两分子炔烃的[3+2+2]环加成反应.在Ni(COD)2/PPh3作用下, 镍和两分子炔烃首先发生环金属化过程得到五元环镍中间体7, 然后与亚甲基环丙烷的双键进行加成, 随后发生β-C消除得到中间体9, 最后还原消除得到环庚二烯产物(Scheme 6). 2006年, 该课题组[12c]又实现了使用含不同取代基的炔烃参与的三组分[3+2+2]环加成反应, 反应能够以优秀的区域选择性得到目标产物.在该反应中, 反应的区域选择性具有很大挑战, 作者通过使用过量的三甲基硅乙炔, 并结合缓慢滴加的操作实现了该反应优秀的区域选择性(Scheme 6).在此研究基础上, 2007年, 该课题组[12d, 12e]利用类似的反应历程, 分别实现了共轭双烯底物与亚甲基环丙烷的[4+3]环加成反应以及1, 7-二炔底物与亚甲基环丙烷的[3+2+2]环加成反应(Scheme 6).
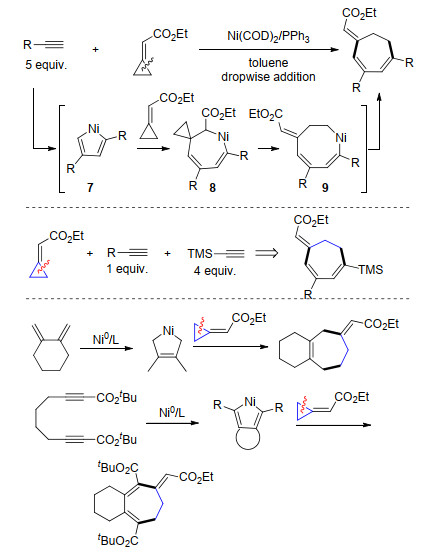
2010年, Saito课题组[13]报道了镍催化含端炔结构的共轭双烯与亚甲基环丙烷的[4+3+2]环加成反应.反应首先通过镍与共轭双烯和炔烃的[4+2+1]环金属化得到七元环镍中间体10, 随后该中间体与亚甲基环丙烷进行加成反应, 然后β-C消除实现三元环开环得到中间体12, 最后还原消除得到环壬二烯产物(Scheme 7).
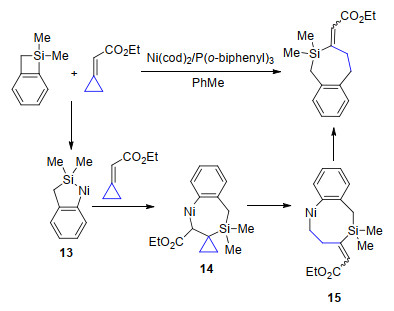
同年, Saito课题组[14]实现了镍催化苯并硅杂环丁烷与亚甲基环丙烷的[4+3]环加成反应, 苯并硅杂环丁烷首先与镍进行氧化加成得到五元环镍中间体13, 接下来与亚甲基环丙烷的双键进行加成反应, 然后β-C消除实现三元环开环得到中间体15, 最后还原消除得到苯并硅杂环庚烷(Scheme 8).
从上面的例子可以看出, 缺电的亚甲基环丙烷常作为Michael受体, 通过C—Ni键对双键的加成后β-C消除实现三元环开环, 而富电的亚甲基环丙烷接受亲核试剂进攻的能力较低, 可以通过零价镍氧化C—C键断裂实现三元环的开环.
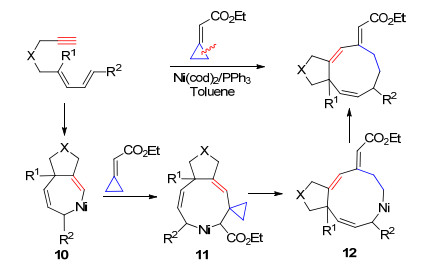
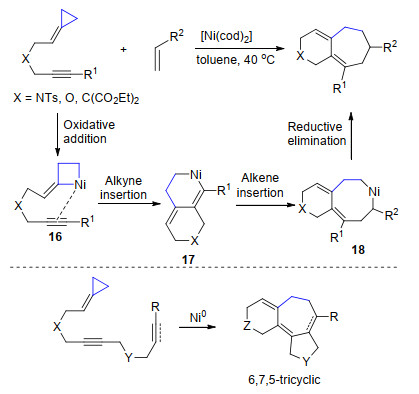
2010年, Lopez和Mascarenas等[15a]报道了镍催化亚甲基环丙烷与烯烃的[3+2+2]环加成反应.作者使用分子内含有炔基的亚甲基环丙烷, 通过镍与三元环的氧化C—C键断裂实现三元环开环得到中间体16, 进一步与分子内的炔烃进行插入反应, 随后与另外一分子烯烃进行加成得到中间体18, 最后还原消除得到6, 7-并环产物. 2014年, 该课题组[15b]进一步将双键(叁键)引入到分子内, 在镍催化剂作用下实现了分子内[3+2+2]环加成反应得到6, 7, 5-并三环产物(Scheme 9).
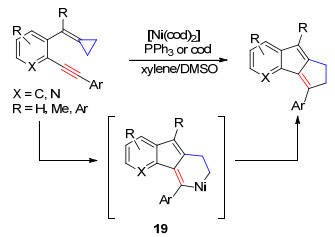
2011年, 张玉红课题组[16]利用类似的策略, 以含芳烃结构的亚甲基环丙烷为底物, 在镍催化剂作用下进行开环反应, 然后与分子内的炔基发生插入反应得到中间体19, 最后还原消除得到环戊[a]茚衍生物, 该方法为茚类化合物的合成提供了新的、简单高效的方法(Scheme 10).
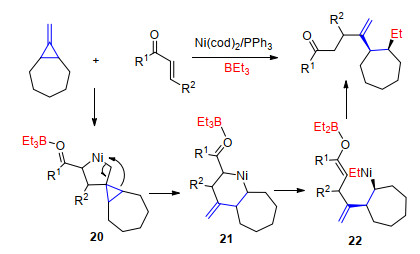
2013年, Ogata课题组[17]报道了镍催化亚甲基环丙烷与α, β-不饱和酮在三乙基硼作用下的三组分偶联反应.作者认为反应首先经过一个环金属化过程得到中间体20, 然后β-C消除发生开环反应, 通过烯醇异构化并使硼烷上的乙基转移到镍金属上得到中间体22, 最后还原消除得到γ-烯基取代的链状酮(Scheme 11).
乙烯基环丙烷被广泛地应用在过渡金属催化的C—C键活化领域, 特别是带有拉电子基团的乙烯基环丙烷底物, 可以在过渡金属作用下发生C—C键断裂生成内离子型的烯丙基金属中间体, 被广泛应用到过渡金属催化的环加成反应中[18], 这里我们将主要阐述近些年镍催化的一些反应体系.
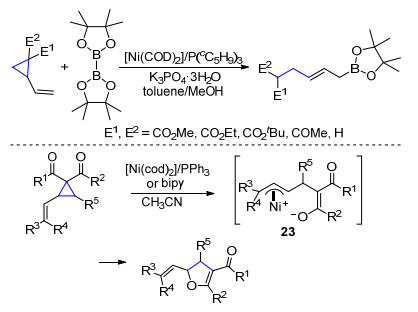
2008年, Yorimitsu等[19a]报道了镍催化乙烯基环丙烷的开环反应.使用硼酸酯作为偶联试剂, 可以高区域选择性地得到烯丙基硼酸酯类化合物(Scheme 12). 2006年Johnson课题组[19b]报道了烯基三元环底物在镍作用下的分子内环化反应.首先乙烯基环丙烷在镍作用下发生开环反应得到内离子型的烯丙基镍中间体23, 然后氧亲核试剂分子内进攻烯丙基镍中间体23得到二氢呋喃类化合物(Scheme 12).
2014年, Matsubara课题组[20]报道了联烯底物参与的镍催化乙烯基环丙烷的开环反应.使用富电子的PMe3为配体, 通过中间体24与联烯的[3+2]环加成得到含环外双键的环戊烷产物(Scheme 13).

与乙烯基环丙烷和亚甲基环丙烷相比, 简单环丙烷的C—C键断裂的能垒较高.人们通过在简单三元环上引入羰基、亚胺或者烯烃等导向基团实现简单环丙烷的开环反应.
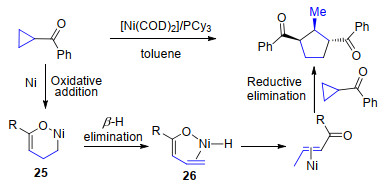
2006年, Ogoshi等[21]报道了以羰基为导向基的环丙烷开环反应.在Ni0/PCy3作用下环丙烷首先发生C—C键断裂并异构化为六元环镍中间体25, 随后该中间体发生β-H消除并分子内加成后异构化为α, β-不饱和酮, 最后在镍作用下与另一分子三元环底物进行[3+2]环加成得到三取代的环戊烷产物(Scheme 14).
2006年, Montgomery小组[22a]实现了该类环丙烷与另一分子α, β-不饱和酮的分子间[3+2]环加成反应.往体系中添加Ti(OiPr)4能够很好地提高反应活性、缩短反应时间并提高产物的收率(Eq. 3).随后该课题组进一步将该催化体系拓展到以亚胺为导向的环丙烷底物与α, β-不饱和酮的[3+2]环加成反应[22c].除了与双键的加成,也实现了炔烃参与的[3+2]环加成反应, 2011年, Ogoshi小组[22d]报道了镍催化以羰基为导向基的环丙烷与炔烃的[3+2]环加成反应, 在添加Me2AlCl的条件下, 以中等到优秀的收率得到环戊烯产物(Eq. 4).
|
|
(3) |
|
|
(4) |
以烯烃为导向的三元环开环反应早期就有一些报道, 该类底物在镍作用下发生C—C键断裂后生成烯丙基镍中间体. 2004年, Louie课题组[23]报道了含烯烃结构的环丙烷在镍作用下的开环反应, 首先环丙烷发生氧化C—C键断裂得到中间体27, 随后异构化得到六元环镍中间体28, 最后还原消除得到环戊烯产物(Scheme 15).
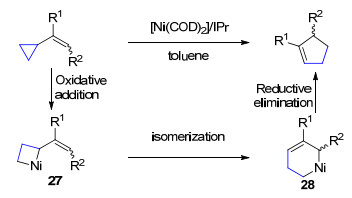
四元环的环张力大小与三元环的环张力大小相比稍有降低, 其参与过渡金属催化的C—C键活化也有较多成功的例子(图 1)[1, 2].
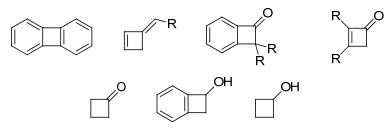
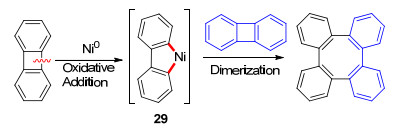
二苯并环丁二烯和环丁酮参与镍催化四元环开环反应的报道较多.由于二苯并环丁二烯的环张力较大, 故该底物主要是通过氧化C—C键断裂过程; 而环丁酮底物主要是经过β-C消除过程. 1990年, Vollhardt等[24]报道了二苯并环丁二烯的二聚反应, 在镍催化剂作用下, 二苯并环丁二烯首先发生氧化C—C键断裂生成五元环镍中间体29, 随后与另一分子二苯并环丁二烯偶联得到四苯并环辛四烯化合物(Scheme 16).
随后, Jones[25a~25c]和Radius[25d, 25e]课题组分别实现了镍催化二苯并环丁二烯与炔烃的[4+2]环加成反应, 分别使用双膦和卡宾为配体, 得到不同取代的菲类化合物(Scheme 17).除了炔烃, 一氧化碳和异氰化合物也可以作为C1合成子参与镍催化的二苯并环丁二烯的C—C键断裂反应, 分别得到9-芴酮和亚胺产物(Scheme 17)[25f].

环丁酮参与的镍催化C—C键断裂反应的报道比较多, 主要是与烯烃或炔烃进行加成反应. 2012年, Ho和Aïasa等[26]报道了镍催化氮杂环丁酮与炔烃的[4+2]环加成反应.在Ni0/PPh3作用下, 通过炔烃、羰基与镍的环金属化过程得到五元环镍中间体31, 然后β-C消除实现四元环开环得到中间体32, 最后还原消除得到哌啶酮产物(Scheme 18).
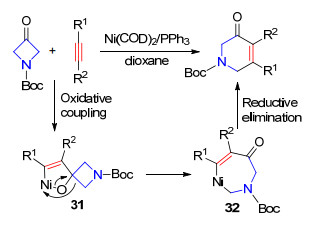
2012年, Louie课题组[27a]报道了镍催化氮杂环丁酮与1, 6-双炔底物的[4+2+2]环加成反应.使用卡宾为配体, 通过与炔烃发生环金属化反应得到中间体33, 现场与另一个炔基进行插入反应后β-C消除得到中间体35, 最后还原消除得到氮杂环辛烯酮产物(Scheme 19). 2013年, 该课题组[27b]进一步实现了1, 3-共轭双烯参与的氮(氧)杂环丁酮的开环反应, 在Ni0/P(p-Tol)3作用下, 经过类似的过程得到氮(氧)杂环辛烯酮(Scheme 19).

2015年, Martin课题组[28]报道了苯并环丁酮与1, 3-共轭双烯的[4+4]环加成反应.在Ni(COD)2/P(4-CF3-C6H4)3作用下, 通过羰基、烯烃和镍的环金属化过程得到中间体36, 随后β-C消除实现C—C键断裂得到烯丙基镍中间体37, 最后还原消除得到苯并环辛烯酮产物(Scheme 20).当使用PPh3为配体时, 在甲苯中加热至100 ℃能够实现炔烃参与的[4+2]环加成反应, 得到萘酮类化合物(Scheme 20).
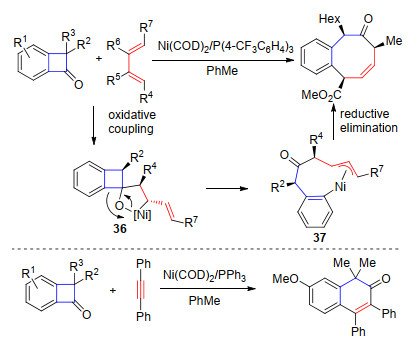
2011年, Harrity课题组[29]报道了镍催化3-苯基环丁烯酮与炔基硼酸酯类化合物的[4+2]环加成反应.反应能够在室温条件下顺利进行, 得到硼酸酯取代的苯酚类化合物, 反应的区域选择性主要由炔烃上取代基的位阻大小控制(Eq. 5).
|
|
(5) |
另外, 环丁酮肟酯是一类常用于过渡金属催化C—C键断裂的反应底物[30].近些年, 人们发展了镍催化环丁酮肟酯的自由基开环反应, 反应首先生成亚胺自由基中间体38, 接下来发生C—C键断裂生成含氰基官能团的烷基自由基中间体39, 进一步与不同的偶联试剂进行反应(Scheme 21). 2017年, 郭丽娜课题组[30b]报道了镍催化芳烃直接C—H键断裂与环丁酮肟酯的自由基加成反应. 2019年, 该课题组[30c]还实现了活泼烯烃参与的加成反应, 通过1, 5氢迁移或者分子内环化得到两类含氮杂环产物. Selander[30d]和梅天胜[30e]课题组分别实现了芳基硼酸和芳基锌试剂参与的加成反应(Scheme 21).另外, 近些年王川课题组[30g]也实现了在添加还原剂条件下环丁酮肟酯的开环反应, 分别实现了酰氯和三氟甲基取代的烯烃参与的加成反应(Scheme 21).

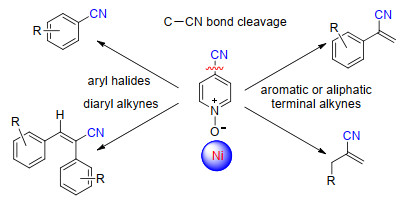
虽然C—CN键具有较高的键能(>418 kJ/mol), 但过渡金属催化的C—CN键断裂反应也有很多成功的例子, 最早可以追溯到1971年[31].氰基参与的C—CN键断裂反应主要是通过氰基上的N原子或者CN的叁键与金属配位, 拉近金属中心与C—CN键的距离从而促进C—CN键的断裂, 可以在低价态金属作用下直接发生氧化C—CN键断裂, 或者在Lewis酸作用下发生C—CN键的插入.
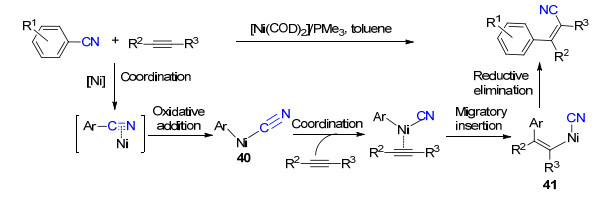
2004年, Nakao和Hiyama等[32]报道了镍催化芳基氰化物与炔烃的加成反应.使用Ni(COD)2为催化剂, 以富电子的三甲基膦为配体, 经过氧化加成得到中间体40, 随后进行炔烃的插入反应得到中间体41, 最后还原消除以优秀的选择性得到顺式加成的烯基腈化合物(Scheme 22).
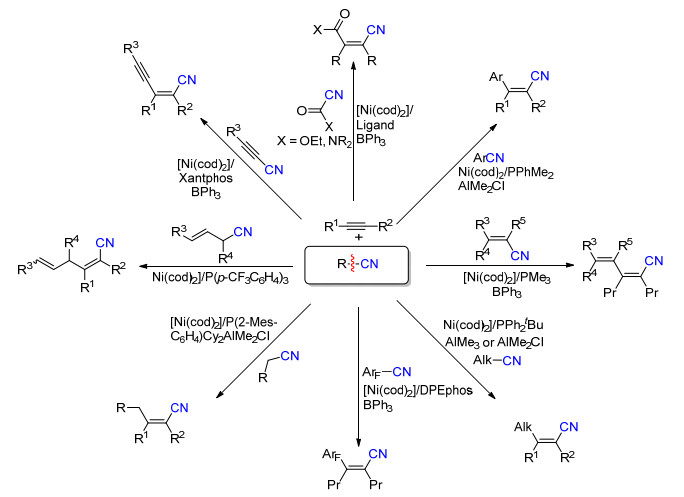
除了芳基腈化合物, 通过使用不同电性的单膦配体、双膦配体并结合添加Lewis酸(铝试剂或硼试剂)的方法, 实现了烷基腈、烯丙基腈、烯基腈、苄基腈、酰腈、炔基腈和其它芳香腈类化合物的C—CN键断裂反应, 在镍催化剂作用下顺利地与炔烃进行加成得到含有各种取代基的烯基腈类化合物(Scheme 23)[33].
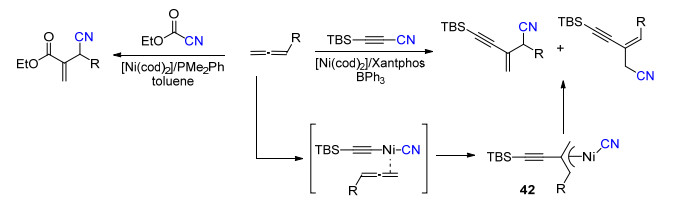
除了炔烃参与的镍催化C—CN键断裂反应, Hiyama等[34]也报道了联烯参与的镍催化C—CN键的断裂反应, 以Ni(COD)2/Xantphos为催化剂, 在添加三苯基硼的条件下, 实现了炔基腈与联烯的加成, 经过中间体42以中等到良好的收率得到烯丙基氰化物.当使用富电的单膦配体时, 氰基甲酸乙酯也可以在镍作用下发生C—CN键断裂反应, 与联烯进行加成反应得到酯基取代的烯丙基氰化物(Scheme 24).
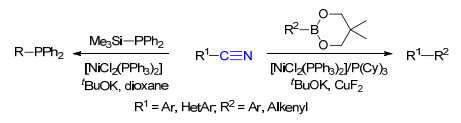
2009年, 施章杰课题组[35a]报道了镍催化芳基氰化物和硼酸酯的Suzuki-Miyaura偶联反应.在添加4 equiv.的叔丁醇钾条件下, 芳基氰化物与硼酸酯化合物顺利地发生偶联反应得到联芳烃类化合物. 2011年, 杨尚东课题组[35b]报道了镍催化芳基氰化物与二苯基(三甲基硅基)膦的偶联反应, 通过镍催化C—CN键的断裂, 为膦化合物的合成提供了一种简单的方法(Scheme 25).
2018年, 韩立彪课题组[36]报道了镍催化C(sp3)—CN键的断裂反应, 在添加叔丁醇钾条件下, 与二芳基氧膦进行偶联反应, 以良好到优秀的收率得到多种类型的磷酸酯类化合物(Eq. 6).
|
|
(6) |
2020年, 廖学斌课题组[37]报道了镍催化4-CN取代吡啶氮氧化合物的氰基转移反应.在镍催化剂作用下, 通过吡啶氮氧底物上的C—CN键断裂反应, 一步与芳基卤化物或者炔烃进行偶联反应, 分别得到芳基氰化物或者烯基氰化物(Scheme 26).
2017年, Chatani等[38]报道了二芳基甲酮的C—C键断裂的反应, 反应使用1 equiv.的镍催化剂, 以卡宾为配体, 在叔丁醇钠作用下, 加热至160 ℃原位脱除CO得到联芳烃类化合物(Eq. 7).
|
|
(7) |
2018年, 魏颢和徐鹏飞课题组[39]首次报道了镍催化链状酮的脱CO反应制备联芳烃产物的方法.利用导向基的策略, 以带有嘧啶导向基的吲哚为底物, 在Ni0/IMes作用下, 通过镍催化氧化C—C键断裂过程, 现场脱除CO后还原消除得到2-芳基取代的吲哚类化合物(Eq. 8).
|
|
(8) |
2019年, 周其林课题组[40]报道了镍催化烯丙基胺与另一分子烯烃的烯基转移反应.在Ni(COD)2/PCy3作用下, 通过形成氮杂环镍中间体后经β-C消除实现了C—C键断裂反应, 为在医药领域有广阔应用价值的烯丙基胺类化合物的合成提供了非常简便的方法(Eq. 9).
|
|
(9) |
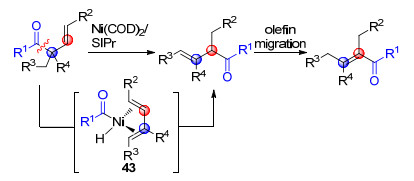
2020年, 魏颢课题组[41]报道了通过镍催化C—C键断裂实现1, 2-酰基转移的反应.在镍催化剂作用下, 以SIPr卡宾为配体, 通过β, γ-不饱和酮的羰基α位C—C键断裂反应得到酰基镍金属中间体43, 随后发生乙酰基与共轭烯烃的插入反应得到酰基转移产物, 最后异构化为稳定的α, β-不饱和酮(Scheme 27).
不对称C—C键活化一直是过渡金属催化C—C键活化领域的一个重要研究内容, 但成功的例子却非常少, 主要是因为C—C键的键能比较高, 难以活化, 反应条件往往比较苛刻, 对映选择性较难控制, 缺乏有效的催化剂来降低C—C键断裂的能垒并很好地控制对映选择性.
经过研究, 近些年镍催化的不对称C—C键活化反应也取得了一些进展. 2013年, Kurahashi等[42]报道镍催化乙烯基环丙烷与亚胺的[3+2]环加成反应.当使用iPr-Duphos为配体时能够以83%的收率和56%的ee值得到四氢吡咯烷产物, 初步实现了该反应的不对称催化(Eq. 10).
|
|
(10) |
2017年, 叶萌春课题组[43]报道了镍催化单羧基取代环丙烷与炔烃的不对称[3+2]环加成反应.使用手性磷氧为配体, 在添加AlMe3的条件下, 可以获得高达99%的收率和94%的对映选择性.作者认为AlMe3的加入不仅可以活化三元环底物, 促进零价镍对三元环开环, 利用生成的Ni-P-O-Al中间体, 促进反应进行并使反应获得高的立体选择性(Eq. 11).
|
|
(11) |
2020年, 李兴伟和白大昌课题组[44]报道了镍催化环丙烯酮和α, β-不饱和酮(亚胺)的不对称[3+2]环加成反应.使用他们设计合成的顺式双苯基取代的手性喹啉噁唑啉配体, 在室温条件下反应能够以优秀的收率和优秀的对映选择性得到含四个取代基手性中心的丁烯酸内酯(内酰胺)类化合物.该反应不仅条件温和、底物使用范围广, 而且反应的效率也很高, 特别是使用缺电子的α, β-不饱和酮时, 反应一般能够在5 min内转化完全, 反应转化数(TON)达到440, 在不对称C—C键活化领域是一个比较高的TON值(Eq. 12).
|
|
(12) |
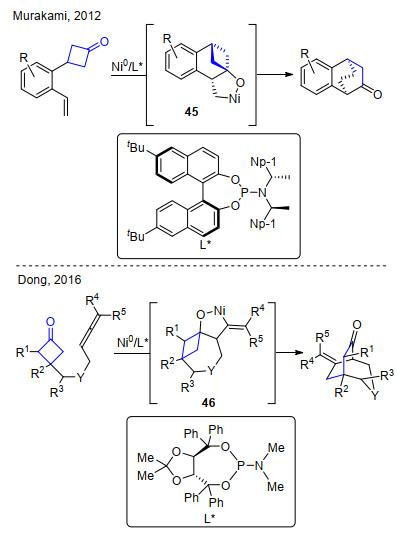
2012年, Murakami小组[45a]报道了镍(0)催化环丁酮与双键的分子内不对称环加成反应.使用手性单膦配体, 通过环丁酮与C=C双键的环金属化后β-C消除得到具有光学活性的桥环产物(Scheme 28). 2016年, 董广斌课题组[45b]实现了环丁酮与联烯的分子内不对称加成反应, 高对映选择性地得到[3.2.2]桥环产物(Scheme 28).
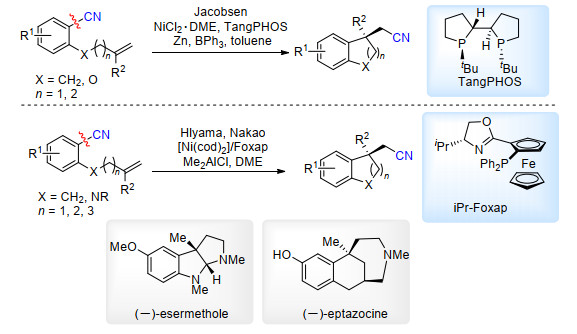
2008年, Jacobsen课题组[46a]实现了镍催化分子内氰基转移反应, 使用TangPHOS配体, 在添加BPh3条件下, 以优秀的对映选择性得到氰基转移的产物(Scheme 29).与此同时, Nakao和Hiyama课题组[46b, 46c]也报道了类似的反应, 以Foxap为配体, 在添加Me2AlCl条件下, 同样以优秀的对映选择性得到氰基转移的产物. Nakao和Hiyama利用该反应实现了(-)-esermethole和(-)-eptazocine的全合成(Scheme 29).
综上所述, 主要介绍了近些年镍催化C—C键断裂反应的研究进展, 根据底物类型分为三元环底物、四元环底物和链状底物, 最后总结了不对称催化体系.这些反应主要经过氧化C—C键断裂过程或者β-C消除过程, 也有一些经过自由基过程的报道.反应的驱动力主要是通过释放环张力或增加反应的熵来促进反应进行, 通常能够生成稳定的过渡金属络合物.随着研究的不断深入, 人们将利用这些反应合成结构更加复杂的小分子化合物, 在生物活性分子和药物分子的合成中获得潜在的应用.
然而, 不可否认的是这一领域还需要进一步的研究, 一方面发生C—C键活化的底物结构类型还比较少, 特别是实现简单底物参与的镍催化C—C键活化的策略还非常少, 发展简单结构底物的C—C键活化非常具有挑战性; 另一方面不对称催化的例子还不多, 有效的手性催化剂还非常少, 特别是手性配体类型、催化体系还有很大的发展空间.因此, 发展新的手性催化剂、研究新的合成方法、实现底物类型多样的C—C键断裂反应体系并研究这些方法在医药和材料等领域的应用具有非常重要的科学意义和实际应用价值.
Reviews: (a) Song, F.-J.; Gou, T.; Wang, B.-Q.; Shi, Z.-J. Chem. Soc. Rev. 2018, 47, 7078.
(b) Deng, L.; Jin, L.; Dong, G. Angew. Chem., Int. Ed. 2018, 57, 2702.
(c) Fumagalli, G.; Stanton, S.; Bower, J. F. Chem. Rev. 2017, 117, 9404.
(d) Chen, P.-H.; Billett, B. A.; Tsukamoto, T.; Dong, G.-B. ACS Catal. 2017, 7, 1340.
(e) Souillart, L.; Cramer, N. Chem. Rev. 2015, 115, 9410.
(f) Liu, H.; Feng, M.-H.; Jiang, X.-F. Chem.-Asian J. 2014, 9, 3360.
(g) Jun, C.-H. Chem. Soc. Rev. 2004, 33, 610.
(h) Liang, Y.-F.; Jiao, N. Acc. Chem. Res. 2017, 50, 1640.
(i) Liu, J.-Z.; Qiu, X.; Huang, X.-Q.; Luo, X.; Zhang, C.; Wei, J.-L.; Pan, J.; Liang, Y.-J.; Zhu, Y.-C.; Qin, Q.-X.; Son, S.; Jiao, N. Nat. Chem. 2019, 11, 94.
(j) Sivaguru, P.; Wang, Z.-K.; Zanoni, G.; Bi, X.-H. Chem. Soc. Rev. 2019, 48, 2615.
(k) Wu, X.-X.; Zhu, C. Chem. Rec. 2018, 18, 587.
(a) Murakami, M.; Ishida, N. J. Am. Chem. Soc. 2016, 138, 13759.
(b) Marek, I.; Masarwa, A.; Delaye, P.-O.; Leibeling, M. Angew. Chem., Int. Ed. 2015, 54, 414.
(c) Li, T.-F.; Xu, F.; Li, X.-C.; Wang, C.-X.; Wan, B.-S. Angew. Chem., Int. Ed. 2016, 55, 2861.
(d) Chen, F.; Wang, T.; Jiao, N. Chem. Rev. 2014, 114, 8613.
(e) Chen, W.-L.; Wu, S.-Y.; Mo, X.-L.; Wei, L.-X.; Liang, C.; Mo, D.-L. Org. Lett. 2018, 20, 3527.
(f) Zhao, H.-P; Liang, G.-C.; Nie, S.-M.; Lu, X.; Pan, C.-X.; Zhong, X.-X.; Su, G.-F.; Mo, D.-L. Green Chem. 2020, 22, 404.
(a) Dai, P.-F.; Ning, X.-S.; Wang, H.; Cui, X.-C.; Liu, J.; Qu, J.-P.; Kang, Y.-B. Angew. Chem., Int. Ed. 2019, 58, 5392.
(b) Sun, T.-W.; Zhang, Y.-N.; Qiu, B.; Wang, Y.-F.; Qin, Y.-T.; Dong, G.-B.; Xu, T. Angew. Chem., Int. Ed. 2018, 57, 2859.
(c) Cao, J.; Fang, R.; Liu, J.-Y.; Lu, H.; Luo, Y.-C.; Xu, P.-F. Chem.-Eur. J. 2018, 24, 18863.
(d) Liu, L.-T.; Guo, Z.-H.; Xu, K.; Hui, S.-S.; Zhao, X.-F.; Zhao, B.-L.; Tan, H.; Chen, C.; Jiao, N.; Shi, Z.-Z. Chin. J Chem. 2018, 36, 995.
(e) Yu, X.-Y.; Chen, J.-R.; Wang, P.-Z.; Yang, M.-N.; Liang, D.; Xiao, W.-J. Angew. Chem., Int. Ed. 2018, 57, 738.
(f) Mao, W.-B.; Zhu, C. J. Org. Chem. 2017, 82, 9133.
Selected examples on nickel catalyzed C—C cleavage: (a) Fan, C.; Lv, X.-Y.; Xiao, L.-J.; Xie, J.-H.; Zhou, Q.-L. J. Am. Chem. Soc. 2019, 141, 2889.
(b) Zhao, T.-T.; Xu, W.-H.; Zheng, Z.-J.; Xu, P.-F.; Wei, H. J. Am. Chem. Soc. 2018, 140, 586.
(c) Morioka, T.; Nishizawa, A.; Furukawa, T.; Tobisu, M.; Chatani, N. J. Am. Chem. Soc. 2017, 139, 1416.
(d) Liu, L.; Montgomery, J. J. Am. Chem. Soc. 2006, 128, 5348.
(e) Lloyd-Jones, G. C. Angew. Chem., Int. Ed. 2006, 45, 67880.
(f) Ogoshi, S.; Nagata, M.; Kurosawa, H. J. Am. Chem. Soc. 2006, 128, 5350.
(g) Jiang, L.-H.; Huang, F.; Wang, Q.; Sun, C.-Z.; Liu, J.-B.; Chen, D.-Z. Org. Chem. Front. 2018, 5, 2332.
(a) Wiberg, K.; Waddell, S. T. J. Am. Chem. Soc. 1990, 112, 2194.
(b) Yu, S. J.; Noble, A.; Bedford, R. B.; Aggarwal, V. K. J. Am. Chem. Soc. 2019, 141, 20325.
(a) Nakamura, M.; Isobe, H.; Nakamura, E. Chem. Rev. 2003, 103, 1295.
(b) Yang, Y.; Zhang, Z.; Zhang, X.; Wang, D.; Wei, Y.; Shi, M. Chem. Commun. 2014, 50, 115.
(c) Li, X.; Han, C.; Yao, H.; Lin, A. Org. Lett. 2017, 19, 778.
Noyori, R.; Umeda, I.; Takaya H. Chem. Lett. 1972, 1, 1189. doi: 10.1246/cl.1972.1189
(a) Baba, A.; Ohshiro, Y.; Agawa, T. Chem. Lett. 1976, 5, 11.
(b) Baba, A.; Ohshiro, Y.; Agawa. T. J. Organomet. Chem. 1976, 110, 121.
Zhao, W.-T.; Gao, F.; Zhao, D.-B. Angew. Chem., Int. Ed. 2018, 57, 6329. doi: 10.1002/anie.201803156
Huang, J. Q.; Ho, C. Y. Angew. Chem., Int. Ed. 2019, 58, 5702. doi: 10.1002/anie.201901255
(a) Noyori, R.; Odagi, T.; Takaya, H. J. Am. Chem. Soc. 1970, 92, 5780.
(b) Noyori, R.; Kumagai, Y.; Umeda, I.; Takaya, H. J. Am. Chem. Soc. 1972, 94, 4018.
(c) Ma, X.-P.; Nong, C.-M.; Zhao, J.; Lu, X.; Liang, C.; Mo, D.-L. Adv. Synth. Catal. 2020, 362, 478.
(a) Saito, S.; Masuda, M.; Komagawa, S. J. Am. Chem. Soc. 2004, 126, 10540.
(b) Saito, S.; Komagawa, S.; Azumaya, I.; Masuda, M. J. Org. Chem. 2007, 72, 9114.
(c) Komagawa, S.; Saito, S. Angew. Chem., Int. Ed. 2006, 45, 2446.
(d) Maeda, K.; Saito, S. Tetrahedron Lett. 2007, 48, 3173.
(e) Saito, S.; Takeuchi, K. Tetrahedron Lett. 2007, 48, 595.
Saito, S.; Maeda, K.; Yamasaki, R.; Kitamura, T.; Nakagawa, M.; Kato, K.; Azumaya, I.; Masu, H. Angew. Chem., Int. Ed. 2010, 49, 1830. doi: 10.1002/anie.200907052
Saito, S.; Yoshizawa, T.; Ishigami, S.; Yamasaki, R. Tetrahedron Lett. 2010, 51, 6028. doi: 10.1016/j.tetlet.2010.09.031
(a) Saya, L.; Bhargava, G.; Navarro, M. A.; Gulías, M.; López, F.; Fernández, I.; Castedo, L.; Mascareñas, J. L. Angew. Chem., Int. Ed. 2010, 49, 9886.
(b) Saya, L.; Fernández, I.; López, F.; Mascareñas, J. L. Org. Lett. 2014, 16, 5008.
Yao, B.; Li, Y.; Liang, Z.; Zhang, Y. Org. Lett. 2011, 13, 640. doi: 10.1021/ol1028628
(a) Ogata, K.; Shimada, D.; Furuya, S.; Fukuzawa, S.-I. Org. Lett. 2013, 15, 1182.
(b) Ogata, K.; Atsuumi, Y.; Fukuzawa, S.-I. Org. Lett. 2010, 12, 4536.
(c) Pan, B.; Wang, C.; Wang, D.; Wu, F.; Wan, B. Chem. Commun. 2013, 49, 5073.
(a) Yamamoto, K.; Ishida, T.; Tsuji, J. Chem. Lett. 1987, 16, 1157.
(b) Wang, Q.; Wang, C.; Shi, W.; Xiao, Y.; Guo, H. Org. Biomol. Chem. 2018, 16, 4881.
(c) Parsons, A. T.; Campbell, M. J.; Johnson, J. S. Org. Lett. 2008, 10, 2541.
(d) Liu, C.-H.; Yu, Z.-X. Angew. Chem., Int. Ed. 2017, 56, 8667.
(e) Wang, Y.-Y.; Wang, J.-H.; Su, J.-C.; Huang, F.; Jiao, L.; Liang, Y.; Yang, D.-Z.; Zhang, S.-W.; Wender, P.-A.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 10060.
(a) Sumida, Y.; Yorimitsu, H.; Oshima, K. Org. Lett. 2008, 10, 4677.
(b) Bowman, R. K.; Johnson, J. S. Org. Lett. 2006, 8, 573.
(a) Tombe, R.; Iwamoto, T.; Kurahashi, T.; Matsubara, S. Synlett. 2014, 25, 2281.
(b) Mori, T.; Nakamura, T.; Kimura, M. Org. Lett. 2011, 13, 2266.
(c) Mita, T.; Tanaka, H.; Higuchi, Y.; Sato, Y. Org. Lett. 2016, 18, 2754.
(d) Tombe, R.; Kurahashi, T.; Matsubara, S. Org. Lett. 2013, 15, 1791.
Ogoshi, S.; Nagata, M.; Kurosawa, H. J. Am. Chem. Soc. 2006, 128, 5350. doi: 10.1021/ja060220y
(a) Liu, L.; Montgomery, J. J. Am. Chem. Soc. 2006, 128, 5348.
(b) Lloyd-Jones, G. C. Angew. Chem., Int. Ed. 2006, 45, 6788.
(c) Liu, L.; Montgomery, J. Org. Lett. 2007, 9, 3885.
(d) Tamaki, T.; Ohashi, M.; Ogoshi, S. Angew. Chem., Int. Ed. 2011, 50, 12067.
(a) Zuo, G.; Louie, J. Angew. Chem., Int. Ed. 2004, 43, 2277.
(b) Nečas, D.; Kotora, M. Org. Lett. 2008, 10, 5261.
(c) Wender, P. A.; Takahashi, H.; Witulski, B. J. Am. Chem. Soc. 1995, 117, 4720.
Schwager, H.; Spyroudis, S.; Vollhardt, K. P. C. J. Organomet. Chem. 1990, 382, 191. doi: 10.1016/0022-328X(90)85227-P
(a) Edelbach, B. L.; Lachicotte, R. J.; Jones, W. D. Organometallics 1999, 18, 4660.
(b) Edelbach, B. L.; Lachicotte, R. J.; Jones, W. D. Organometallics 1999, 18, 4040.
(c) Müller, C.; Lachicotte, R. J.; Jones, W. D. Organometallics 2002, 21, 1975.
(d) Schaub, T.; Radius, U. Chem.-Eur. J. 2005, 11, 5024.
(e) Schaub, T.; Backes, M.; Radius, U. Organometallics 2006, 25, 4196.
(f) Iverson, C. N.; Jones, W. D. Organometallics 2001, 20, 5745.
Ho, K. Y. T.; Aïssa, C. Chem.-Eur. J. 2012, 18, 3486. doi: 10.1002/chem.201200167
(a) Kumar, P.; Zhang, K.; Louie, J. Angew. Chem., Int. Ed. 2012, 51, 8602.
(b) Thakur, A.; Facer, M. E.; Louie, J. Angew. Chem., Int. Ed. 2013, 52, 12161.
(a) Juliá-Hernández, F.; Ziadi, A.; Nishimura, A.; Martin, R. Angew. Chem., Int. Ed. 2015, 54, 9537.
(b) Yang, S.; Xu, Y.; Li, J. Org. Lett. 2016, 18, 6244.
Auvinet, A.-L.; Harrity, J. P. A. Angew. Chem., Int. Ed. 2011, 50, 2769. doi: 10.1002/anie.201007598
(a) Boivin, J.; Fouquet, E.; Zard, S. Z. J. Am. Chem. Soc. 1991, 113, 1055.
(b) Gu, Y.-R.; Duan, X.-H.; Yang, L.; Guo, L.-N. Org. Lett. 2017, 19, 5908.
(c) Tang, Y.-Q.; Yang, J.-C.; Wang, L.; Fan, M.-J.; Guo, L.-N. Org. Lett. 2019, 21, 5178.
(d) Yang, H.-B.; Pathipati, S.-R.; Selander, N. ACS Catal. 2017, 7, 8441.
(e) Shuai, B.; Li, Z.-M.; Qiu, H.; Fang, P.; Mei, T.-S. Chin. J. Org. Chem. 2020, 40, 651 (in Chinese).
(帅斌, 李兆明, 裘晖, 方萍, 梅天胜, 有机化学, 2020, 40, 651.)
(f) Ding, D.-C.; Wang, C. ACS Catal. 2018, 8, 11324.
(g) Ding, D.-C.; Lan, Y.; Lin, Z.-Y.; Wang, C. Org. Lett. 2019, 21, 2723.
Gerlach, D. H.; Kane, A. R.; Parshall, G. W.; Jesson, J. P.; Muetterties, E. L. J. Am. Chem. Soc. 1971, 93, 3543. doi: 10.1021/ja00743a050
Nakao, Y.; Oda, S.; Hiyama, T. J. Am. Chem. Soc. 2004, 126, 13904. doi: 10.1021/ja0448723
(a) Nakao, Y.; Oda, S.; Yada, A.; Hiyama, T. Tetrahedron 2006, 62, 7567.
(b) Ohnishi, Y.-Y.; Nakao, Y.; Sato, H.; Nakao, Y.; Hiyama, T.; Sakaki, S. Organometallics 2009, 28, 2583.
(c) Hirata, Y.; Yukawa, T.; Kashihara, N.; Nakao, Y.; Hiyama, T. J. Am. Chem. Soc. 2009, 131, 10964.
(d) Nakao, Y.; Yada, A.; Ebata, S.; Hiyama, T. J. Am. Chem. Soc. 2007, 129, 2428.
(e) Yada, A.; Yukawa, T.; Nakao, Y.; Hiyama, T. Chem. Commun. 2009, 3931.
(f) Yada, A.; Yukawa, T.; Idei, H.; Nakao, Y.; Hiyama, T. Bull. Chem. Soc. Jpn. 2010, 83, 619.
(g) Nakao, Y.; Yada, A.; Hiyama, T. J. Am. Chem. Soc. 2010, 132, 10024.
(h) Nakao, Y.; Hirata, Y.; Tanaka, M.; Hiyama, T. Angew. Chem., Int. Ed. 2008, 47, 385.
(i) Hirata, Y.; Tanaka, M.; Yada, A.; Nakao, Y.; Hiyama, T. Tetrahedron 2009, 65, 5037.
(j) Nakao, Y.; Ebata, S.; Yada, A.; Hiyama, T.; Ikawa, M.; Ogoshi, S. J. Am. Chem. Soc. 2008, 130, 12874.
(k) Minami, Y.; Yoshiyasu, H.; Nakao, Y.; Hiyama, T. Angew. Chem., Int. Ed. 2013, 52, 883.
(l) Nakai, K.; Kurahashi, T.; Matsubara, S. Org. Lett. 2013, 15, 856.
(a) Nakao, Y.; Hirata, Y.; Tanaka, M.; Hiyama, T. Angew. Chem., Int. Ed. 2008, 47, 385.
(b) Hirata, Y.; Tanaka, M.; Yada, A.; Nakao, Y.; Hiyama, T. Tetrahedron 2009, 65, 5037.
(c) Hirata, Y.; Yada, A.; Morita, E.; Nakao, Y.; Hiyama, T.; Ohashi, M.; Ogoshi, S. J. Am. Chem. Soc. 2010, 132, 10070.
(d) Hirata, Y.; Inui, T.; Nakao, Y.; Hiyama, T. J. Am. Chem. Soc. 2009, 131, 6624.
(e) Nakao, Y.; Hirata, Y.; Hiyama, T. J. Am. Chem. Soc. 2006, 128, 7420.
(f) Hirata, Y.; Inui, T.; Nakao, Y.; Hiyama, T. J. Am. Chem. Soc. 2009, 131, 6624.
(a) Yu, D.-G.; Yu, M.; Guan, B.-T.; Li, B.-J.; Zheng, Y.; Wu, Z.-H.; Shi, Z.-J. Org. Lett. 2009, 11, 3374.
(b) Sun, M.; Zhang, H.-Y.; Han, Q.; Yang, K.; Yang, S.-D. Chem.-Eur. J. 2011, 17, 9566.
Zhang, J.-S.; Chen, T.-Q.; Zhou, Y.-B.; Yin, S.-F.; Han, L.-B. Org. Lett. 2018, 20, 6746. doi: 10.1021/acs.orglett.8b02854
Chen, H.; Sun, S.-H.; Liu, Y.-H.; Liao, X.-B. ACS Catal. 2020, 10, 1397. doi: 10.1021/acscatal.9b04586
Morioka, T.; Nishizawa, A.; Furukawa, T.; Tobisu, M.; Chatani, O. J. Am. Chem. Soc. 2017, 139, 1416. doi: 10.1021/jacs.6b12293
Zhao, T.-T.; Xu, W.-H.; Zheng, Z.-J.; Xu, P.-F.; Wei, H. J. Am. Chem. Soc. 2018, 140, 586. doi: 10.1021/jacs.7b11591
Fan, C.; Lv, X.-Y.; Xiao, L.-J.; Xie, J.-H.; Zhou, Q.-L. J. Am. Chem. Soc. 2019, 141, 7, 2889.
Jiang, C.; Lu, H.; Xu, W.-H.; Wu, J.-N.; Yu, T.-Y.; Xu, P.-F.; Wei, H. ACS Catal. 2020, 10, 1947. doi: 10.1021/acscatal.9b04112
Tombe, R.; Kurahashi, T.; Matsubara, S. Org. Lett. 2013, 15, 1791. doi: 10.1021/ol4005068
(a) Liu, Q.-S.; Wang, D.-Y.; Yang, Z.-J.; Luan, Y.-X.; Yang, J.-F.; Li, J.-F.; Pu, Y.-G.; Ye, M.-C. J. Am. Chem. Soc. 2017, 139, 18150.
(b) Wang, Y.-X.; Ye, M.-C. Sci. China, Chem. 2018, 61, 1004.
Bai, D.-C.; Yu, Y.-J.; Guo, H.-M.; Chang, J.-B.; Li, X.-W. Angew. Chem., Int. Ed. 2020, 59, 2740. doi: 10.1002/anie.201913130
(a) Liu, L.; Ishida, N.; Murakami, M. Angew. Chem., Int. Ed. 2012, 51, 2485.
(b) Zhou, X.; Dong, G.-B. Angew. Chem., Int. Ed. 2016, 55, 15091.
(c) Murakami, M.; Ashida, S.; Matsuda, T. J. Am. Chem. Soc. 2005, 127, 6932.
(a) Watson, M. P.; Jacobsen, E. N. J. Am. Chem. Soc. 2008, 130, 12594.
(b) Nakao, Y.; Ebata, S.; Yada, A.; Hiyama, T.; Ikawa, M.; Ogoshi, S. J. Am. Chem. Soc. 2008, 130, 12874.
(c) Hsieh, J.-C.; Ebata, S.; Nakao, Y.; Hiyama, T. Synlett 2010, 1709.

图式 1 (A) 氧化加成或(B)β-C消除实现C—C键断裂
Scheme 1 C—C cleavage by (A) C—C oxidative addition or (B) β-carbon elimination

图式 2 镍催化[1.1.1]螺桨烷参与的烯烃环丙烷化反应
Scheme 2 Nickel-catalyzed cyclopropanation of alkenes with [1.1.1]propellane

图式 4 环丙烯酮与联烯酮或亚磺酰亚胺的环加成反应
Scheme 4 Cycloaddition of diphenylcyclopropenone with N-sulfinylamine or ketenes

图式 5 亚甲基环丙烷参与的镍催化C—C键活化
Scheme 5 Nickel-catalyzed C—C bond activations of alkylidenecyclopropanes

图式 6 酯基取代的亚甲基环丙烷参与的镍催化C—C键活化
Scheme 6 Nickel-catalyzed C—C bond activations of cyclopropylideneacetate

图式 7 镍催化双烯炔底物与亚甲基环丙烷的[4+3+2]环加成反应
Scheme 7 Nickel(0)-catalyzed [4+3+2] cycloaddition reaction of ethyl cyclopropylideneacetate and dienynes

图式 8 镍催化苯并硅杂环丁烷与亚甲基环丙烷的[4+3]环加成反应
Scheme 8 Nickel(0)-catalyzed [4+3] cycloaddition of ethyl cyclopropylideneacetate and benzosilacyclobutenes

图式 9 炔基取代的亚甲基环丙烷参与的镍催化C—C键活化
Scheme 9 Nickel-catalyzed C—C bond activations of alkynylidenecyclopropanes

图式 10 镍催化分子内亚甲基环丙烷与炔烃的环加成反应
Scheme 10 Ni(0)-catalyzed intramolecular cycloaddition of methylenecyclopropanes with alkynes

图式 11 镍催化亚甲基环丙烷与α, β-不饱和酮的烷基化偶联反应
Scheme 11 Nickel-catalyzed alkylative coupling of enone with methylenecyclopropane

图式 12 乙烯基三元环参与的镍催化C—C键活化
Scheme 12 Nickel-catalyzed C—C bond activations of vinylcyclopropanes

图式 13 镍催化乙烯基环丙烷与联烯的环加成发应
Scheme 13 Nickel-catalyzed cycloaddition of vinyl cyclopropanes with allenes

图式 15 镍催化非活化乙烯基环丙烷的三元环开环反应
Scheme 15 Nickel-catalyzed three-membered ring-opening of unactivated vinyl cyclopropanes

图 1 四元环底物参与的镍催化C—C键活化
Figure 1 Nickel-catalyzed C—C bond activations of fourmembered rings

图式 17 二苯并环丁二烯参与的镍催化C—C键活化
Scheme 17 Nickel-catalyzed C—C bond activations of biphenylene

图式 18 镍催化氮杂环丁酮与炔烃的[4+2]环加成反应
Scheme 18 Nickel-catalyzed [4+2] cycloaddition of 3-azetidinones with alkynes

图式 19 镍催化氮(氧)环丁酮与双炔或共轭双烯的环加成反应
Scheme 19 Nickel-catalyzed cycloaddition of 3-azetidinones (or 3-oxetanones) with diynes or 1, 3-dienes

图式 20 镍催化苯并环丁酮的开环反应
Scheme 20 Nickel-catalyzed ring-opening reactions of benzocyclobutanones

图式 21 环丁酮肟酯参与的镍催化C—C键活化
Scheme 21 Nickel-catalyzed C—C bond activations of cycloketone oxime esters

图式 22 镍催化芳基氰化物与炔烃的加成反应
Scheme 22 Nickel-catalyzed C—C bond activation of aryl cyanide with alkynes

图式 23 炔烃参与的镍催化C—CN键活化
Scheme 23 Nickel-catalyzed C—C bond activations of C—CN bond with alkynes

图式 24 联烯参与的镍催化C—CN键活化
Scheme 24 Nickel-catalyzed C—C bond activations of C—CN bond with 1, 2-dienes

图式 25 硅膦试剂或者硼酸酯参与的镍催化C—CN键活化
Scheme 25 Nickel-catalyzed C—C bond activations of C—CN bond with silicon phosphine reagent or borate

图式 26 镍催化4-CN取代吡啶氮氧化合物的C—CN键断裂和氰基转移反应
Scheme 26 Nickel-catalyzed C—CN bond cleavage and cyano tranfer of 4-cyanopyridine N-oxide

图式 27 通过镍催化C—C键活化实现1, 2酰基迁移反应
Scheme 27 Ni-catalyzed 1, 2-acyl migration triggered by C—C bond cleavage

图式 28 镍催化环丁酮参与的不对称C—C键活化反应
Scheme 28 Nickel-catalyzed asymmetric C—C bond activation reactions of cyclobutanones

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:












 下载:
下载:



