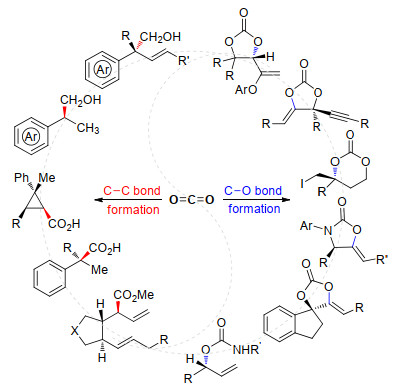
Scheme 1.
Enantioselective synthesis of small molecules via CO2 fixation by asymmetric catalysis
Recent Advances of CO2 Fixation via Asymmetric Catalysis for the Direct Synthesis of Optically Active Small Molecules
Xiao Guo , Yazhou Wang , Jie Chen , Gongqiang Li , Ji-Bao Xia

The current global economic development has accelerated carbon emission, and the amount of CO2 generated by human activity is estimated to be 35 billion tons in 2015.[1] The relentless rise of atmospheric CO2 is thought to be a main cause of global warming and abnormal climate changes. However, CO2 is an attractive C1 building block in synthetic chemistry and chemical industry because of its abundance, non-toxicity, and low cost. Therefore, conversion of CO2 into valuable products will meet the demand of sustainable chemistry, although it earns a small proportion from carbon emission at the present stage. In this respect, a lot of research has been conducted on the development of new transformations using CO2 as a C1 source, including transition metal catalysis, photocatalysis, electrocatalysis, ionic liquids catalysis, etc.[2-18] Industrial synthetic applications of CO2 have also been realized in the synthesis of salicylic acid, urea, methanol, and polycarbonates.[18-21]
In general, reactive reagents or harsh conditions are needed in the transformation of CO2 because of its thermodynamic stability and/or kinetic inertness. First, the thermodynamic stability of CO2 imposes an input of energy to convert it into fuels or chemicals. The standard reduction potential (ER0) is a measure of the spontaneity of a given reaction relative to the hydrogen evolution reaction (HER) as equation ΔG°=-nFErxn0. Due to the relative stability of gaseous CO2 (ΔGf0 =-394.4 kJ•mol-1), enough energy must be introduced into the reaction to drive its transformation to the products. Second, catalysts are usually required to ensure that the activation barriers remain as low as possible in the presence of chemical transformation processes. Thus, the overall carbon balance for CO2 utilization is not hampered by thermal loading needed to overcome high energy transition states.[22-23] Traditional methods for the transformation of CO2 generally need high-energy substrates and reagents, such as strong nucleophilic organometallic reagents (the Grignard reagents or organolithiums), small-ring heterocycles with high ring strain (epoxides or aziridines), and highly reactive reductants, etc.[24-26] Physical energy such as light or electricity was also employed to solve the issues brought by thermodynamic stability of CO2.[27-28] However, these traditional methods usually require multi-steps, resulting in waste or undesirable by-products.
Over the past decades, catalysts have been widely used to activate CO2 and facilitate its transformations. A variety of transition-metal-catalyzed carboxylations of alkenes, alkynes, dienes, allenes, organic halides, organometallic reagents, or C—H compounds have been reported for the synthesis of carboxylic acids and their derivatives.[29-37] On the other hand, chemists have also utilized organocatalysts, such as N-heterocyclic carbenes, phosphines, amines, and frustrated Lewis pairs, to activate CO2 in its conversion to small molecules.[38-40] The efficiency and applicability of CO2 transformations were thus improved greatly in organic synthesis. CO2 fixation and subsequent catalytic conversion to fine chemicals, particularly to chiral molecules, represents a highly value-added process. Enantiopure carbonates, carbamates and carboxylic acid derivatives are versatile synthons and widespread in natural products, drug molecules, and advanced materials. Catalytic enantioselective methods for the synthesis chiral carbonates and carbamates with CO2 via kinetic resolution of racemic epoxides and aziridines have been well explored. These achievements have been summarized in previous publications [41-43] and will not be discussed here. In 2017, a general review on the enantioselective synthesis of carboxylic acid derivatives and carbamates with CO2 was reported in broad content, including asymmetric carboxylation, chiral transfer, and ring-opening of epoxides, etc.[44] Herein, we would like to focus on the advances of CO2 fixation by catalytic asymmetric via carbon-carbon (C—C) and carbon–oxygen (C—O) bonds formation (Scheme 1). The interaction between catalyst, CO2 and substrate will be emphasized in this review to inspire the design of new catalytic systems for asymmetric CO2 transformations.
The development of new C—C bond forming reactions using sustainable chemical feedstock is rather attractive in synthetic chemistry. In this respect, conversions of CO2 into useful chemicals have gained considerable attention in recent years. One of the major challenges to synthesize chemicals with CO2 as a raw material is to construct C—C bonds with high efficiency and selectivity, including chemo-, regio-, and stereo-selectivity, expecially enantioselectivity. Because of the high thermodynamic and kinetic stability of CO2, most of the C—C bond-forming reactions with CO2 were carried out under harsh conditions, such as high pressure of CO2 and high reaction temperature. It is difficult to control the enantioselectivity in these reactions. Recently, transition-metal-catalyzed CO2 fixation to form C—C bond has emerged to be a mild and powerful tool to convert CO2 into useful chemicals. However, highly enantioselective C—C bond-forming reactions with CO2 as C1 source by asymmetric catalysis is still challenging and rare.
In 1981, an asymmetric CO2 fixation reaction was reported by Sato and co-workers[45] but with low enantioselectivity control. Asymmetric carboxylation of π-allylti- tanium complex tethered with chiral cyclopentadienyl ligand was achieved and 2-methylbut-3-enoic acid was obtained in 70% yield with 19% ee.
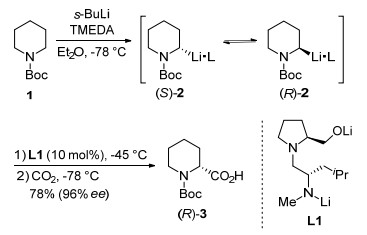
In 2010, Gawley and co-workers[46] reported a catalytic dynamic resolution of rac-2-lithio-N-Boc-piperidine (2), which was obtained by deprotonation of N-Boc-piperidine (1) with s-BuLi and N, N, N', N'-tetramethylethylenediamine (TMEDA) at a low temperature (Scheme 2). Catalytic dilithiodiaminoalkoxide (L1) was found to be an efficient chiral catalyst in this reaction. The authors investigated the plot of ΔG versus temperature for racemization of 2 and dynamic thermodynamic resolution of 2•L1 in the presence of TMEDA. The result revealed that the barrier for dynamic thermodynamic resolution of 2•L1 is lower than racemization of it below -27 ℃. Lithiation of 1 at -78 ℃, followed by ligand exchange at -45 ℃ and quenching with various electrophiles at -78 ℃ afforded 2-substituted N-Boc-piperidines with excellent enantioselectivities. When CO2 was used as an electrophile, (R)-N-Boc-pipe- colic acid (3) was obtained in 78% yield with 96% ee.
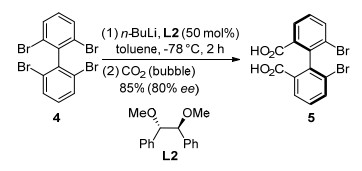
Later, Alexakis and co-workers[47] reported an asymmetric desymmetrization of 2, 2'-dibromobiphenyls to synthesize biphenyl molecules with axial chirality via an enantio- selective bromine-lithium exchange process. They investigated the influence factors on the backbone of the diether ligand. The increase of enantioselectivity was observed by decreasing the hindrance of the backbone of chiral ligand. With methyl-protected diol derivative L2 as a chiral ligand, 2, 2', 6, 6'-tetrabromobiphenyl (4) was converted to atropoisomeric 1, 1'-biphenyl-2, 2'-dicarboxylic acid (5) in 85% yield with 80% ee via bromine-lithium exchange followed by quenching with CO2 (Scheme 3). This was the first highly selective (up to 82% ee) bromine-lithium exchange with a catalytic amount of ligand (0.5 equiv., 25% per BrLi exchange). Recently, Hamashima and co-workers[48] disclosed that (S)-1, 1'-binaphthyl-2, 2'-dicarboxylic acid could be easily accessed by carboxylation of 2, 2'-dilithium-1, 1'- binaphthalene generated from lithiation of (S)-BINOL bis(diethylphosphate) with lithium/naphthalene. Notably, the reaction could be carried out on gram-scale and the corresponding di-acid was obtained with 99% ee.
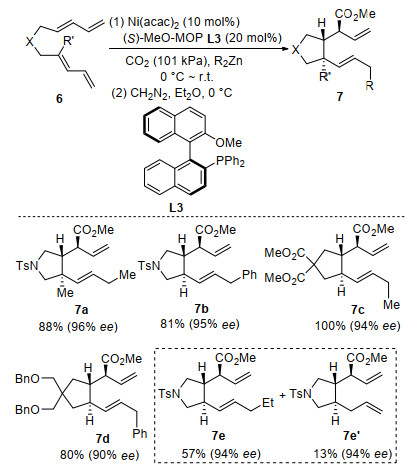
In 2002, a highly regio- and stereo-selective ring-closing carboxylation of bis-1, 3-dienes with CO2 was reported by Mori and co-workers.[49] After that, a breakthrough in asymmetric transition-metal-catalyzed C—C bond-forming reaction with CO2 was reported by the same group in 2004 (Scheme 4).[50] In the presence of 10 mol% of Ni(acac)2 and 20 mol% of (S)-MeO-MOP (L3), a highly regio-, diastereo-, and enantio-selective ring-closing carboxylation of bis-1, 3-diene (6) was realized under atmospheric pressure of CO2. Trans-disubstituted pyrrolidines or cyclopentanes 7 were obtained in high yields with 90%~96% ee. Chiral allylic carboxylic acid esters with three continuous stereocenters were synthesized efficiently in one-step. Single diastereoisomer was obtained with dimethylzinc or diphenylzinc as coupling reagents. When diethylzinc was used as a coupling reagent, cyclized products 7e and 7e' were obtained simultaneously with the same enantiomeric excess, indicating that they were generated from the same intermediate.
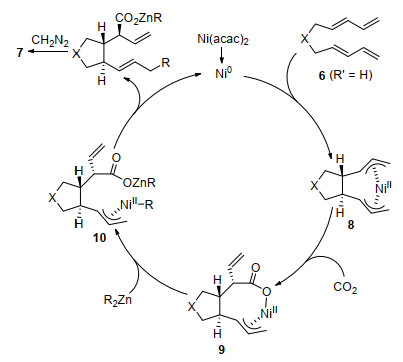
The authors proposed a plausible mechanism in Scheme 5. The reaction is initiated by oxidative cyclometalation of bis-1, 3-diene 6 with Ni0 complex generating cyclized bis-π-allylnickel species 8. Then insertion of CO2 into the Ni—C bond produces carboxylates 9. After transmetallation with an organozinc reagent, π-allylnickel intermediate 10 is generated. Reductive elimination from 10 provides the corresponding carboxylates and regenerates the Ni0 catalyst. The final product 7 is obtained after quenching with diazomethane. When diethylzinc is used as coupling reagent, complex 10 can undergo β-hydride elimination to afford π-allylnickel hydride complex. Then the byproduct 7e' is generated after reductive elimination.
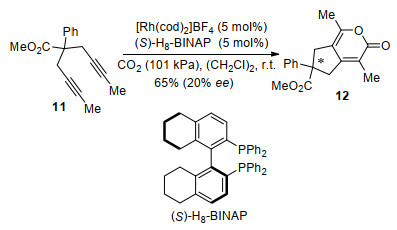
In 2014, Tanaka and co-workers[51] reported an enantioselective cyclization reaction of prochiral 1, 6-diyne 11 with CO2 via desymmetrization (Scheme 6). The [2+2+2] cycloaddition product 12 was obtained in good yield with a promising enantioselectivity (20% ee) using catalytic cationic [Rh(cod)2]BF4 and (S)-H8-BINAP.
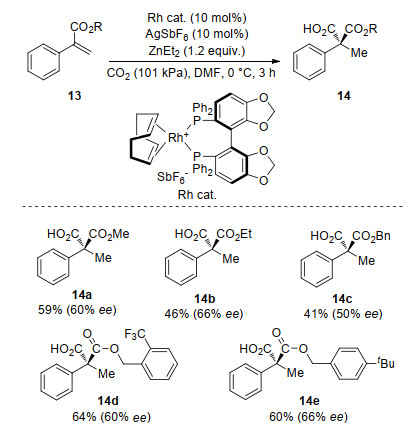
Recently, catalytic hydrocarboxylation of alkenes with CO2 has been reported using transition-metal-catalyst, such as Ni, [52] Fe, [53] or Co, [54, 55] with diethylzinc or ethylmagnesium halides as a hydride source. In 2016, Mikami and co-workers[56] reported a Rh-catalyzed hydrocarboxylation of alkenes with CO2. Using [RhCl(cod)]2 as catalyst and diethylzinc as hydride source, the corresponding carboxylic acids were obtained in moderate to excellent yields. They also investigated the asymmetric version of this reaction. Enantioselective hydrocarboxylation of α-aryl acrylates 13 afforded the corresponding acid 14 bearing a quaternary carbon center with moderate ee using cationic (S)-Segphos-Rh complex as catalyst and catalytic AgSbF6 as cocatalyst (Scheme 7). In this reaction, the steric and electronic effect on substituent of the ester (R group) has moderate effect on the enantioselectivity of the product. The authors didn't show the influence of aryl group on the enantioselectivity of the product.
The plausible reaction mechanism is displalyed in Scheme 8. First, RhI—Et complex 15 is generated by transmetalation of the ethyl group between diethylzinc and RhI precursor. Second, β-hydride elimination from 15 generates the RhI—H species 16 and releases ethylene. Then insertion of styrene derivatives into 16 affords benzyl rhodium intermediate 17, which undergoes insertion of CO2, followed by transmetalation with diethylzinc producing 19 and regenerates RhI—Et complex 15. Finally, acids 14 are obtained after hydrolysis of 19.
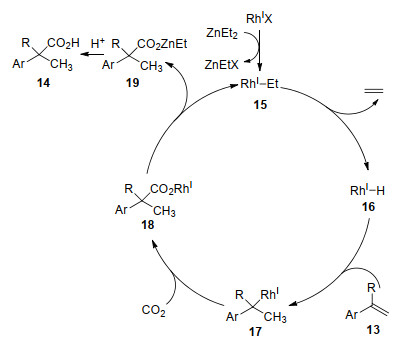
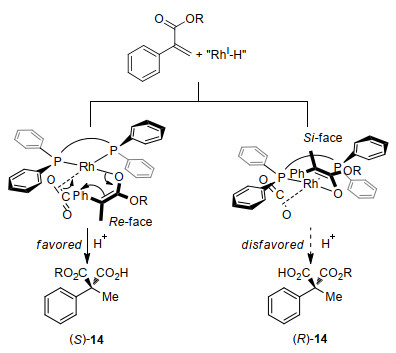
An enantioselective induction model has been proposed by the authors based on the absolute configuration of product 14 (Scheme 9). The corresponding (Z)-RhI-enolate is generated by insertion of α, β-unsaturated esters with RhI-H species in an s-trans fashion. Attack of CO2 with the enolate by the Si face is prevented by the equatorial phenyl group on the phosphorus atom in the chiral ligand. Thus, attack of CO2 from the Re face of the rhodium side would be favored to afford the corresponding product (S)-14.
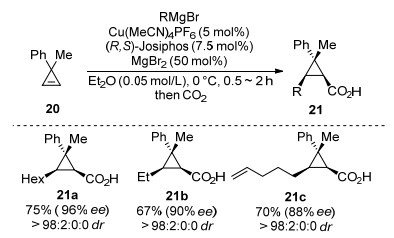
In 2017, Marek and co-workers[57] reported a highly diastereo- and enantio-selective Cu-catalyzed carboxylation of poly-substituted cyclopropenes with CO2 and Grignard reagents. Cyclopropylmetal species was first obtained through a Cu-catalyzed carbomagnesiation reaction, followed by quenching with CO2 as an electrophile affording the cyclopropyl acid product 21 in good yields with 88%~96% ee (Scheme 10). It was shown that the cyclopropylmagnesium species are configurationally stable. The addition of this cyclopropylmagnesium species to electrophile, such as CO2, leads to the product with clean retention of configuration.
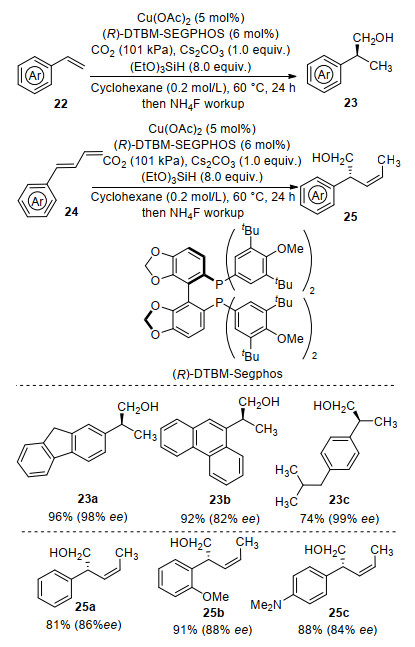
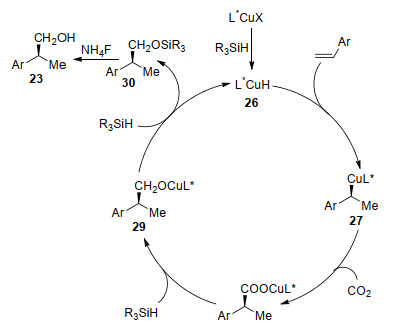
In 2017, Yu and co-workers[58] recently reported the first enantioselective Cu-catalyzed reductive hydroxymethylation of alkenes with CO2, which provides an efficient method for the synthesis of chiral homobenzylic/allylic alcohols (Scheme 11). Using Cu(OAc)2 as a catalyst, bulky (R)-DTBM-Segphos as a chiral ligand, and hydrosilane as a hydride source, styrene derivatives 22 were converted to a variety of chiral homobenzylic alcohols 23 in high yields with excellent enantioselectivities. Moreover, a series of chiral homoallylic alcohols 25 were also generated from aryl substituted 1, 3-dienes 24 with high regioselectivity and enantioselectivity under similar conditions. Notably, the homoallylic alcohols were obtained with high Z-selectivity for the alkene. The chiral benzylic alcohol 23c was easily transformed into the anti-inflammatory drug (S)-(+)-ibu- profen.
The proposed reaction mechanism is described in Scheme 12. At the beginning, active catalyst L*CuH 26 was generated by reaction of ligand coordinated pre-catalyst with silanes. Then, insertion of alkenes into the Cu—H bond of 26 affords alkyl copper species 27 with excellent regio- and enantio-selectivity. Subsequent carboxylation of 27 with CO2 generates copper carboxylate 28. Next, reduction of 28 by hydrosilanes leads to copper alkoxide 29. Further transmetalation of 29 with hydrosilane generates 30 and releases catalyst 26. Finally, desilylation of 30 affords the corresponding alcohol 23 by the treatment with NH4F.
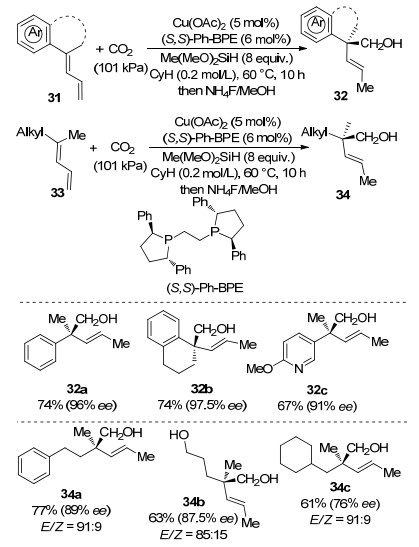
Continuing the previous research, Yu and co-workers[59] recently reported a Cu-catalyzed highly enantioselective hydroxymethylation of 1, 3-dienes with CO2 to construct chiral all-carbon quaternary stereocenter in acyclic molecules. In this studies, homoallylic alcohols 32 or 34 were obtained in moderate to good yields with excellent ees using (S, S)-Ph-BPE as chiral phosphine ligand and Me- (MeO)2SiH as hydride source (Scheme 13).
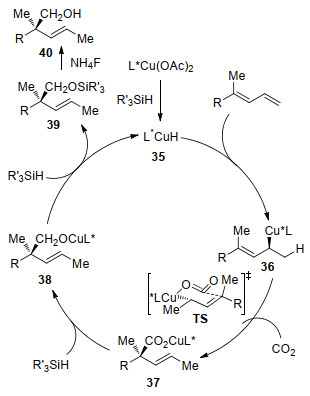
The mechanism of this transformation was shown in Scheme 14. First, a complex between the copper precatalyst and the chiral phosphine ligand reacts with silane generating a catalytically active chiral L*Cu—H species 35. Then a chiral allylic copper intermediate 36 is formed via a highly regio- and stereo-selective 1, 2-syn-addition of Cu—H species 35 into 1, 3-diene. Steric repulsion might prevent the energy-disfavored 1, 3-migration process to generate an isomeric allylcuprate. Through a possible six-membered ring chair like transition state in the nucleophilic addition of 36 to CO2, the copper carboxylate intermediate 37 is formed. Further reduction of 37 by silane leads to copper alkoxide 38. Then, σ-bond metathesis of 38 with silane affords silyl ether 39 and regenerates the active catalyst 35. Finally, the desired alcohol 40 is formed after treatment of 39 with ammonium fluoride.
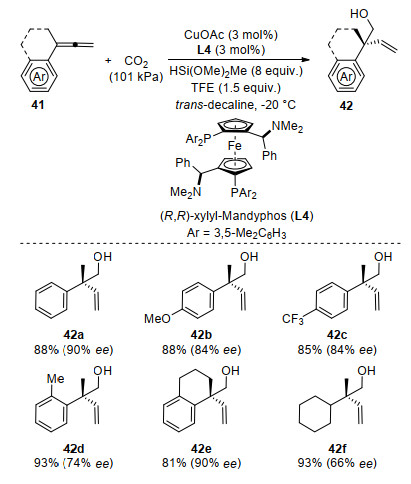
At the same time, Ding and co-workers[60] reported a Cu-catalyzed highly enantioselective hydroxymethylation of 1, 1-disubstituted allenes with CO2 to construct all- carbon chiral quaternary centers. Under the optimized reaction conditions of Cu/Mandyphos (L4) catalyst, the corresponding homoallylic alcohol product 42 was obtained in good yields with excellent enantioselectivities from 1, 1- disubstituted allenes 41 (Scheme 15).
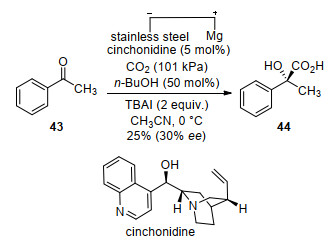
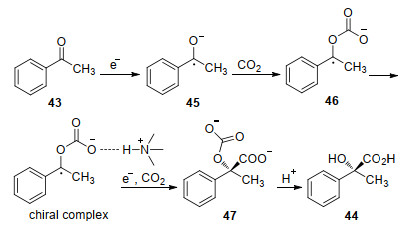
Another noteworthy achievement in CO2 fixation via asymmetric catalysis is enantioselective electrochemical carboxylation. In 2004, the diastereoselective electrochemical CO2 fixation using chiral auxiliaries was reported by Feroci, Inesi and co-workers.[61-62] Moderate diastereoselectivity was obtained in electrochemical carboxylation of alkyl halide and cinnamic acid derivatives with CO2. In 2009, Lu and co-workers[63] reported an inspiring catalytic asymmetric electrochemical reductive carboxylation of acetophenone (43) with CO2 giving 2-hydroxy-2-phenyl- propionic acid (44) (Scheme 16). The product was obtained in 25% yield with 30% ee using cinchonidine as chiral catalyst, butanol as co-catalyst, tetrabutylammonium iodide (TBAI) as supporting electrolyte on stainless steel cathodes. Recently, the efficiency of this reaction was improved by using phenol as co-catalyst and tetrahexyl-am- monium iodide as supporting electrolyte (41% yield, 49% ee) under similar conditions.[64]
The authors proposed a possible reaction pathway as shown in Scheme 17. First, a ketyl radical anion 45 is generated from 43 after receiving an electron from the cathode. Next, nucleophilic addition of 45 on CO2 affords the radical carbonate anion intermediate 46. A possible chiral proton-donating complex is then generated after 46 bonding to protonated cinchonidine. Further single electron reduction and subsequent carboxylation produces 47. At last, the final product 44 is formed by decarboxylation and protonation.
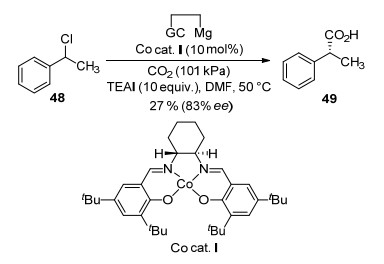
Electrochemical reduction of organic halides has been investigated for several decades. Electrochemical carboxylation of organic halides with CO2 could produce corresponding carboxylic acids.[65-66] In 2014, Wang, Lu and co-workers[67] reported an asymmetric CO2 fixation by electrochemical carboxylation of 1-phenylethyl chloride (48) (Scheme 18). In the presence of 5 mol% (R, R)-salen- Co complex (Co cat. I) and tetrabutylammonium iodide, racemic 1-phenylethyl chloride 48 was electrocarboxylated affording optically active 2-phenylpropionic acid 49 in 27% yield with 83% ee on a glassy carbon (GC) cathode. The electrogenerated [(R, R)-salen-CoⅠ)] species is considered to be the key catalytic species and alkyl CoⅡ complex is thought to be reaction intermediate. Recently, Wang, Lu and co-workers further expanded this reaction into a heterogeneous system.[68] They synthesized a [Co]@Ag composite by entrapment of (R, R)-salen-CoⅡ complex within silver nanoparticles. Using the chiral [Co]@Ag composite as cathode, electrocarboxylation of racemic 1-phenylethyl bromide with CO2 afforded 49 in 58% yield with 73% ee, without adding additional chiral source. Moreover, the catalyst [Co]@Ag showed remarkable stability and efficiency. The product could be obtained without significant loss of enantioselecitivity even after recycle the catalyst for 7 times.
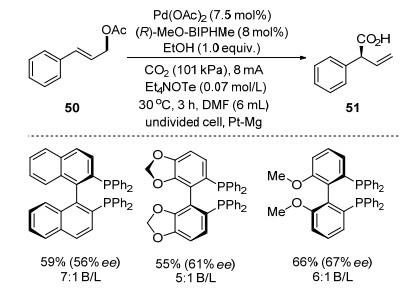
Recently, Mei and co-worker[69] reported an enantioselective Pd-catalyzed electrochemical carboxylation of allylic acetates with CO2 (Scheme 19). They found that moderate to good enantioselectivities could be obtained using Pd(OAc)2 as catalyst and chiral bidentate phosphine as ligands. The carboxylation reaction was performed with Pd(OAc)2 as catalyst, Et4NOTs as electrolyte, EtOH as additive, and DMF as solvent at 30 ℃ under constant current electrolysis conditions of 8.0 mA (J=8.0 mA• cm-2) and 3 F/mol. After screening several chiral bidentate phosphine ligands, carboxylation of 50 with CO2 gave allylic acid 51 in 66% yield with 67% ee using (R)-MeO- BIPHMe as ligand.
The central carbon atom of CO2 bears strong electrophilicity and the oxygen atoms act as a Lewis base with weak nucleophilicity. The structure property of CO2 makes it both an electrophile at carbon atom and a nucleophile at oxygen atom. Accordingly, cyclic carbonates and carbamates have been synthesized by reaction of CO2 with epoxides and aziridines.[70-72] In the other hand, nucleophiles such as alcohols and amines can attack the central carbon atom of CO2 generating carbonate and carbamate anion. Subsequent anion trapping with electrophiles affords carbonate and carbamate. By design of proper substrates and choosing of suitable catalysts, chemists have developed several efficient catalytic systems for the enantioselectvie synthesis of carbonate and carbamate with CO2. This type of asymmetric CO2 fixation reaction is summarized as below.
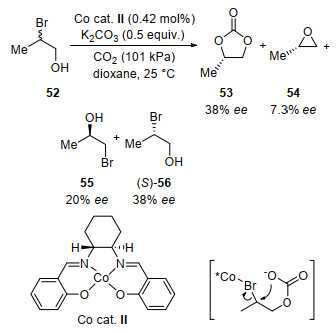
In 1987, Takeichi and co-workers[73] reported a Co-cat- alyzed kinetic resolution of propylene bromohydrin (Scheme 20). Chiral propylene carbonate 53 was obtained in promising ee via cyclization under mild conditions. Based on the configuration of the products, this reaction was considered via an attack of alcoholate anion on CO2 followed by an intromolecular nucleophilic substitution reaction.
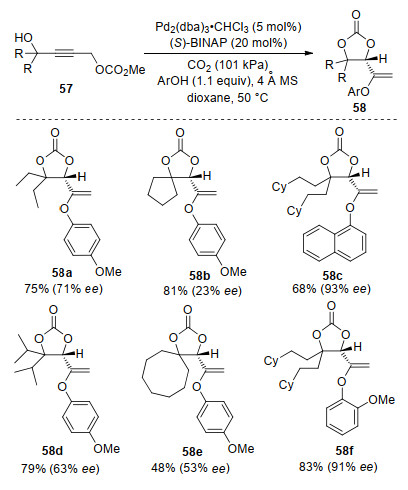
In 2003, Yoshida, Ihara and co-workers[74] reported a Pd-catalyzed CO2 recycling reaction for the preparation of chiral cyclic carbonates based on novel CO2 elimination-fixation strategy (Scheme 21). With Pd2(dba)3•CHCl3 as catalyst and (S)-BINAP as ligand, the asymmetric CO2 elimination-fixation reaction of propargylic carbonates 57 with phenols afforded the corresponding cyclic carbonate 58 in 48%~94% yields with 23%~93% ee. The authors found that the reactions with longer chain substrates leaded to cyclic carbonates in higher enantiomeric purities. Thus, bulky substituents at the β-position of alkyl side chain resulted in increased enantioselectivities.
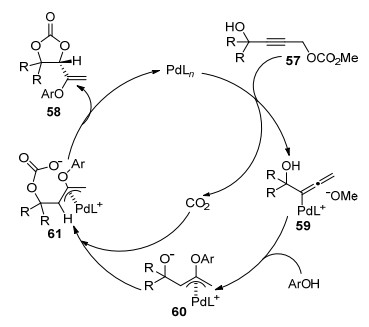
A plausible mechanism for this CO2 elimination-fixation is shown in Scheme 22. The key of this transformation is the π-allyl-Pd intermediate 60 generated by nucleophilic attack of allenylpalladium methoxide 59 by phenols. When this reaction is conducted under a CO2 atmosphere, the released and external CO2 promotes the reaction efficiently to generate carbonate. The direct cyclization of 60 leads to the dihydrofuran and epoxide byproducts. A new chiral center is formed by intramolecular nucleophilic attack by the carbonate anion 61 in the presence of (S)-BINAP-Pd complex. To examine whether CO2 dissociates from the substrate in this reaction, the reaction was conducted in both the presence and the absence of added CO2. While the reaction under argon atmosphere gave cyclic carbonate 58 in 85% yield, the process carried out under 101 kPa of CO2 leaded to 58 in 96% yield. In addition, when the reaction is run under bubbling argon to remove the resulting CO2, 58 was formed in only 21% yield together with by-product dihydrofuran (32%) and epoxide (11%). The results suggested that the process proceeded via a pathway involving decarboxylation-followed fixation of the liberated CO2.
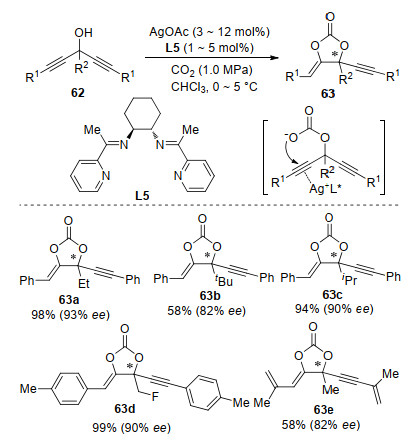
In 2010, another asymmetric CO2 fixation with propargyl alcohols towards chiral cyclic carbonates is reported by Yamada and co-workers.[75] Asymmetric desymmetrization of bispropargylic alcohols 62 was realized efficiently with chiral silver complex as a catalyst (Scheme 23). The alkyne moiety of bispropargylic alcohols 62 was activated by the π-Lewis acidic Ag-catalyst. Thus, the intramolecular nucleophilic addition of alkyne with the carbonate anion was facilitated from the opposite side of alkyne.[76] By the combination of AgOAc and chiral Schiff base ligand L5, a range of α-alkylidene cyclic carbonates 63 were obtained in good yield with moderate to high enantioselectivities (47%~93% ee). Furthermore, stereospecific hydrolyzation of such products was readily performed to afford the α-hydroxyketone bearing a chiral quaternary carbon center without loss of optical purity.
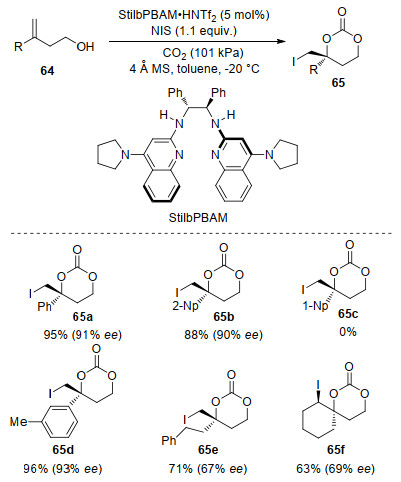
In 2015, Johnston and co-workers[77] described a organocatalytic iodocarbonation of homoallylic alcohols 64 with N-iodosuccinimide and CO2 to produce chiral cyclic carbonates 65 based on dual Brønsted acid/base activation (Scheme 24). They used StilbPBAM•HNTf2 salt as a bifunctional hydrogen-bond donor and acceptor catalyst. The acid/base combination lowered the barrier of CO2 incorporation and assisted the stabilization of the resulting carbonic acid. A variety of styrene derivatives were tested under the optimal conditions affording the corresponding carbonates in high yields with good to excellent enantioselectivities. However, no reaction occurred when using styrene derivatives with the bulky aryl group, such as 2-methylphenyl and 1-naphthyl (65c). High yield and moderate ee were obtained with homoallylic alcohol bearing aliphatic substituents (65e). Spirocyclic carbonate could be synthesized with trisubstituted alkene with promising enantioselectivity (65f).
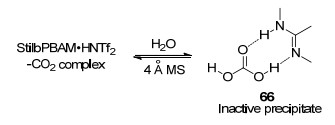
In the course of condition optimization, both efficiency and enantioselectivity were found to be sensitive to a variety of parameters, including concentration, temperature, stirring rate, counterion, and ratio of acid/base. The latter two factors were thought to influence the selectivity-determining hydrogen-bonding network. Moreover, 4Å MS was also crucial to the chemical yield, which attributed to maintaining the active complex formed by catalyst salt and CO2 (Scheme 25). In the presence of water, carbonic acid salt 66 was formed which consumed the active catalyst. This offers an insight into the interaction between hydrogen-bond donor/acceptor catalyst and CO2 along with water. Very recently, This group prepared six-membered cyclic carbamates through an high enantioselective amine CO2-capture/cyclization reaction with MeO-StilbPBAM•HNTf2 salt as catalyst and analogous amine as substrate.[78] And mechanism-guided analysis of the reaction provides insight to various dominant substrate- reagent combinations in the solution.
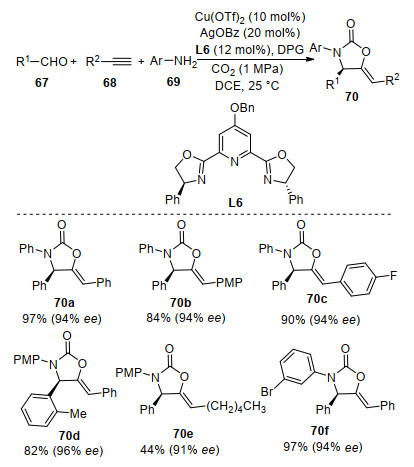
In 2017, Zhou and co-workers[79] reported a Cu/Ag cocatalyzed tandem asymmetric aldehyde-alkyne-amine coupling-carboxylative cyclization with CO2 for the highly enantioselective synthesis of chiral N-aryl 2-oxazolidinones 70 (Scheme 26). In this reaction, the chiral propargylamine was formed first with Cu(OTf)2 as catalyst and PYBOX L6 as ligand. Then Ag-catalyzed cyclization of propargylamine with CO2 afforded N-aryl 2-oxazolidinones in excellent yields with 90%~96% ee.[80] This process is a rare example of multicatalyst-promoted asymmetric tandem reaction using CO2 as a C1 synthon.
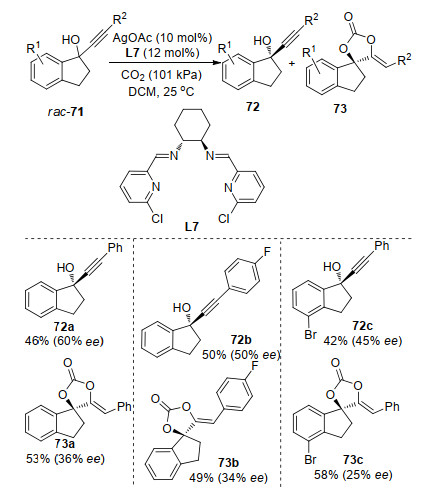
Very recently, Zhou and co-workers[81] reported an Ag- catalyzed enantioselective carboxylative cyclization of propargylic alcohols and CO2 via kinetic resolution under mild conditions. The reaction enabled the synthesis of chiral propargylic alcohols 72 and cyclic carbonates 73 with promising yield and enantioselectivity simultaneously (Scheme 27). They found that the substituents on the pyridyl ring had great influence on this reaction when they used the 2-pyridinecarboxyaldehyde derived Schiff base ligand. Chiral ligand L7 derived from ortho-chloro-substituted pyridine was demonstrated to be the best one.
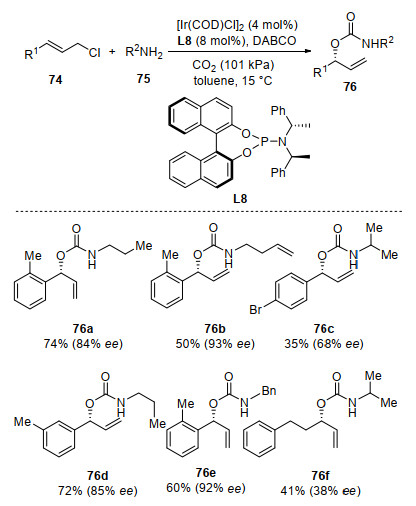
Ir-catalyzed asymmetric allylic amination with alkyl amine has been widely reported for the synthesis of chiral allyl amines.[82-92] Interestingly, Zhao and co-workers[93-94] reported an Ir-catalyzed asymmetric allylation reaction to synthesize optically active carbamates with CO2 and amine that involving both C—O and C—N bonds formation (Scheme 28). With linear allylic chlorides 74, amines 75, and CO2 as substrates, Ir-catalyzed three-component domino reaction was developed. By addition of DABCO as base and using slight excess of allyl chlorides, the chiral carbamates were obtained as major products comparing with chiral allyl amines. A variety of alkyl amines and cinnamyl chlorides were employed affording carbamates 76 in moderate yield with high regioseletivity (branched/linear > 9/1) and moderate to good enantioselectivities (48%~94% ee). Notably, this reaction featured with excellent ortho substituent tolerance of aryl substituent in allylic chlorides, which is difficult in the preparation of branched allyl alcohols.[95] They found that aryl substituted allyl chlorides with the electron-donating groups on the phenyl ring gave the branched allyl carbamates in fair to good yields with a high level of both regio- and enantio-selectivities. Aryl substituted allyl chlorides with the electron-withdrawing groups on the phenyl ring led to a good yields and excellent regioselectivities but with lower enantioselectivities. For substrate bearing an aliphatic group, it provided the corresponding allyl carbamate with lower ee value.
We conclude the recent advances in the CO2 fixation via asymmetric catalytic reactions for the synthesis of chiral small molecules. Although transformations of CO2 into valuable chemicals have been widely reported over the last decade, the CO2 fixation via asymmetric catalysis is still less developed. In general, transition metal catalysis is broadly applied in the CO2 fixation via asymmetric cataly- sis. In comparison, there are just several successful examples of organocatalytic asymmetric CO2 fixation. Notably, electrocatalysis could also be used in the asymmetric CO2 fixation, albeit with moderate enantioselectivities.
Carboxylic acids and their derivatives with an alpha chirality center are widely found in biological active natural products and medicinal molecules. In order to synthesize these compounds, asymmetric carboxylation of carbon nucleophiles with CO2 as a building block is an attractive method. Although several efficient methodologies have been developed for the enantioselective synthesis of carboxylic acid derivatives via catalytic C—C bond formation with CO2, special substrates such as styrenes, 1, 3-diene, allenes are needed. Using readily available substrates such as simple alkenes and alkanes will be highly desirable in the asymmetric carboxylation with CO2 in the future.
Chiral carbonates and carbamates are important chemicals and intermediates in synthetic chemistry. Asymmetric synthesis of these compounds by ring opening of epoxides and aziridines with CO2 has been well developed. Beyond these achievements, several new asymmetric CO2 fixation reactions for the synthesis of carbonates and carbamates have been developed with metal-catalysis or organocatalysis. However, the successful reactions are still limited and new types of asymmetric reactions are needed to be developed in this area.
In summary, significant progress has been made on the asymmetric CO2 fixation in recent years. Investigation of the mechanism of CO2 activation is important for the development of CO2 fixation reaction via asymmetric catalysis. It is anticipated that more researches will emerge in this area. The fixation of CO2 with high efficiency and excellent stereoselectivities is a longstanding goal under with mild conditions, such as low catalyst loading, room temperature, and atmospheric pressure of CO2. In this regard, converting CO2 and H2O to chiral carbohydrate with light by photosynthesis in nature is a perfect process. Thus, enzyme-catalyzed CO2 fixation might inspire the development of new catalytic systems on asymmetric catalysis with CO2 as a sustainable C1 synthon.
Scott, A. Chem. Eng. News 2015, 93, 10. doi: 10.1021/cen-09237-bus1
Aresta, M. Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010.
Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Nat. Commun. 2015, 6, 5933. doi: 10.1038/ncomms6933
Fiorani, G.; Guo, W.; Kleij, A.-W. Green Chem. 2015, 17, 1375. doi: 10.1039/C4GC01959H
Yu, B.; He, L.-N. ChemSusChem 2015, 8, 52. doi: 10.1002/cssc.201402837
Maeda, C.; Miyazaki, Y.; Ema, T. Catal. Sci. Technol. 2014, 4, 1482. doi: 10.1039/c3cy00993a
Tlili, A.; Frogneux, X.; Blondiaux, E.; Cantat, T. Angew. Chem., Int. Ed. 2014, 53, 2543. doi: 10.1002/anie.201310337
Yuan, G.; Qi, C.; Wu, W.; Jiang, H. Curr. Opin. Green Sust. Chem. 2017, 3, 22. doi: 10.1016/j.cogsc.2017.03.003
张文珍, 张宁, 郭春晓, 吕小兵, 有机化学, 2017, 37, 1309.Zhang, W.; Zhang, N.; Guo, C.-X.; Lü, X. Chin. J. Org. Chem. 2017, 37, 1309(in Chinese).
Zhang, Z.; Ju, T.; Ye, J.-H.; Yu, D.-G. Synlett 2017, 28, 741. doi: 10.1055/s-0036-1588403
Gui, Y.-Y.; Zhou, W.-J.; Ye, J.-H.; Yu, D.-G. ChemSusChem 2017, 10, 1337. doi: 10.1002/cssc.201700205
Cao, Y.; He, X.; Wang, N.; Li, H.-R.; He, L.-N. Chin. J. Chem. 2018, 36, 644. doi: 10.1002/cjoc.201700742
Zhao, Y., Liu, Z.-M. Chin. J. Chem. 2018, 36, 455 doi: 10.1002/cjoc.201800014
Hu, J.; Liu, H.; Han, B. Sci. China Chem. 2018, 61, 1486. doi: 10.1007/s11426-018-9396-3
Tan, F.; Yin, G. Chin. J. Chem. 2018, 36, 545. doi: 10.1002/cjoc.201800011
张振, 龚莉, 周晓渝, 颜思顺, 李静, 余达刚, 化学学报, 2019, 77, 783.Zhang, Z.; Gong, L.; Zhou, X.-Y.; Yan, S.-S.; Li, J.; Yu, D.-G. Acta Chim. Sinica 2019, 77, 783(in Chinese).
程磊, 谢建华, 有机化学, 2020, 40, 247.Cheng, L.; Xie, J.-H. Chin. J. Org. Chem. 2020, 40, 247(in Chinese).
Kolbe, H. Justus Liebigs Ann. Chem. 1860, 113, 125. doi: 10.1002/jlac.18601130120
Lindsey, A.-S.; Jeskey, H. Chem. Rev. 1957, 57, 583. doi: 10.1021/cr50016a001
Luo, J.; Preciado, S.; Xia, P.; Larrosa, L. Chem.-Eur. J. 2016, 22, 6798. doi: 10.1002/chem.201601114
Zhang, W.-Z.; Li, H.; Zeng, Y.; Tao, X.; Lu, X. Chin. J. Chem. 2018, 36, 112. doi: 10.1002/cjoc.201700581
Sakakura, T.; Choi, J.-C.; Yasuda, H. Chem. Rev. 2007, 107, 2365. doi: 10.1021/cr068357u
White, J.-L.; Baruch, M.-F.; Pander Ⅲ, J.-E.; Hu, Y.-I.; Fortmeyer, C.; Park, J.-E.; Zhang, T.; Liao, K.; Gu, J.; Yan, Y.; Shaw, T.-W.; Abelev, E.; Bocarsly, A.-B. Chem. Rev. 2015, 115, 12888. doi: 10.1021/acs.chemrev.5b00370
Wang, J.-L.; Miao, C.-X.; Dou, X.-Y.; Gao, J.; He, L.-N. Curr. Org. Chem. 2011, 15, 621. doi: 10.2174/138527211794518952
Tlili, A.; Blondiaux, E.; Frogneux, X.; Cantat, T. Green Chem. 2015, 17, 157. doi: 10.1039/C4GC01614A
Li, Y.; Cui, X.; Dong, K.; Junge, K.; Beller, M. ACS Catal. 2017, 7, 1077. doi: 10.1021/acscatal.6b02715
Liu, M.; Qin, T.; Zhang, Q.; Fang, C.; Fu, Y.; Lin, B.-L. Sci. China Chem. 2015, 58, 1524. doi: 10.1007/s11426-015-5405-y
Zhang, L.; Zhao, Z.-J.; Gong, J. Angew. Chem., Int. Ed. 2017, 56, 11326. doi: 10.1002/anie.201612214
Yu, D.; Teong, S.-P.; Zhang, Y. Coord. Chem. Rev. 2015, 293-294, 279. doi: 10.1016/j.ccr.2014.09.002
Liu, A.-H.; Yu, B.; He, L.-N. Greenhouse Gas Sci. Technol. 2015, 5, 17. doi: 10.1002/ghg.1461
Yeung, C.-S.; Dong, V.-M.; Top. Catal. 2014, 57, 1342. doi: 10.1007/s11244-014-0301-9
Zhang, W.; Lu, X. Chin. J. Catal. 2012, 33, 745. doi: 10.1016/S1872-2067(11)60390-2
Tsuji, Y.; Fujihara, T. Chem. Commun. 2012, 48, 9956. doi: 10.1039/c2cc33848c
Ackermann, L. Angew. Chem., Int. Ed, .2011, 50, 3842. doi: 10.1002/anie.201007883
Huang, K.; Sun, C.-L.; Shi, Z.-J. Chem. Soc. Rev. 2011, 40, 2435. doi: 10.1039/c0cs00129e
Börjesson, M.; Moragas, T.; Gallego, D.; Martin, R. ACS Catal. 2016, 6, 6739. doi: 10.1021/acscatal.6b02124
Chen, Y.-G.; Xu, X.-T.; Zhang, K.; Li, Y.-Q.; Zhang, L.-P.; Fang, P.; Mei, T.-S. Synthesis 2018, 50, 35. doi: 10.1055/s-0036-1590908
Riduan, S.-N.; Zhang, Y.; Ying, J.-Y. Angew. Chem., Int. Ed. 2009, 48, 3322. doi: 10.1002/anie.200806058
Gomes, C.-D.-N.; Jacquet, O.; Villiers, C.; Thuery, P.; Ephritikhine, M.; Cantat, T. Angew. Chem., Int. Ed. 2012, 51, 187. doi: 10.1002/anie.201105516
Xin, Z.; Lescot, C.; Friis, S. D.; Daasbjerg, K.; Skrydstrup, T. Angew. Chem., Int. Ed. 2015, 54, 6862. doi: 10.1002/anie.201500233
Kielland, N.; Whiteoak, C.-J.; Kleij, A.-W. Adv. Synth. Catal. 2013, 355, 2115. doi: 10.1002/adsc.201300422
Lu, X.-B.; Darensbourg, D.-J. Chem. Soc. Rev. 2012, 41, 1462. doi: 10.1039/C1CS15142H
Decortes, A.; Castilla, A.-M.; Kleij, A.-W. Angew. Chem., Int. Ed. 2010, 49, 9822. doi: 10.1002/anie.201002087
Vaitla, J.; Guttormsen, Y.; Mannisto, J.-K.; Nova, A.; Repo, T.; Bayer A.; Hopmann, K.-H. ACS Catal. 2017, 7, 7231. doi: 10.1021/acscatal.7b02306
Sato, F.; Iijima, S.; Sato, M. J. Chem. Soc., Chem. Commun. 1981, 180.
Beng, T.-K.; Gawley, R.-E. J. Am. Chem. Soc. 2010, 132, 12216. doi: 10.1021/ja105772z
Perrona, Q.; Alexakis, A. Adv. Synth. Catal. 2010, 352, 2611. doi: 10.1002/adsc.201000517
Egami, H.; Sato, K.; Asada, J.; Kawato, Y.; Hamashima, Y.; Tetrahedron 2015, 71, 6384. doi: 10.1016/j.tet.2015.05.041
Takimoto, M.; Mori, M. J. Am. Chem. Soc. 2002, 124, 10008 doi: 10.1021/ja026620c
Takimoto, M.; Nakamura, Y.; Kimura, K.; Mori, M. J. Am. Chem. Soc. 2004, 126, 5956. doi: 10.1021/ja049506y
Ishii, M.; Mori, F.; Tanaka, K. Chem.-Eur. J. 2014, 20, 2169. doi: 10.1002/chem.201304623
Williams, C.-M.; Johnson, J.-B.; Rovis, T. J. Am. Chem. Soc. 2008, 130, 14936. doi: 10.1021/ja8062925
Greenhalgh, M.-D.; Thomas, S. P. J. Am. Chem. Soc. 2012, 134, 11900. doi: 10.1021/ja3045053
Hayashi, C.; Hayashi, T.; Kikuchi, S.; Yamada, T. Chem. Lett. 2014, 43, 565. doi: 10.1246/cl.131163
Hayashi, C.; Hayashi, T.; Yamada, T. Bull. Chem. Soc. Jpn. 2015, 88, 862. doi: 10.1246/bcsj.20150043
Kawashima, S.; Aikawa, K.; Mikami, K.; Eur. J. Org. Chem. 2016, 19, 3166.
Dian, L.; Müller, D.-S.; Marek, I. Angew. Chem., Int. Ed. 2017, 56, 6783. doi: 10.1002/anie.201701094
Gui, Y.-Y.; Hu, N.; Chen, X.-W.; Liao, L.-L.; Ju, T.; Ye, J.-H.; Zhang, Z.; Li, J.; Yu, D.-G. J. Am. Chem. Soc. 2017, 139, 17011. doi: 10.1021/jacs.7b10149
Chen, X.-W.; Zhu, L.; Gui, Y.-Y.; Jing, K.; Jiang, Y.-X.; Bo, Z.-Y.; Lan, Y.; Li J.; Yu, D.-G. J. Am. Chem. Soc. 2019, 141, 18825. doi: 10.1021/jacs.9b09721
Qiu, J.; Gao, S.; Li, C.; Zhang, L.; Wang, Z.; Wang, X.; Ding, K.-L. Chem.-Eur. J. 2019, 25, 13874. doi: 10.1002/chem.201903906
Feroci, M.; Orsini, M.; Palombi, L.; Sotgiu, G.; Colapietro, M.; Inesi, A. J. Org. Chem. 2004, 69, 487. doi: 10.1021/jo0343836
Orsini, M.; Feroci, M.; Sotgiuand. G.; Inesi, A. Org. Biomol. Chem. 2005, 3, 1202. doi: 10.1039/b500570a
Zhang, K.; Wang, H.; Zhao, S.-F.; Niu D.-F.; Lu, J.-X. J. Electroanal. Chem. 2009, 630, 35. doi: 10.1016/j.jelechem.2009.02.013
Chen, B.-L.; Tu, Z.-Y.; Zhu, H.-W.; Sun, W.-W.; Wang, H.; Lu, J.-X. Electrochim. Acta 2014, 116, 475. doi: 10.1016/j.electacta.2013.11.001
He, Q.; O'Brien, J.-W.; Kitselman, K.-A.; Tompkins, L.-E.; Curtis G.-C.-T.; Kerton, F.-M. Catal. Sci. Technol. 2014, 4, 1513. doi: 10.1039/C3CY00998J
Sakakura, T.; Kohnoa, K. Chem. Commun. 2009, 45, 1312.
Chen, B.-L.; Zhu, H.-W.; Xiao, Y.; Sun, Q.-L.; Wang, H.; Lu, J.-X. Electrochem. Commun. 2014, 42, 55. doi: 10.1016/j.elecom.2014.02.009
Yang, H.-P.; Yue, Y.-N.; Sun, Q.-L.; Feng, Q.; Wang, H.; Lu, J.-X. Chem. Commun. 2015, 51, 12216. doi: 10.1039/C5CC04554A
Jiao, K.-J.; Li, Z.-M.; Xu, X.-T.; Zhang, L.-P.; Li, Y.; Zhang, K.; Mei, T.-S. Org. Chem. Front. 2018, 5, 2244. doi: 10.1039/C8QO00507A
Song, Q.-W.; Liu, P.; Han, L.-H.; Zhang K.; He, L.-N. Chin. J. Chem. 2018, 36, 147. doi: 10.1002/cjoc.201700572
Niu, D.-F.; Xiao, L.-P.; Zhang, A.-J.; Zhang, G.-R.; Tan, Q.-Y.; Lu, J.-X. Tetrahedron 2008, 64, 10517. doi: 10.1016/j.tet.2008.08.093
Isse, A.; Gennaro, A.; Vianello, E. J. Chem. Soc., Dalton Trans. 1996, 1613.
Takeichi, T.; Ozaki, Y.; Takayama, Y. Chem. Lett. 1987, 1137.
Yoshida, M.; Fujita, M.; Ishii, T.; Ihara, M. J. Am. Chem. Soc. 2003, 125, 4874. doi: 10.1021/ja0340681
Yoshida, S.; Fukui, K.; Kikuchi, S.; Yamada, T. J. Am. Chem. Soc. 2010, 132, 4072. doi: 10.1021/ja1007118
Yamada, W.; Sugawara, Y.; Cheng, H.-M. Ikeno, T.; Yamada, T. Eur. J. Org. Chem. 2007, 2007, 2604. doi: 10.1002/ejoc.200700169
Vara, B.-A.; Struble, T.-J.; Wang, W.; Dobish, M.-C.; Johnston, J.-N. J. Am. Chem. Soc. 2015, 137, 7302. doi: 10.1021/jacs.5b04425
Yousefi, R.; Struble, T.-J.; Payne, J.-L.; Vishe, M.; Schley, N.-D.; Johnston, J.-N. J. Am. Chem. Soc. 2019, 141, 618. doi: 10.1021/jacs.8b11793
Gao, X.-T.; Gan, C.-C.; Liu, S.-Y.; Zhou, F.; Wu, H.-H.; Zhou, J. ACS Catal. 2017, 7, 8588. doi: 10.1021/acscatal.7b03370
Gao, X.-T.; Xie, S.-L.; Zhou, F.; Wu, H.-H.; Zhou, J. Chem. Commun. 2019, 55, 14303. doi: 10.1039/C9CC07671A
Xie, S.; Gao, X.; Zhou, F.; Wu, H.; Zhou, J. Chin. Chem. Lett. 2020, 31, 324. doi: 10.1016/j.cclet.2019.05.060
Hartwig, J.-F.; Pouy, M.-J. Top. Organomet. Chem. 2011, 34, 169.
Liu, W.-B.; Xia, J.-B.; You, S.-L. Top. Organomet. Chem. 2012, 38, 155. doi: 10.1021/ja210923k
Qu, J.; Helmchen, G. Acc. Chem. Res. 2017, 50, 2539. doi: 10.1021/acs.accounts.7b00300
Zhang, X.; Liu, W.-B.; Cheng, Q.; You, S.-L. Organometallics 2016, 35, 2467. doi: 10.1021/acs.organomet.6b00339
Zhuo, C.-X.; Zhang X.; You, S.-L. ACS Catal. 2016, 6, 5307. doi: 10.1021/acscatal.6b01585
Ye, K.-Y.; He, H.; Liu, W.-B.; Dai, L.-X.; Helmchen, G.; You, S.-L. J. Am. Chem. Soc. 2011, 133, 19006. doi: 10.1021/ja2092954
Roggen, M.; Carreira, E.-M. J. Am. Chem. Soc. 2010, 132, 11917. doi: 10.1021/ja105271z
Xia, J.-B.; Liu, W.-B.; Wang, T.-M.; You, S.-L. Chem.-Eur. J. 2010, 16, 6442. doi: 10.1002/chem.201000467
Yamashita, Y.; Gopalarathnam, A; Hartwig, J.-F. J. Am. Chem. Soc. 2007, 129, 7508. doi: 10.1021/ja0730718
Nemoto, T.; Sakamoto, T.; Matsumoto T.; Hamada, Y. Tetrahedron Lett. 2006, 47, 8737. doi: 10.1016/j.tetlet.2006.10.003
Welter, C.; Moreno, R.-M.; Streiff, S.; Helmchen, G. Org. Biomol. Chem. 2005, 3, 3266. doi: 10.1039/b508634e
Zheng, S.-C.; Zhang, M.; Zhao, X.-M. Chem.-Eur. J. 2014, 20, 7216. doi: 10.1002/chem.201402388
Zhang, M.; Zhao X.; Zheng, S. Chem. Commun. 2014, 50, 4455. doi: 10.1039/c4cc00413b
Liu, W.-B.; He, H.; Dai, L.-X.; You, S.-L. Synthesis 2009, 2076.

Scheme 1 Enantioselective synthesis of small molecules via CO2 fixation by asymmetric catalysis

Scheme 3 Asymmetric bromine-lithium exchange to synthesize axially chiral di-acid with CO2

Scheme 4 Ni-catalyzed enantioselective carboxylative cyclization of bis-1, 3-dienes with CO2

Scheme 5 Mechanism of Ni-catalyzed carboxylative cyclization of bis-1, 3-dienes with CO2

Scheme 9 Plausible reaction model for asymmetric induction of rhodium-catalyzed hydrocarboxylation

Scheme 11 Cu-catalyzed enantioselective hydroxymethylation of styrenes and 1, 3-dienes with CO2

Scheme 13 Cu-catalyzed enantioselective reductive hydroxy- methylation of 1, 3-dienes to construct all-carbon quaternary stereocenter with CO2

Scheme 14 Mechanism of Cu-catalyzed enantioselective reductive hydroxymethylation of 1, 3-dienes with CO2

Scheme 15 CuI-catalyzed enantioselective reductive hydroxy- methylation of 1, 1-disubstituted allenes with CO2

Scheme 21 Pd-catalyzed enantioselective CO2 elimination- fixation of propargylic carbonates

Scheme 22 Plausible mechanism of Pd-catalyzed elimination-fixation of propargylic carbonates

Scheme 23 Ag-catalyzed enantioselectivede symmetrization of bispropargylic alcohols with CO2

Scheme 27 Enantioselective carboxylative cyclization of propargylic alcohol with CO2 under mild conditions

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:














