图 1.
配位辅助的烯烃不对称转化
Figure 1.
Substrate-directed asymmetric alkene functionalization

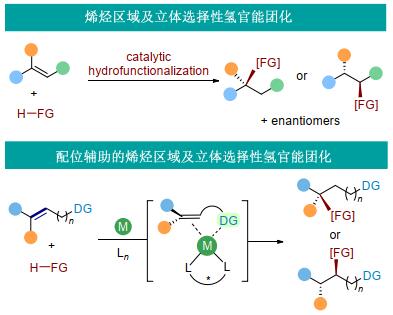
烯烃是有机分子中最常见的官能团之一.烯烃的转化是有机合成中极为重要的官能团转换过程[1].另一方面, 烯烃是廉价易得的化工原料, 烯烃转化也是化学工业的基础过程之一.因此, 不论是在有机合成还是工业生产中, 烯烃的催化转化长期以来都是非常重要的研究方向.高效、实用的烯烃转化方法的发展持续受到化学家的关注, 新颖的烯烃转化方法的发现可能为有机合成及化工生产带来变革性影响.
烯烃(特别是内烯烃)的不对称催化转化具有相当的挑战性[2].首先, 内烯烃位阻较端位烯烃更大, 反应性能更低.其次, 烯烃有两个反应位点, 反应的区域选择性需要得到很好的控制.再者, 如果反应生成了两个手性中心, 还涉及到非对映选择性的控制.此外, 对于不对称的催化转化过程, 还要叠加对映选择性控制的问题.因此, 烯烃的区域及立体选择性催化转化是化学家们长期面临的挑战.此类研究的关键在于如何开发高效的过渡金属催化体系, 在发展新反应的同时实现区域选择性、非对映选择性及对映选择性的控制(图 1).
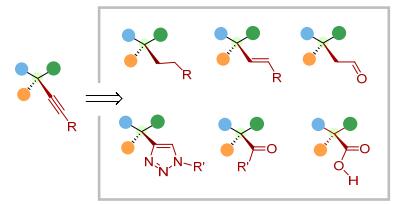
针对这一问题, 我们采用配位辅助策略[3], 探索烯烃尤其是内烯烃的不对称催化转化.配位基团起到多个方面的作用.首先, 通过金属与配位基团的配位, 加快了底物与催化剂的络合过程, 从而更有利于反应进行.其次, 利用配位基团及烯烃与金属中心形成双位点配位模式, 控制烯烃转化中的选择性包括区域选择性及对映选择性.此外, 配位基团本身作为一个官能团存在于反应产物中, 可以进行后续的转化.基于此策略, 我们以烯烃不对称炔氢化作为模型转化研究多取代烯烃、非活化烯烃不对称催化转化中的选择性控制.选择炔氢化[4, 5]作为研究的切入点基于几个方面的考虑.一方面, 炔烃同样是有机合成中常见的官能团, 原料廉价易得, 基于炔烃的方法学有重要的合成价值[6].更为重要的是, 炔氢化产物中的炔烃可以进行多种类型的转化[7].例如, 通过氢化、硼氢化、水合、Click反应等, 炔基能转化为烷基、烯基、羰基、杂环等官能团.因此, 构建含有炔基的手性中心, 再通过后续炔基的转化, 可以合成一系列含有各种官能团的手性分子砌块(图 2).
在早期含烯基的醇类化合物的环丙烷化[8]及环氧化[9]中, 人们发现羟基通过与金属中心的配位起到了控制面选择性的作用.这一策略在后来的烯烃催化氢化中得到了进一步应用[10], 很多配位基团包括羟基、羧酸、酰胺等官能团都可以与金属中心配位, 从而控制烯烃氢化中的面选择性[11].以此为基础, 陆续发展了配位辅助的烯烃硼氢化[12]、硅氢化[13]、酰基氢化[14]等反应.与此相比, 配位辅助的烯烃不对称催化转化发展较为缓慢, 该类反应主要集中在烯烃不对称氢化.配位辅助的烯烃不对称氢官能团化有待进一步发展.除了配位辅助的烯烃官能团化以外, 配位辅助的碳氢键官能团化领域也得到了广泛关注[15].
通过配位辅助策略, 我们研究了过渡金属催化的烯烃不对称氢官能团化过程, 发展了多取代及非活化烯烃的区域及立体选择性炔氢化, 构建单个、多个手性中心包括季碳手性中心, 实现了烯烃催化转化中区域选择性、非对映选择性及对映选择性的高效控制.此外, 通过实验和计算相结合的方法, 研究了配位辅助的烯烃不对称炔氢化反应机理, 揭示了该类反应的区域及立体选择性决定因素, 探索了反应区域及立体选择性与配体间的相互关系(图 3).
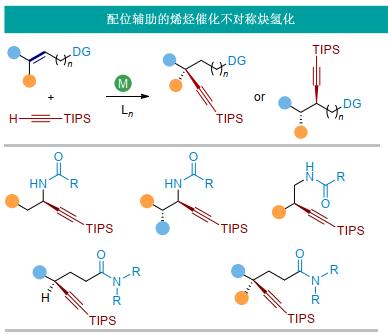
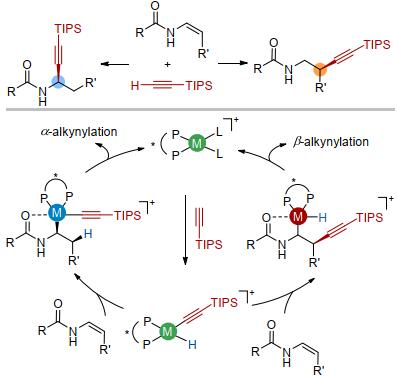
我们的研究起始于烯基酰胺类化合物的不对称炔氢化.烯基酰胺是一类常见的化合物[16], 可以通过烯基卤代物的偶联反应及炔烃的氢胺化反应制备[17].烯基酰胺两个反应位点的炔基化分别生成炔丙基胺及高炔丙基胺化合物, 两者都是有用的合成单元.此外, 烯基酰胺中的酰胺基团可以与过渡金属进行配位, 起到稳定过渡金属中间体或降低反应过渡态能量的作用(图 4).
此类反应的难点在于烯基酰胺是富电子烯烃, 而端炔形式上是亲核试剂, 二者之间的反应在电性上并不有利.在我们的反应设计中, 通过端炔与阳离子低价金属的氧化加成生成一个高价的亲电性金属物种, 从而有利于后续与富电子烯基酰胺的反应.该炔基金属氢物种与烯基酰胺有两种可能的反应途径.一是先发生烯烃对金属氢的迁移插入, 再通过后续的碳碳键还原消除得到相应的炔丙基胺化合物(Chalk-Harrod机理)[18].另一个途径是先发生烯烃对金属碳键的迁移插入, 再通过后续的碳氢键还原消除得到相应的高炔丙基胺化合物(修正的Chalk-Harrod机理)[19].因此, 迁移插入过程决定了烯烃炔氢化反应的选择性.
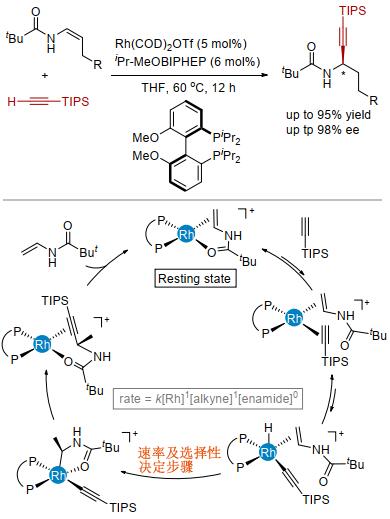
研究发现, 当我们使用阳离子铑作催化剂、富电子双膦配体iPr-MeOBIPHEP作配体时, 以单一的区域选择性和很高的对映选择性得到手性的炔丙基酰胺产物[20].该方法为手性炔丙基胺类化合物的制备提供了一种新的思路[21].该反应对于一系列的硅基、大位阻的烷基和芳基取代的端炔能够顺利发生; 但是, 小位阻的端炔反应性较低.一系列不同官能团取代的烯基酰胺均有很好的反应性和较高的对映选择性(图 5).
我们研究了炔氢化的非对映选择性, 结果表明该炔基化过程具有立体专一性.炔烃与烯基酰胺的反应是同面加成过程, 因而排除了经由亚胺中间体的机理.
进一步的机理研究表明, 催化剂以烯基酰胺配位的阳离子铑为主要存在形式.动力学测试表明该反应对催化剂是一级, 对炔烃也是一级, 而对烯胺是零级反应.氘代炔烃的动力学同位素实验结果表明, KIE为1.5±0.2.以上这些结果均支持烯烃的迁移插入而非炔烃的C—H键断裂是反应的决速步.
计算化学的结果表明, 烯烃的迁移插入步骤具有最高的活化能, 这一不可逆的过程是反应决速步及对映选择性决定步骤.生成主要对映异构体的过渡态中, 配体与底物间的排斥更小, 并且配体与底物形成的C—H···O相互作用较强.因此, 配体结构在控制烯烃迁移插入步骤的能量起到了关键作用.
烯基烯胺类化合物炔氢化的另一个可能的产物是经由β位炔基化得到高炔丙基胺.在前述反应设计的分析中, 如果能够使烯烃迁移插入的方式发生改变, 则可以实现区域选择性的逆转.我们设想, 将金属中心从铑改变为铱时, 由于金属铱中心较难发生碳碳键还原消除[22], 从而使得烯烃迁移插入至金属氢键的途径不利, 反应有可能按照烯烃迁移插入至金属碳键的途径进行.
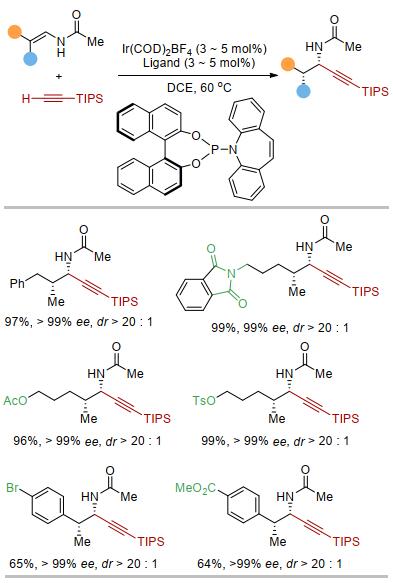
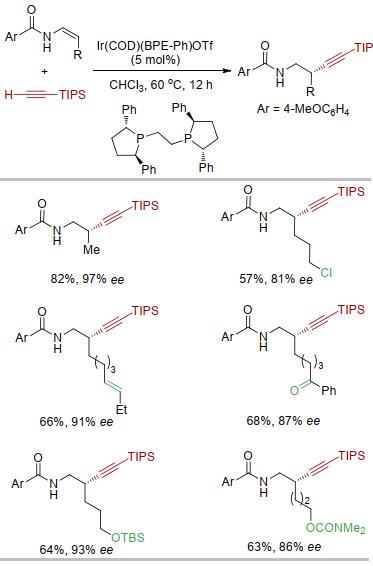
实验发现, 当使用阳离子铱作催化剂、富电子双膦配体BPE-Ph作配体时, 以单一的区域选择性和很高的对映选择性得到手性的高炔丙基酰胺产物, 反应中并没有观察到任何的炔丙基胺产物[23].该方法提供了一种合成β位取代的高炔丙基胺的新策略[24].反应对官能团有较好的兼容性.产物中的炔基可以进行多种类型的转换, 从而得到一系列手性的胺类化合物(图 6).
林正扬课题组[25]对这一体系的机理进行了计算.计算结果表明, 烯烃对铱氢键的插入可以顺利发生, 但后续铱中心碳碳键的还原消除过渡态能量很高, 不能顺利进行.相比而言, 烯烃对于铱碳键的迁移插入虽然比烯烃对于铱氢键的迁移插入能量稍高, 但后续铱中心碳氢键还原消除的能量比相应碳碳键还原消除的能量低很多.因此, 最终反应朝着烯烃迁移插入到铱碳键的方向进行, 从而得到β位炔基化的产物.
在单取代烯基酰胺不对称炔氢化构建单个手性中心的基础上, 我们进一步发展新的催化体系以实现二取代烯基酰胺的不对称炔氢化构建连续手性中心.二取代烯基酰胺较单取代烯基酰胺位阻更大, 因此其反应性更低.此外反应产生了两个手性中心, 因而涉及到非对映选择性的控制.我们发展的低价金属催化的不对称炔氢化反应具有同面加成的立体化学特征, 其固有的立体专一性即能保证非对映选择性得到很好的控制.
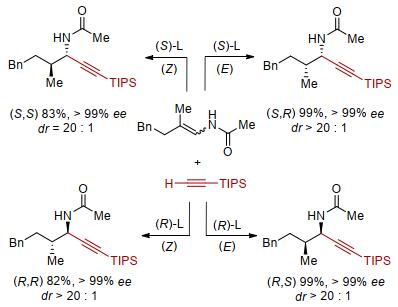
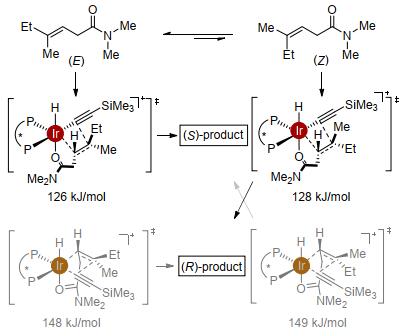
从β, β-二取代的烯基酰胺出发, 我们尝试了阳离子铑络合物与一系列的配体以实现其α位炔基化, 但均没有得到目标产物, 这也反映了β, β-二取代的烯基酰胺比单取代烯基酰胺更低的反应性.我们转而尝试阳离子铱催化剂.研究发现, 以阳离子铱作催化剂、Carreira类型亚磷酰胺[26]为配体, 实现了β, β-双取代烯基酰胺的不对称炔氢化, 以单一的α位选择性、>20:1的非对映选择性及高达99% ee值的对映选择性得到炔丙基胺类化合物, 高效构建两个手性中心[27].亚磷酰胺配体对反应的进行至关重要, 许多其他类型的富电子膦配体均只有很低的反应性.基于该催化体系的炔氢化选择性发生在α位, 说明除了催化中心金属的特性以外, 配体也在很大程度上参与了反应选择性的调控(图 7).
由于该催化炔氢化过程是立体专一性的, 因此通过改变烯基酰胺的构型就能实现非对映选择性的控制.从E型烯基酰胺出发, 在标准条件下得到syn型炔氢化产物.改变烯基酰胺的构型, 从Z型烯基酰胺出发, 同样条件下即能得到单一的anti-产物.使用另一个构型的配体, 从E和Z-烯基酰胺进行炔氢化则得到以上两个产物的对映体(图 8).因此, 利用烯烃构型及配体构型的组合, 即可以高非对映选择性、高对映选择性实现四个立体异构体的立体多样性合成[28].
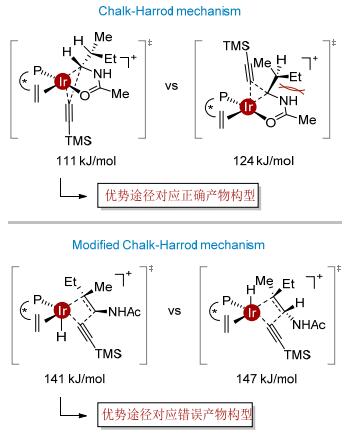
通过测试不同催化剂与配体比例的催化活性及非线性效应的实验结果, 表明催化体系中金属与配体以1:1形式配位.在此基础上, 我们进行了计算化学的研究以分辨催化过程是通过Chalk-Harrod机理还是修正的Chalk-Harrod机理进行[18, 19].两组关键的数据支持该反应是通过Chalk-Harrod机理进行的.首先, 两个催化途径的能量相比, Chalk-Harrod机理最高过渡态的能量(111 kJ/mol)比修正的Chalk-Harrod机理最高过渡态的能量(141 kJ/mol)低, 而且与实验条件(温度为60 ℃)更加吻合.其次, 通过Chalk-Harrod机理计算得到的主要对映体与实验结果相一致, 而通过修正的Chalk-Harrod机理预测的主要对映体则与实验结果相反.在Chalk-Harrod机理中, 决速步铱中心的碳碳键还原消除步骤能量较低, 配体对此起到了关键作用.亚磷酰胺类配体比双膦配体更贫电子, 因此金属中心的电子云密度降低, 从而有利于铱中心发生还原消除(图 9).
以上铱催化的烯基酰胺不对称炔氢化机理研究的结果可用于指导新型烯烃不对称炔氢化反应的设计.例如, 从非活化烯烃β, γ-不饱和酰胺出发, 使用铱催化剂及较富电子双磷配体, 反应经由修正的Chalk-Harrod机理即可得到羰基γ位炔基化的产物.若该反应成功实现, 将为羰基远端手性中心的构建提供新的方法[29], 并为配位辅助的非活化烯烃的区域及立体选择性氢官能团化提供新的思路.
实验结果表明, 当使用阳离子铱作催化剂、富电子双膦配体DM-Segphos作配体时, 以单一的区域选择性和较高的对映选择性得到手性的γ-炔基酰胺[30].进一步监测反应体系发现, 部分β, γ-不饱和酰胺异构化为α, β-不饱和酰胺, 但最终二者都转化为相应的γ-炔基酰胺产物(图 10).相应的β, γ-不饱和酯及α, β-不饱和酯类化合物不能发生相应的反应, 说明酰胺的配位作用对反应的进行至关重要.
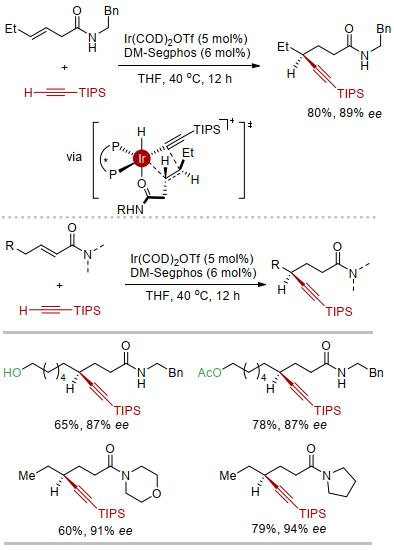
因此, 我们直接从α, β-不饱和酰胺出发, 在同样的条件下即可实现其γ位不对称炔基化反应, 形式上实现了氧化还原中性的α, β-不饱和酰胺γ位不对称碳氢键官能团化[31].特别需要指出的是, 在该催化体系中, 并没有发生共轭加成的炔基化产物, 我们认为是由于中间体中铱中心难以直接发生碳碳键还原消除, 转而进行β-氢消除得到烯烃异构化的产物再发生后续的碳碳键迁移插入及碳氢键还原消除(Chalk-Harrod机理), 从而选择性的得到酰胺远端炔基化的产物.
全碳季碳手性中心的高效构建是不对称合成中的挑战之一[32].在实现了酰胺辅助的烯烃不对称炔氢化构建三级碳中心的基础上, 我们发展新的催化体系, 进一步实现了全碳季碳手性中心的构建.
从三取代非活化烯烃出发通过不对称炔氢化构建季碳手性中心面临几个方面的挑战.首先, 三取代烯烃较二取代烯烃位阻更大, 形成季碳手性中心更加困难.其次, 炔氢化的区域选择性需要加以控制, 否则反应有可能发生在位阻更小的一端.另外, 还需抑制烯烃异构化过程, 因为原料烯烃构型的变化将直接导致对映选择性的下降.
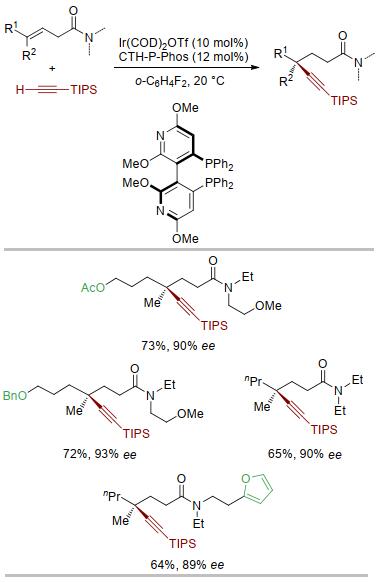
实验结果表明(图 11), 以三取代烯烃β, γ-不饱和酰胺为原料, 当使用阳离子铱作催化剂、双膦配体CTH-P-Phos作配体时, 以单一的γ位区域选择性和较高的对映选择性得到手性的炔基化产物, 从非官能团化的原料出发高效构建全碳季碳手性中心[33].在这个催化体系中, 三级酰胺比二级酰胺表现出更好的反应性, 具体原因还有待进一步研究.
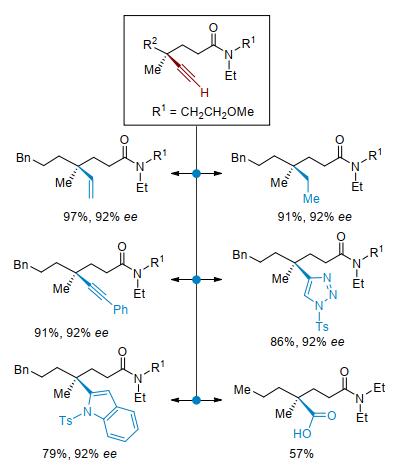
虽然目前我们发展的几类炔氢化反应中炔烃主要局限于硅基端炔, 但硅基非常容易脱除, 所得到的端炔可以进行多种转化.例如, 含全碳季碳手性中心的产物脱除硅基后再进行多种类型的后续转化, 从而合成一系列含季碳手性中心的分子.炔基进行半氢化、氢化、Sonogashira偶联、Click反应、吲哚环化、氧化断裂等, 分别得到含烯基、烷基、炔基、杂环及羧基的季碳手性中心, 实现了手性分子的多样性合成(图 12).
催化体系中催化剂主要存在形式及动力学同位素效应实验的研究表明, 烯烃迁移插入至铱碳键是反应的决速步.进一步的计算结果也支持了这一结论; 但计算预测反应的对映选择性应在99% ee以上, 这与实验结果有一定差别.后续的研究发现, 原料中烯烃的异构化是导致对映选择性下降的原因.监测反应体系发现, 随着反应的进行, 原料的E/Z比例逐渐下降, 产物的ee值也逐步下降, 二者之间存在一定的相关性, 说明烯烃迁移插入步骤的面选择性及烯烃异构化过程共同控制了反应的对映选择性(图 13).
配位基团酰胺的存在对反应的进行及立体选择性的控制至关重要, 相应含酯基的烯烃底物则没有反应性.通过计算相应酰胺与金属中心不进行配位的烯烃迁移插入过渡态发现, 此类迁移插入过渡态的能量比相应酰胺与金属中心进行配位的过渡态能量高出很多, 说明酰胺通过与金属中心的配位大大降低了烯烃迁移插入这一决速步的过渡态能量.
配位辅助是烯烃不对称催化转化中实现选择性控制的一类十分有效的策略.通过底物中配位基团与金属中心的配位作用, 提升烯烃的反应性, 并调控烯烃的反应位点及反应面, 实现烯烃转化中区域选择性及立体选择性的高效控制.通过使用配位辅助策略, 我们发展了烯烃催化转化的新型反应, 实现了温和条件下烯烃高区域选择性、高立体选择性的催化转化, 并通过实验与计算相结合, 研究了烯烃转化中区域选择性及立体选择性的控制因素, 为烯烃的高效利用提供科学依据.
虽然配位辅助的烯烃不对称转化领域取得了一些进展, 但目前该方向仍然有许多问题有待进一步深入研究.例如, 基于配位辅助的不对称碳碳键形成反应类型还十分有限, 氢官能团化(hydrofunctionalization)相比于质子官能团化(protofunctionalization)反应[34]更加稀少; 配位基团的实用性有待进一步提升, 如羟基、胺基、羰基等官能团作为配位基团的烯烃转化需要进一步开发; 另外, 反应机理的研究仍需进一步深入, 并基于机理的研究进而指导新型、高效催化体系的发现.相信经过化学家的持续努力, 配位辅助的烯烃不对称转化领域将取得更大的进展, 并催生烯烃不对称催化新方法、新策略的发现及应用.
Wang, J. Stereoselective Alkene Synthesis; Springer-Verlag, Berlin/Heidelberg, 2012.
(a) Coombs, J. R.; Morken, J. P. Angew. Chem. Int. Ed. 2016, 55, 2636.
(b) Chen, J. H.; Lu, Z. Org. Chem. Front. 2018, 5, 260.
(c) Dhungana, R. K.; Kc, S.; Basnet, P.; Giri, R. Chem. Rec. 2018, 18, 1314.
(d) Zhang, J.-S.; Liu, L.; Chen, T.; Han, L.-B. Chem. Asian J. 2018, 13, 2277.
(e) Ping, Y.; Li, Y.; Zhu, J.; Kong, W. Angew. Chem., Int. Ed. 2019, 58, 1562.
(f) Li, S.; Li, Z.; You, C.; Lü, H.; Zhang, X. Chin. J. Org. Chem. 2019, 39, 1568.
(g) Cheng, L.; Xie, J. Chin. J. Org. Chem. 2020, 40, 247.
(h) Zhou, Q.; Feng, X, ; Yang, J.; Du, H. Chin. J. Org. Chem. 2019, 39, 2188.
Wang, Z.-X.; Bai, X.-Y.; Li, B.-J. Chin. J. Chem. 2019, 37, 1174.
(a) Knöpfel, T. F.; Zarotti, P.; Ichikawa, T.; Carreira, E. M. J. Am. Chem. Soc. 2005, 127, 9682.
(b) Nishimura, T.; Guo, X.-X.; Uchiyama, N.; Katoh, T.; Hayashi, T. J. Am. Chem. Soc. 2008, 130, 1576.
(c) Yazaki, R.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2010, 132, 10275.
(d) Shirakura, M.; Suginome, M. Angew. Chem. Int. Ed. 2010, 49, 3827.
(e) Fan, B.-M.; Yang, Q.-J.; Hu, J.; Fan, C.-L.; Li, S.-F.; Yu, L.; Huang, C.; Tsang, W. W.; Kwong, F. Y. Angew. Chem., Int. Ed. 2012, 51, 7821.
(f) Sawano, T.; Ou, K.; Nishimura, T.; Hayashi, T. Chem. Commun. 2012, 48, 6106.
(g) Blay, G.; Pedro, J. R.; Sanz-Marco, A. Synthesis 2018, 50, 3281.
(h) Zhi, Y.; Huang, J.; Liu, N.; Lu, T.; Dou, X. Org. Lett. 2017, 19, 2378.
(a) Fu, L.; Zhou, S.; Wan, X.; Chen, P.; Liu, G. J. Am. Chem. Soc. 2018, 140, 10965.
(b) Yeqiang, H.; Ding, Y.; Zhou, T.; Yan, S.-Y.; Song, H.; Shi, B.-F. J. Am. Chem. Soc. 2019, 141, 4558.
(c) Dong, X.-Y.; Zhang, Y.-F.; Ma, C.-L.; Gu, Q.-S.; Wang, F.-L.; Li, Z.-L.; Jiang, S.-P.; Liu, X.-Y. Nat. Chem. 2019, 11, 1158.
(d) Chen, Q.; Tang, Y.; Huang, sT.; Liu, X.; Lin, L.; Feng, X. Angew. Chem., Int. Ed. 2016, 55, 5286.
(a) Trost, B. M., Li, C.-J. Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations, Wiley, New York, 2014.
(b) Li, Y.; Liu, X.; Jiang, H.; Liu, B.; Chen, Z.; Zhou, P. Angew. Chem., Int. Ed. 2011, 50, 6341.
(c) Sivaguru, P.; Wang, Z.; Zanoni, G.; Bi, X. Chem. Soc. Rev. 2019, 48, 2615.
(d) Hong, F.-L.; Wang, Z.-S.; Wei, D.-D.; Zhai, T.-Y.; Deng, G.-C.; Lu, X.; Liu, R.-S.; Ye, L.-W. J. Am. Chem. Soc. 2019, 141, 16961.
(e) Zhu, D.; Chen, L.; Zhang, H.; Ma, Z.; Jiang, H.; Zhu, S. Angew. Chem., Int. Ed. 2018, 57, 12405.
(f) Ren, R.; Wu, Z.; Xu, Y.; Zhu, C. Angew. Chem., Int. Ed. 2016, 55, 2866.
(g) Shen, T.; Wang, T.; Qin, C.; Jiao, N. Angew. Chem., Int. Ed. 2013, 52, 6677.
(h) Xu, G.; Zhao, H.; Fu, B.; Cang, A.; Zhang, G.; Zhang, Q.; Xiong, T.; Zhang, Q. Angew. Chem., Int. Ed. 2017, 56, 13130. (i) Wang, Q.; Yu, X.; Jin, J.; Wu, Y.; Liang, Y. Chin. J. Chem. 2018, 36, 223. (j) Jiang, Z.; Lu, P.; Wang, Y. Org. Lett. 2012, 14, 6266.
(k) Zhu, C.; Chu, H.; Li, G.; Ma, S.; Zhang, J. J. Am. Chem. Soc. 2019, 141, 19246.
Diederich, F.; Stang, P. J.; Tykwinski, R. R. Acetylene Chemistry, Wiley-VCH, Weinheim, 2005. https://www.researchgate.net/publication/296408661_Acetylene_Chemistry_Chemistry_Biology_and_Material_Science
(a) Simmons, H. E.; Smith, R. D. J. Am. Chem. Soc. 1959, 81, 4256.
(b) Winstein, S.; Sonnenberg, J.; De Vries, L. J. Am. Chem. Soc. 1959, 81, 6523.
(c) Winstein, S.; Sonnenberg, J. J. Am. Chem. Soc. 1961, 83, 3235.
Henbest, H. B.; Wilson, R. A. L. J. Chem. Soc. 1957, 1958. https://www.ingentaconnect.com/content/stl/jcr/2003/00002003/00000012/art00018
(a) Thompson, H. W.; McPherson, E. J. Am. Chem. Soc. 1974, 96, 6232.
(b) Crabtree, R. H.; Davis, M. W. J. Org. Chem. 1986, 51, 2655.
(a) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029.
(b) Zhu, S.-F.; Zhou, Q.-L. Acc. Chem. Res. 2017, 50, 988.
(a) Evans, D. A.; Fu, G. C. J. Am. Chem. Soc. 1991, 113, 4042.
(b) Rubina, M.; Rubin, M.; Gevorgyan, V. J. Am. Chem. Soc. 2003, 125, 7198.
(c) Bochat, A. J.; Shoba, V. M.; Takacs, J. M. Angew. Chem., Int. Ed. 2019, 58, 9434.
Kawasaki, Y.; Ishikawa, Y.; Igawa, K.; Tomooka, K. J. Am. Chem. Soc. 2011, 133, 20712. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM22142167
(a) Willis, M. C. Chem. Rev. 2010, 110, 725.
(b) Murphy, S. K.; Bruch, A.; Dong, V. M. Angew. Chem., Int. Ed. 2014, 53, 2455.
(a) Albrecht, M. Chem. Rev. 2010, 110, 576.
(b) Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147.
(c) He, J.; Wasa, M.; Chan, K. S. L.; Shao, Q.; Yu, J.-Q. Chem. Rev. 2017, 117, 8754.
(d) Li, B.-J.; Shi, Z.-J. Chem. Soc. Rev. 2012, 41, 5588.
Wang, M.-X. Chem. Commun. 2015, 51, 6039. doi: 10.1039/C4CC10327K
(a) Jiang, L.; Job, G. E.; Klapars, A.; Buchwald, S. L. Org. Lett. 2003, 5, 3667.
(b) Gooßen, L. J.; Rauhaus, J. E.; Deng, G. Angew. Chem., Int. Ed. 2005, 44, 4042.
Sakaki, S.; Mizoe, N.; Sugimoto, M. Organometallics 1998, 17, 2510. https://www.researchgate.net/publication/231735206_Theoretical_Study_of_Platinum0-Catalyzed_Hydrosilylation_of_Ethylene_Chalk-Harrod_Mechanism_or_Modified_Chalk-Harrod_Mechanism
Sakaki, S.; Sumimoto, M.; Fukuhara, M.; Sugimoto, M.; Fujimoto, H.; Matsuzaki, S. Organometallics 2002, 21, 3788. https://www.researchgate.net/publication/32136868_Why_Does_the_Rhodium-Catalyzed_Hydrosilylation_of_Alkenes_Take_Place_through_a_Modified_Chalk-Harrod_Mechanism_A_Theoretical_Study
Bai, X. Y.; Zhang, W. W.; Li, Q.; Li, B.-J. J. Am. Chem. Soc. 2018, 140, 506. doi: 10.1021/jacs.7b12054
Peshkov, V. A.; Pereshivko, O. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2012, 41, 3790. doi: 10.1039/c2cs15356d
Hartwig, J. F. Organotransition Metal Chemistry:From Bonding to Catalysis, University Science Books, Sausalito, CA, 2010. https://www.researchgate.net/publication/263946699_Organotransition_Metal_Chemistry_From_Bonding_to_Catalysis
Bai, X.-Y.; Wang, Z.-X.; Li, B.-J. Angew. Chem., Int. Ed. 2016, 55, 9007. doi: 10.1002/anie.201601792
Ding, C. H.; Hou, X. L. Chem. Rev. 2011, 111, 1914. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21344874
Yu, Z.; Meng, L.; Lin, Z. Organometallics 2019, 38, 2998. doi: 10.1021/acs.organomet.9b00343
Rössler, S. L.; Petrone, D. A.; Carreira, E. M. Acc. Chem. Res. 2019, 52, 2657. https://www.researchgate.net/publication/333889753_Iridium-Catalyzed_Asymmetric_Synthesis_of_Functionally_Rich_Molecules_Enabled_by_PhosphoramiditeOlefin_Ligands
Zhang, W.-W.; Zhang, S.-L.; Li, B.-J. Angew. Chem., Int. Ed. 2020, 59, 6874. doi: 10.1002/anie.201916088
(a) Krautwald, S.; Carreira, E. M. J. Am. Chem. Soc. 2017, 139, 5627.
(b) Huo, X.; He, R.; Zhang, X.; Zhang, W. J. Am. Chem. Soc. 2016, 138, 11093.
(c) Wei, L.; Zhu, Q.; Xu, S.-M.; Chang, X.; Wang, C.-J. J. Am. Chem. Soc. 2018, 140, 1508.
(d) Zhang, Q.; Yu, H.; Shen, L.; Tang, T.; Dong, D.; Chai, W.; Zi, W. J. Am. Chem. Soc. 2019, 141, 14554.
(a) Mei, T.-S.; Patel, H. H.; Sigman, M. S. Nature 2014, 508, 340.
(b) Liu, W.-B.; Okamoto, N.; Alexy, E. J.; Hong, A. Y.; Tran, K.; Stoltz, B. M. J. Am. Chem. Soc. 2016, 138, 5234.
(c) Guo, C.; Fleige, M.; Janssen-Muller, D.; Daniliuc, C. G.; Glorius, F. J. Am. Chem. Soc. 2016, 138, 7840.
Wang, Z.-X.; Bai, X.-Y.; Yao, H.-C.; Li, B.-J. J. Am. Chem. Soc. 2016, 138, 14872. http://www.istic.ac.cn/suoguan/detailed.htm?dbname=xw_qk&wid=0220170600476719
Li, Y.; Wu, D.; Cheng, H.-G.; Yin, G. Angew. Chem., Int. Ed. 2020, 59, 7990.
(a) Quasdorf, K. W.; Overman, L. E. Nature 2014, 516, 181.
(b) Feng, J.; Holmes, M.; Krische, M. J. Chem. Rev. 2017, 117, 12564.
(c) Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev. 2016, 116, 7330.
Wang, Z.-X.; Li, B.-J. J. Am. Chem. Soc. 2019, 141, 9312. doi: 10.1021/jacs.9b03027
(a) Wang, H.; Bai, Z.; Jiao, T.; Deng, Z.; Tong, H.; He, G.; Peng, Q.; Chen, G. J. Am. Chem. Soc. 2018, 140, 3542.
(b) Liu, Z.; Li, X.; Zeng, T.; Engle, K. M. ACS Catal. 2019, 3260.
(c) Shen, H.-C.; Zhang, L.; Chen, S.-S.; Feng, J.; Zhang, B.-W.; Zhang, Y.; Zhang, X.; Wu, Y.-D.; Gong, L.-Z. ACS Catal. 2019, 9, 791.
(d) Nimmagadda, S. K.; Liu, M.; Karunananda, M. K.; Gao, D. W.; Apolinar, O.; Chen, J. S.; Liu, P.; Engle, K. M. Angew. Chem., Int. Ed. 2019, 58, 3923.
(e) Vanable, E. P.; Kennemur, J. L.; Joyce, L. A.; Ruck, R. T.; Schultz, D. M.; Hull, K. L. J. Am. Chem. Soc. 2019, 141, 739.
(f) Tang, C.; Zhang, R.; Zhu, B.; Fu, J.; Deng, Y.; Tian, L.; Guan, W.; Bi, X. J. Am. Chem. Soc. 2018, 140, 16929.

图 3 配位辅助的烯烃炔氢化小结
Figure 3 Overview of substrate-directed asymmetric hydroalkynylations

图 5 烯基酰胺不对称α炔氢化及机理
Figure 5 Asymmetric α-hydroalkynylation of enamide and its mechanism

图 7 烯基酰胺不对称α炔氢化构建连续手性中心
Figure 7 Asymmetric α-hydroalkynylation of enamide to create vicinal stereocenters

图 8 不对称炔氢化用于立体多样性合成
Figure 8 Asymmetric hydroalkynylation for stereodivergent synthesis

图 9 铱催化不对称α炔氢化机理
Figure 9 Mechanism of Ir-catalyzed asymmetric α-hydroal- kynylation of enamide

图 10 铱催化不饱和酰胺不对称γ炔氢化
Figure 10 Ir-catalyzed asymmetric γ-hydroalkynylation of unsaturated amides

图 11 铱催化三取代烯烃炔氢化构建季碳中心
Figure 11 Ir-catalyzed asymmetric hydroalkynylation of tri-substituted alkene to construct quaternary carbon centers

图 12 炔基取代季碳中心的转化
Figure 12 Transformations of alkyne substituted quaternary carbon centers

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



