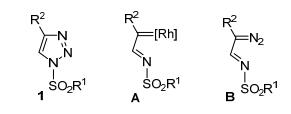
图 1.
N-磺酰三唑及其开环中间体结构
Figure 1.
Structures of N-sulfonyltriazole and its ring opening intermediates

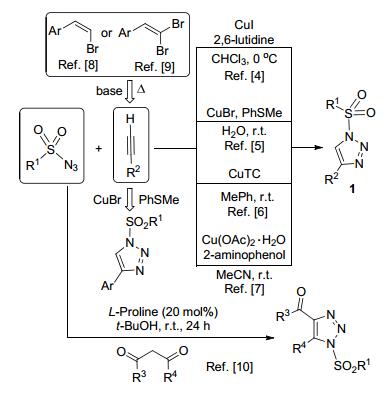
N-磺酰-1, 2, 3-三唑1是一类非常有价值的有机物.该类化合物通过开环产生的Rh-卡宾亚胺中间体A和重氮亚胺B被广泛应用于多取代含氮杂环化合物的合成(图 1)[1].因此, 发现和建立有效的合成N-磺酰-1, 2, 3-三唑的方法是非常有必要的. Cu(I)催化的炔-叠氮环加成反应(CuAAC)是构建1, 2, 3-三唑最重要和有效的方法[2]. 但是, 经典的CuAAC反应显然不适用于含有磺酰基等吸电子基团的有机叠氮类反应底物.磺酰基的强吸电子性会降低N-磺酰-1, 2, 3-三唑铜盐中间体的稳定性.该铜盐中间体会快速转化为非预期的磺酰亚胺中间体[3].所以, 有关N-磺酰-1, 2, 3-三唑合成方法的报道相对较少[4~10].已有的综述文献[1b~1j]对2017年以前该领域的研究工作进行了系统全面总结.本文将对N-磺酰-1, 2, 3-三唑的主要合成方法和2018~2019年期间在有机合成中的应用最新研究进展进行综述.
Fokin和Chang等[4]报道了在低温条件下, 通过添加2, 6-二甲基吡啶, 实现了CuI催化下磺酰叠氮和炔之间的环加成反应, 以较满意的收率得到N-磺酰-1, 2, 3-三唑1.付华等[5]以CuBr-PhSMe体系作为高效催化剂, 在温和条件下合成了化合物1. Fokin等[6]发现噻吩-2-甲酸铜(I) (CuTC)可以作为磺酰叠氮和炔的[3+2]环加成反应的高效催化剂.该反应可以在室温下水相中或以甲苯为溶剂顺利完成, 成为迄今为止最为有效和广泛应用的磺酰三唑1合成方法之一.王歆燕和胡跃飞等[7]利用一水合醋酸铜/2-氨基苯酚作为合成N-磺酰-1, 2, 3-三唑1的催化剂, 收率最高可达96%.本课题组尝试了分别以廉价易得的(Z)-芳乙烯基溴化合物[8]和1, 1-二溴芳基乙烯[9]为原料, 通过筛选合适的消除反应条件, 得到芳基乙炔中间体.该炔中间体在Fu等[5]报道的催化体系促进下可以被磺酰叠氮捕捉, 通过“一锅法”反应得到N-磺酰-4-芳基-1, 2, 3-三唑类化合物.最近, Anbarasan等[10]报道了一种以1, 3-二羰基化合物和磺酰叠氮为原料, 20~40 mol% L-脯氨酸为催化剂, 在叔丁醇等质子性溶剂合成N-磺酰基-4-酰基-5-取代-1, 2, 3-三唑的新方法.以1, 3-戊二酮为原料, 可得到最高86%的收率(Scheme 1).
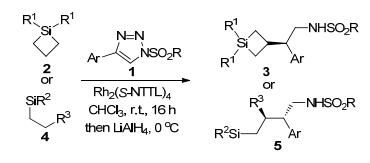
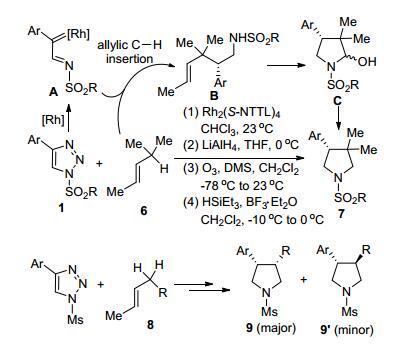
Davies等[11]研究了N-磺酰三唑1和硅取代环烷烃2及硅取烷烃4的C(sp3)—H功能化反应(Scheme 2).这是首次报道N-磺酰三唑原位产生的Rh-卡宾亚胺中间体和硅烷底物β-位C原子之间的C—H插入反应.该反应选择环状或链状取代硅烷为底物, 以理想的区域和对映选择性分别得到相应产物3和5, 最高收率可达95%, 最高ee值达到97%.
Davies等[12]通过Rh2(S-NTTL)4催化N-磺酰-1, 2, 3-三唑1和烯烃底物6的分子间C(sp3)—H功能化、亚胺的双键还原、烯烃双键的臭氧化、分子内半氨缩醛化和三乙基硅烷还原脱羟基等多步骤反应, 立体选择性地合成了β-芳基吡咯烷类化合物7 (Scheme 3).反应起始于Rh-卡宾亚胺A和烯丙基C—H的插入反应和随即进行的LiAlH4还原.各种C4-芳基取代的N-磺酰-1, 2, 3-三唑1都可以参与反应, 获得中等到高收率的预期产物.但是苯环上连有CF3等吸电子基团或邻位基团的底物会由于电子效应和空间位阻因素导致产物收率下降.烯键α-位为仲碳的底物8可以在相似的优化条件下得到具有两个手性中心且具有高的dr和优秀ee值的产物9.为了控制环化过程的差向异构化导致的dr选择性下降, 第四步反应需在更低的反应温度下(从-10 ℃降至-65 ℃)进行.在优化的反应条件下, 多种底物8都可以获得满意的收率和dr/ee值.该合成策略虽然经历四步反应, 但仅需要一次柱层析分离, 即可获得相应的产物7或9.
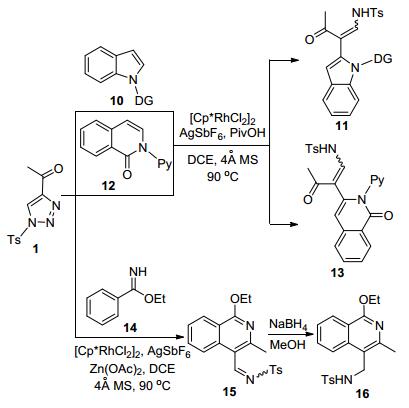
李兴伟等[13]研究发现, 4-酰基-N-磺酰-1, 2, 3-三唑1作为一种通用的Rh-卡宾前体, 可以和吲哚10、异喹啉- 2-酮12等不同结构的芳烃底物发生邻位-选择性的C—H活化偶联反应, 生成高立体选择性的相应芳烃烯基化产物11和13 (Scheme 4).该反应体系中, Rh-催化剂、反应溶剂二氯乙烷(DCE)和添加物PivOH的选择至关重要.用Rh2(OAc)4代替[Cp*RhCl2]2, 吲哚底物10则会以低产率和立体选择性得到吲哚C3-烯基化产物.机理研究表明, 催化剂通过邻位螯合作用促进了C—H活化和反应的效率.该反应对取代吲哚类底物10具有很好的结构普适性, 含有C4、C5和C6位各种类型取代基的N-嘧啶或N-吡啶底物都可以高收率和选择性地声称相应产物11.但是C7位取代的底物则会导致产率下降.调整磺酰三唑底物1中的C4-酰基结构也可以获得满意的结果.将该反应体系扩展到N-吡啶取代异喹啉-2-酮底物12, 其烯化产物13的立体选择性会普遍下降, 且吡啶环上含有取代基时也会导致产物收率的大幅下降. 在Zn(OAc)2促进下, 底物14则在发生C—H插入烯基化后接着进行了缩合环化反应, 得到的是产物15.该产物经过NaBH4一锅法还原, 高效率地转化为产物16, 总收率最高可达98%.
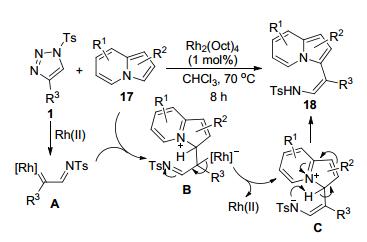
Dawande等[14]报道了N-磺酰三唑1和吲哚嗪17之间的Rh(II)-催化C3-功能化反应, 以较为满意的收率(53%~86%)立体选择性地合成了一系列的产物18 (Scheme 5).底物17和由N-磺酰三唑1原位产生的Rh-卡宾亚胺中间体A首先发生C—H插入形成中间体B.中间体B脱去Rh-催化剂后转化为中间体C.该中间体通过质子转移发生芳构化得到了相应的目标产物18.
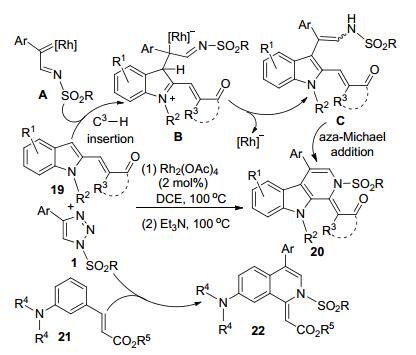
Anbarasan等[15]以Rh-催化剂和手性芳胺有机催化剂“接力催化”(relay catalysis), 通过N-磺酰-1, 2, 3-三唑1和含有α, β-不饱和酮的吲哚-2-取代底物19的一锅法反应, 以中等到较好的转化率和高立体选择性(多数产物的er>92:8)合成了二氢-β-咔啉类化合物20 (Scheme 6).该合成策略包含了Rh-卡宾亚胺中间体A与吲哚的C3—H的选择性插入和有机催化剂促进下中间体C的分子内N-Michael加成反应, 具有高的原子经济性和步骤经济性等优点.萘取代或大位阻芳基取代磺酰三唑底物的转化率相对较低.将Michael受体更换为连有芳胺的底物21, 在同样的反应条件下得到的则是二氢异喹啉类化合物22.中间体A对苯环上氨基对位的C—H插入是转化为产物22的关键步骤.
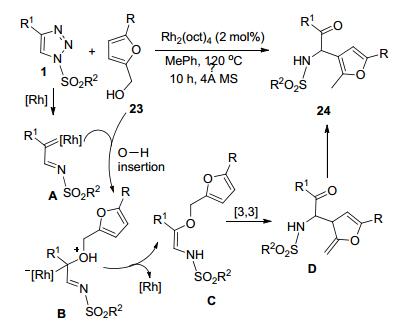
宋金娜等[16]报道了一种通过N-磺酰-4-芳基-1, 2, 3-三唑1和糠醇23之间的Rh-催化反应合成多取代呋喃24的方法. Rh-卡宾亚胺A和糠醇之间通过串联的O—H插入脱去Rh形成中间体C.该中间体发生[3, 3]-σ-迁移和质子转移芳构化等重排过程, 得到最终的产物24 (Scheme 7).该反应主要适用于包括5-Br和5-AcO等C5-取代的糠醇底物, 可获得72%~91%的收率. 5-萘糠醇同样可以顺利完成转化.以C3或C4-取代糠醇和磺酰三唑反应, 不能得到相应的预期产物.以1-(2-苯并呋喃)乙醇为底物时, 大位阻效应导致产物收率急剧下降(31%).各种C4-芳基取代的磺酰三唑底物都以最高85%的收率获得产物.但是, N-磺酰-4-环丙基-1, 2, 3-三唑则不能和糠醇反应.
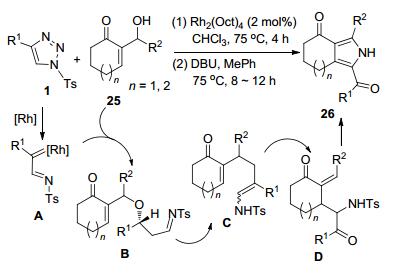
余星昕和邓卫平等[17]以N-磺酰-三唑1和α, β-不饱和环酮的MBH加成物25为原料, 通过Rh-催化的O—H插入等多步骤“一锅法”反应, 合成了一类3, 4-并吡咯衍生物26.该反应过程首先由磺酰三唑前体产生的Rh-卡宾亚胺中间体A和底物1之间发生O—H插入和异构化形成中间体C.该中间体发生分子内3, 3-迁移重排得到中间体D.中间体D再经历迈克尔加成和氧化芳构化过程构建为目标产物3, 4-并吡咯26 (Scheme 8).各种C4-芳基取代的N-磺酰基-1, 2, 3-三唑1都以中等或较高的收率完成反应, 苯环邻、间或对位MeO、F和Cl等的存在对反应效率均没有显著影响. C4-杂芳环(如噻吩和吲哚)-N-磺酰基-1, 2, 3-三唑同样可获得满意的结果.但是, C4-烷基N-磺酰-三唑参与反应只能得到痕量的产物.对MBH加成物25的考察也得到较好的结果.当R2为芳基时, 苯环上邻、间和对位存在的OMe、F、Br、Cl、CN、和CF3等取代基都很少影响转化.当R2为萘环、呋喃或噻吩环时, 同样得到预期产物.但是, R2为i-Pr或Cy等烷基时, 则会导致相应产率明显下降(31%、35%).此外, 底物25的不饱和环酮可以是六元或七元环酮.
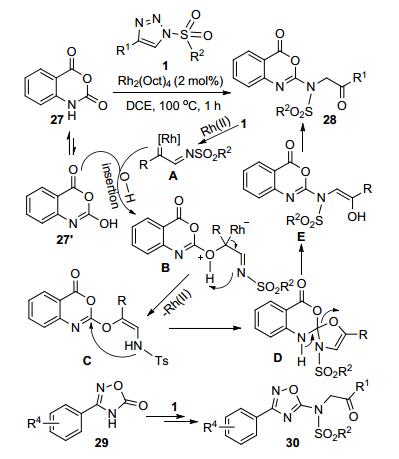
Volla等[18]以N-磺酰-1, 2, 3-三唑1和靛红酸酐27为原料, 通过Rh-催化的O—H插入反应和后续串联的重排过程, 高效简便地合成了2-磺酰氨基苯并噁唑嗪酮类化合物28. Rh-卡宾亚胺中间体A和底物27的烯醇异构体27'发生O—H插入反应先后形成了中间体B和其脱去Rh-催化剂的中间体C.中间体C通过分子内C—N键的形成和C—O键的断裂转化为中间体D和E.中间体E发生重排反应得到最终的目标产物28.底物1中的取代基团R1和R2的适用范围非常宽泛, 产物收率最高可达93%.用3-芳基-1, 2, 4-噁二唑-5(4H)-酮29代替底物27, 在同样的反应条件下也以满意的收率(81%~92%)获得产物30 (Scheme 9).
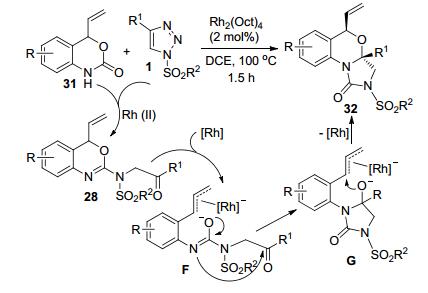
Volla等[19]发现, 用4-烯基苯并噁嗪酮31代替Scheme 9中的底物27, 也可以和N-磺酰-1, 2, 3-三唑1反应, 但得到的产物是2-咪唑酮类衍生物32 (Scheme 10).该产物是由Scheme 9中的产物28进一步发生Rh-催化环重构过程得到的.中间产物28和Rh-催化剂配位首先得到中间体F.该中间体发生分子内亲核进攻得到中间体G, 脱去Rh-催化剂生成最终的产物32.该反应操作条件简便易控, 收率最高可达93%, 且可以放大到“克级”规模.
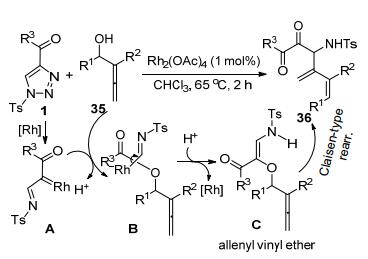
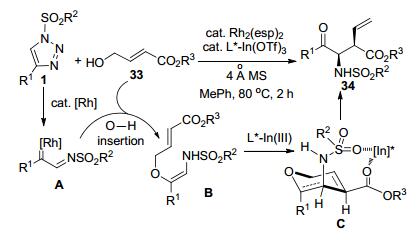
冯小明等[20]以N-磺酰三唑1和烯丙醇33为原料, 以Rh(II)-催化剂和手性N, N'-二氧化物-In(III)-配合物组成双金属催化体系, 通过“接力催化”合成了一系列的不饱和手性β/γ-氨基酸衍生物34 (Scheme 11).由磺酰三唑1原位产生的Rh-卡宾亚胺中间体A和烯丙醇2通过O—H插入反应得到中间体B.中间体B发生In(III)-促进的不对称Claisen-[3,3]-迁移重排反应, 实现了目标产物34的合成.该合成方法不仅具有最高可达99%的收率和优秀的立体选择性(最高95:5的dr和98:2的er), 而且还具催化效率高和底物结构宽泛等优点.随着底物33中R3的基团的体积从Me到t-Bu逐渐增大, 相应生成产物的dr略有降低.底物1的结构对立体选择性也有一定的影响. C4-芳基-N-磺酰-三唑底物比相应的C4-烷基底物可以获得更高的dr和er值, 而芳环上取代基的电子和空间效应则对立体选择性影响有限.
Alcaide和Almendros等[21]以N-磺酰-4-酰基-三唑1和丙二烯基醇35为原料, 合成了一组化合物36 (Scheme 12).多数底物在优化条件下可以获得中等程度的收率(31%~74%).底物35的醇羟基对Rh-卡宾亚胺中间体A的O—H插入反应形成了中间体B.该中间体脱去Rh-催化剂成为中间体C.中间体C发生的Claisen重排转化为相应的产物36.
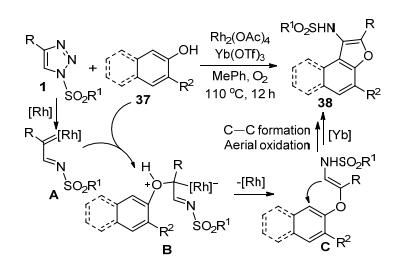
Anbarasan等[22]报道了一种由Rh(II)-和Yb(III)-催化剂协同催化, 通过磺酰三唑1和β-萘酚37“环交换”得到取代萘并呋喃类化合物38的反应.该串联反应包括了Rh-卡宾亚胺中间体A和萘酚37的O—H插入反应和Yb(III)-催化下中间体C的C—C偶联闭环和同步的氧化芳构化过程(Scheme 13).作者考察了各种C4-芳基取代的N-磺酰基-1, 2, 3-三唑的反应通用性, 该双金属催化反应对苯环上的烷基或卤素取代基都具有耐受性, 但强吸电子的NO2和强供电子的Me会导致反应活性降低.磺酰基R1的改变对反应活性没有明显的影响.对萘酚的底物扩展实验表明, 多数β-萘酚都可以有效参与反应.总体来看, 萘环上存在吸电子基团如酯基, 可以使反应收率提高, 而苄氧基的供电子性会导致收率下降.该反应还可扩展到苯酚类的底物, 得到取代苯并呋喃类产物, 但收率低于相应的萘酚类底物.
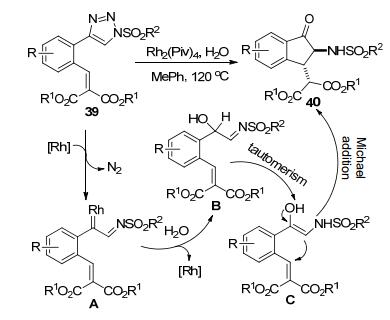
邢思洋等[23]以含有缺电子烯烃基团的N-磺酰三唑39为原料, 实现了2-氨基茚酮40的立体选择性合成(dr 83:17~95:5) (Scheme 14).底物39原位产生的Rh-卡宾亚胺中间体A和H2O之间发生O—H插入反应再经异构化得到中间体B.该中间体通过异构化转化为中间体C.后者再通过分子内C-Michael加成反应构建了产物40的茚酮结构单元, 总反应收率40%~79%.
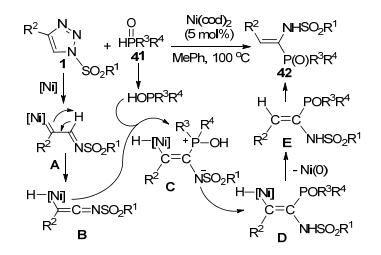
江俊等[24]通过Ni-催化N-磺酰-1, 2, 3-三唑1和二烷基氧膦41偶联构建C(sp2)—P键, 得到立体选择性好、C(sp2)—P键位置选择性单一的α-氨基烯基膦氧化物42, 产率最高达到80% (Scheme 15).磺酰三唑1在Ni-催化体系中脱去氮气原位产生Ni-卡宾亚胺中间体A.该中间体异构化成为Ni-烯酮亚胺中间体B.烯酮亚胺B接受二烷基氧膦底物的酸式异构体二烷基亚膦酸的亲核进攻产生两性中间体C.后者异构化为P—H插入中间体D, 再脱去Ni-催化剂异构化后得到目标产物42.相对于以前报道的金属-卡宾亚胺中间体表现的1, 1-和1, 3-反应活性, 这是第一例关于金属-卡宾亚胺中间体通过异构化位烯酮亚胺中间体而表现出1, 2-反应活性的报道.该反应的二烷基氧膦底物结构对反应活性有明显的影响, R为供电子基团时可以取得较为满意的收率, 而吸电子的三氟甲基苯基或氟苯基则会降低反应效率. N-磺酰-1, 2, 3-三唑底物中的各种4-位取代基R2都可以取得中等收率的产物, 唯一的例外是当R2为4-三氟甲苯基时, 无法得到预期的产物.而磺酰基中的各种取代基则对反应转化没有明显的影响.
夏飞和胡文浩等[25]发展了一种Rh(II)-催化下N-磺酰三唑1、吲哚衍生物43和多聚甲醛之间的三组分反应, 实现了α-氨基-β-吲哚取代酮类化合物44的高效简便合成(Scheme 16).由底物1产生的Rh-卡宾亚胺中间体A和H2O结合形成O-ylide中间体B.该中间体通过1, 4-H迁移转化为α-亚胺烯醇中间体C.中间体C被由吲哚43和多聚甲醛经酸催化原位产生的烯基亚胺离子中间体D捕获, 终止了传统的烯醇-酮转化模式, 从而在脱去H+后得到目标产物44.当R1为CF3等吸电子基团或R为MeO等供电子基团时, 该反应只能得到痕量或收率低于10%的目标产物.其余各种基团组合的反应底物1和43都可以获得中等收率的化合物44.
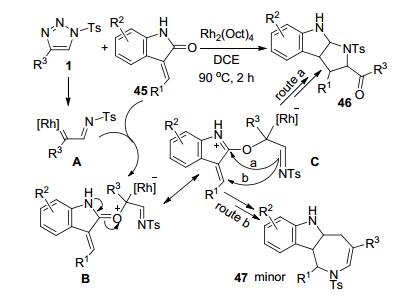
王杭祥等[26]通过Rh-催化下N-磺酰-1, 2, 3-三唑(1)和3-苄亚基-吲哚-2-酮45的环化反应, 以中等到高收率合成了一组吡咯烷并吲哚类化合物46 (Scheme 17).中间体A接受45的亲核进攻形成氧鎓离子中间体B及其共振中间体C.中间体C的亚胺基团进攻吲哚环的C2得到一个不稳定的螺环中间体, 再经过C—O断裂和分子内环化重排后得到产物46 (route a).若中间体C的亚胺基团进攻苄亚甲基的C原子, 则会直接闭环得到副产物47 (route b).主产物46的产率以及其和副产物47的相对比率随苄亚基芳环R1和吲哚苯环上取代基R2的电子特性而改变. R1为供电子基团4-Me-C6H4时, 比R1为4-Cl-C6H4时更有利于得到主产物46.但是R1上强供电子MeO基团的存在则导致反应失败. R1为噻吩环的底物45可以获得70%的单一产物46.尝试使用R1为H或n-Pr等烷基的底物时, 反应没有得到预期产物.底物45中吲哚环上的取代基R2对产物选择性也有显著的影响. R2为吸电子的Cl或酯基取代基时, 以满意的收率获得单一的产物.吲哚环上存在的强供电子的MeO基团导致反应收率明显降低.磺酰三唑底物1中的R3可以是各种芳基或烷基, 大多数情况下对反应转化没有显著的影响, 但R3为噻吩环的磺酰三唑会导致产物46的收率显著下降.
徐泽锋和李传莹等[27]通过4-酰氧甲基-N-磺酰- 1, 2, 3-三唑1和烯基醚类化合物48的Rh(II)-催化区域选择性串联反应, 建立了一种多取代哌啶衍生物49的新合成方法, 最高收率达到96% (Scheme 18). Rh-卡宾亚胺中间体A发生分子内O-亲核进攻形成了O-ylide中间体B.该中间体脱去Rh(II)-催化剂开环产生了酰氧基迁移的N-取代二烯中间体C.中间体C和底物48之间的Diels-Alder反应得到选择性单一的产物49.该反应历程的合理性通过中间体C的分离和结构鉴定得到了进一步的证实.
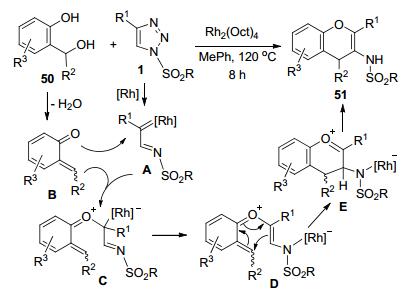
据Anbarasan等[28]的报道, N-磺酰-1, 2, 3-三唑1和2-羟基苄醇50可以发生Rh-催化的环交换反应, 以中等到较高的收率获得苯并吡喃类化合物51 (Scheme 19).由苄醇50脱水原位产生的邻亚甲基苯醌中间体B和磺酰-1, 2, 3-三唑1原位产生的Rh-卡宾亚胺中间体A之间发生O-亲核进攻形成羰基氧叶立德中间体C.该中间体接着发生6π-电环化和异构化过程, 经历中间体D和E得到产物51.三唑底物1中的R1主要为取代苯基, 苯基上烷基和卤素等多数取代基对收率无显著影响, 但是强供电子的MeO和强吸电子的NO2导致收率下降.该反应对苄醇底物50中的R2和R3具有好的耐受性, 可以获得中等以上的产物收率.
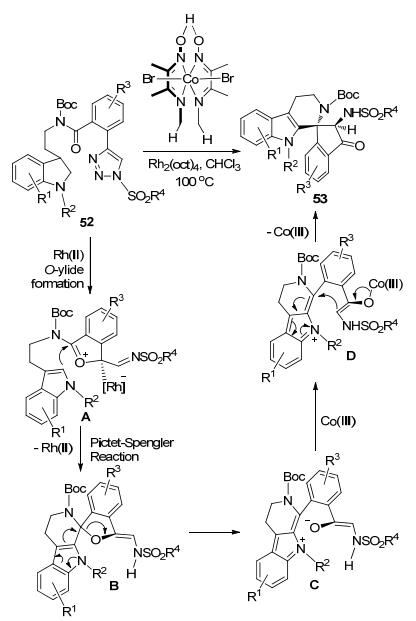
李云等[29]以苯甲酰胺链接的N-磺酰-1, 2, 3-三唑52为起始原料, 通过Rh(II)/Co(III)-双金属催化, 一步法合成了含有吲哚环和茚酮结构的高立体选择手性螺环化合物53 (Scheme 20).原料52在Rh-催化下原位产生的亚胺卡宾和酰胺羰基氧之间形成O-ylide中间体A.中间体A再通过Pictet-Spengler反应得到中间体B.后者经重排过程和Co-催化的Mannich反应形成最终的螺环产物53.该反应的底物范围很宽泛, 能够容忍吲哚环、苯环和磺酰基上各种结构和电性的取代基团.总体来看, 除了吲哚环上C5-位置上的供电子基团如MeO和苯环上吸电子性较强的F取代基会不同程度地降低反应的效率, 其余情况下大多可以获得满意的合成收率.
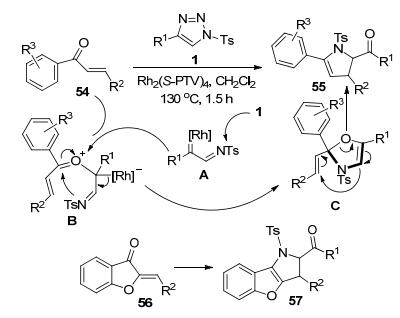
马学骥和王杭祥等[30]以N-磺酰三唑1和α, β-不饱和酮54为原料, 通过Rh-催化的环加成反应合成了2, 3-二氢吡咯衍生物55 (Scheme 21).羰基O原子对Rh-卡宾亚胺中间体A的亲核进攻首先形成O-ylide中间体B.该中间体B发生分子内N-亲核进攻得到中间体C.该不稳定中间体发生C—O键断裂重排为目标产物55.含有苯并呋喃结构单元的α, β-不饱和酮底物56在该反应体系下的反应效率更高, 以最高达92%的收率获得呋喃并吡咯多环化合物57.
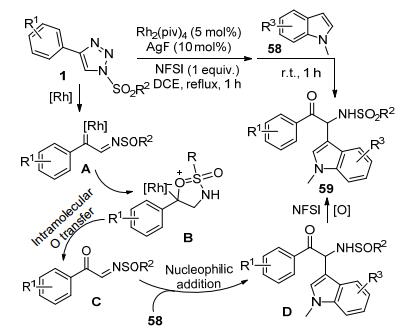
李传莹等[31]发现, N-磺酰-1, 2, 3-三唑1原位产生的Rh-卡宾亚胺中间体A在Rh2(piv)4和AgF协同催化下可以形成O-ylide中间体B.中间体B通过O-迁移形成亚磺酰亚胺酮中间体C.该中间体在室温条件下接受N-甲基吲哚58的C3亲核进攻和氟代双苯磺酰亚胺(NFSI)氧化亚磺酰基, 得到一类功能化的α-氨基酮类化合物59 (Scheme 22).该合成方法具有非常好的底物普适性. R2为包括取代苯基、萘取代和烷基的各种磺酰基三唑都可以顺利完成反应, 获得中等到较高的转化率. R1为C4-芳基取代的各种磺酰三唑同样可以收获满意结果, 但是C4-烷基取代的相应底物则无法完成转化.吲哚底物的普适性试验同样获得满意效果.当吲哚底物58上的R3为Br和BnO时,反应可以顺利完成, R3为邻位基团的位阻因素对收率影响也很有限.
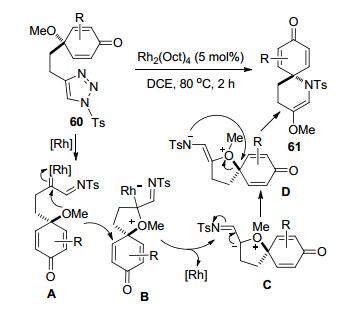
施敏等[32]报道了一种合成1-氮杂-[5, 5]环十一烷螺环结构化合物61的合成方法.该方法以连有3-(4-甲氧基-2, 5-环己二烯酮)丙基的4-取代-N-磺酰三唑60为底物, 通过Rh-卡宾亚胺中间体A接受甲氧基的分子内进攻形成闭环的氧鎓离子中间体B.氧鎓离子再经历异构化和C—O键断裂、甲氧基迁移和分子内C—N成键等重排过程, 得到相应的目标产物61 (Scheme 23).醌羰基的α-位引入Me、CF3等烷基和卤素官能团的反应底物都可以在优化条件下获得中等程度到较高的收率.但是α-甲氧基、酯基和芳杂环的存在会导致反应失败. α-芳基取代底物的总体反应效率低于烷基取代底物, 但是苯环上的供电子、缺电子或中性基团对反应收率影响并不显著.可能是位阻因素以及对Rh-中间体过渡态的稳定性有不利影响, 导致羰基β-位含有取代基的底物60不能发生反应.
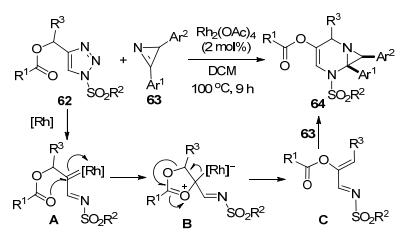
徐泽锋等[33]以N-磺酰基-4-酰氧甲基-三唑62和亲二烯体2H-azirine 63为起始原料, 通过Rh-催化反应合成了一组多取代N-磺酰基嘧啶衍生物64 (Scheme 24). Rh-卡宾亚胺中间体A通过羰基氧的分子内进攻形成五元环O-ylide中间体B.该中间体通过O-1, 2-迁移得到可作为1-N-[4C]合成子的中间体C.中间体C和底物63发生环加成反应得到产物64.该azirine并嘧啶产物还可以扩环为七元的1, 4-二氮杂环庚烷衍生物衍生物, 显示了其在N-杂环化合物合成中的良好应用前景.底物扩展实验表明, R2为烷基和R1为取代苯基的底物62可以获得更高的产物收率, 反之则会导致收率显著下降.考察R1苯环上的取代基发现, 位于邻位或对位的Me或卤素取代基对反应活性影响较小, 而苯环对位含有硝基时, 只能检测到痕量的产物.作者还考察了底物63的结构对反应的影响. Ar1和Ar2为未取代的苯环时反应效率最高, 而Ar1和Ar2都含有取代基时反应的活性最低, 只能得到较低收率的预期产物.
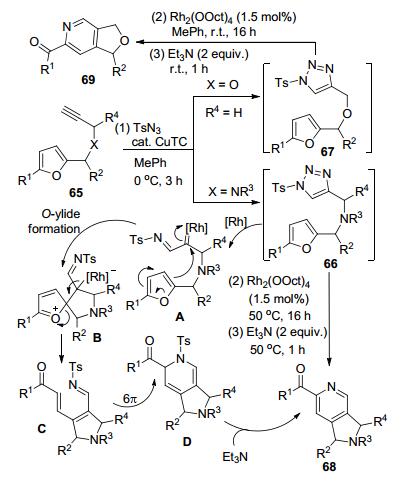
Uchuskin和Hashmi等[34]以底物65为原料, 通过CuTC催化的click反应得到两种含有呋喃环侧链的N-磺酰-1, 2, 3-三唑66和67.该两种底物再经过Rh-催化的加热或室温条件下开环-闭环的多步骤串联反应, 分别以较高反应收率得到2-酰基吡啶类产物68和69 (Scheme 25).以产物68的生成为例, 反应的关键步骤是Rh-卡宾亚胺中间体A发生分子内重构得到O-ylide中间体B.该中间体再经历开环及6π-N-电环化过程得到中间体D.中间体D在三乙胺的作用下发生消除芳构化反应得到最终的产物.底物结构中呋喃环上的R1和侧链上的R2、R3和R4对反应都有一定程度的影响.例如当R2为Ph或R4为i-Pr时, 几乎无法获得相应的产物68.当R1为t-Bu-或4-Cl-C6H4时, 可能是受到大位阻因素的影响, 也无法得到相应产物68, 只能检测到呋喃开环中间体C的生成.
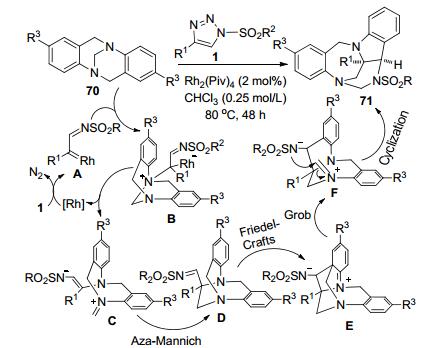
Lacour等[35]通过Rh(II)-催化N-磺酰三唑1和Tröger碱70的分子间反应, 合成了一组有价值的吲哚啉-苯并二氮杂环庚烷类多环化合物71 (Scheme 26).该反应包含了[1,2]-Stevens、Friedel-Crafts、Grob和aminal的形成等多步骤串联反应, 产物的总收率最高可达85%, dr>49:1. N-磺酰三唑1在Rh(II)-催化下原位产生的Rh-卡宾亚胺中间体A和Tröger碱70反应形成N-ylide中间体B.季铵化的中间体B发生N-桥键断裂开环转化为中间体C.中间体C发生aza-Mannich反应产生中间体D, 从而完成了[1,2]-Stevens重排过程.中间体D再先后完成了Friedel-Crafts、Grob和aminal形成等多步骤转化, 得到了最终的产物71.该产物可以再插入一个Rh-卡宾亚胺, 构建含有三氮环壬烷的多环化合物, 但是产率相对较低(29%~34%).
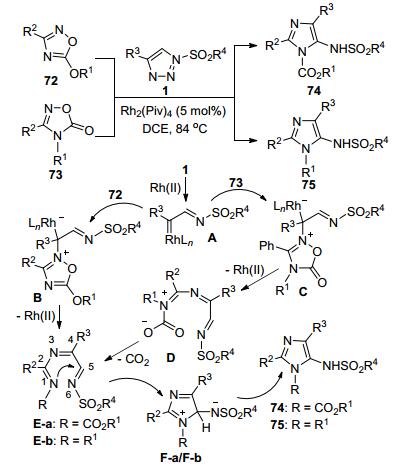
Novikov等[36]通过N-磺酰三唑1和1, 2, 4-噁二唑类底物72或73之间的Rh(II)-催化“环交换”反应, 高效地合成了一系列5-磺酰氨基全取代的咪唑类化合物74或75, 最高收率高达98% (Scheme 27).来自磺酰三唑1的Rh-卡宾亚胺中间体A进攻72或73分别形成N-ylide中间体B或C.中间体B开环脱去Rh(II)-催化剂得到中间体E-a.中间体C开环脱去Rh(II)-催化剂后再发生脱羧反应得到中间体E-b.中间体E闭环获得了相应的目标产物74或75.该反应通过第一例的金属卡宾和N, N, O-五元杂环底物反应, 实现了具有生理活性的5-氨基咪唑类化合物的高区域选择性合成.
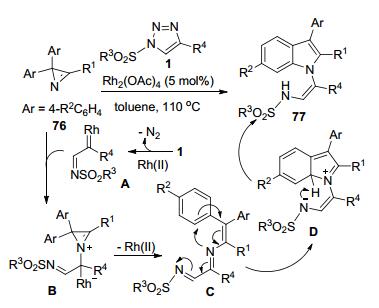
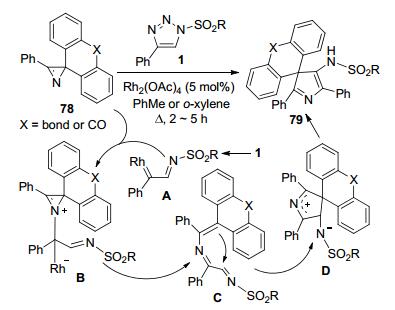
Novikov等[37]发展了N-磺酰三唑1和2, 2-二芳基-氮丙啶76之间的Rh(II)-催化反应, 立体选择性地合成了一类1-(2-磺酰氨乙烯基)吲哚化合物77 (Scheme 28).氮丙啶76进攻由底物1原位生成的Rh-卡宾亚胺中间体A形成N-ylide中间体B.中间体B脱去Rh(II)-催化剂并开环得到中间体C.中间体C通过分子内的N-亲核进攻环化和质子转移反应构建了产物77中的吲哚环结构.氮丙啶底物76上的R1为Me时, 产物收率相对较低(37%~59%), 而R1为Ph的多数其它底物均可获得满意的收率(58%~98%).此外, Novikov等[38]还将二芳基-氮丙啶底物延伸到含有芴或蒽酮框架的原料78, 在类似的反应条件下实现了含有4-氨基-3H-吡咯的螺环化合物79的合成(Scheme 29).但是由于受底物78结构复杂的局限, 所报道的产物只有4种, 且收率也相对较低(35%~68%).
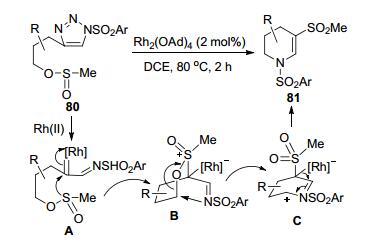
Urabe等[39]通过4-[3-(甲亚磺酰氧)丙基]-N-(芳基磺酰)-1, 2, 3-三唑80的Rh(II)-催化分子内环化反应, 合成了一组2, 3-脱氢-N-芳磺酰基-3-甲磺酰基哌啶类化合物81 (Scheme 30).底物80在Rh2(OAd)4的作用下形成Rh-卡宾亚胺中间体A.甲磺酰基S原子的分子内亲核进攻形成了S-ylide中间体B.该中间体通过C—O键断裂和C—N键的形成重构化为中间体C.中间体C脱去Rh-催化剂后转化为相应的产物81.三唑侧链1、2号位的各种烷基R和芳磺酰基苯环上的各种取代基存在下的底物都可以获得中等到相对较为满意的产物收率(43%~76%).但是侧链中的亚磺酰氧基团的位阻效应明显, 尝试使用位阻较大的芳基或t-Bu代替体积较小的Me时, 反应都难以顺利完成.
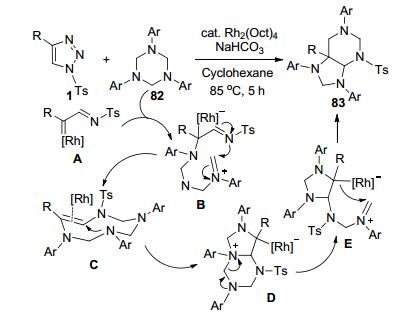
鲍晓光等[40]报道了一种通过Rh-催化下N-磺酰- 1, 2, 3-三唑1和1, 3, 5-三嗪82的环加成反应, 以中等到较好收率构建多氢嘌呤类衍生物83的方法(Scheme 31).三嗪N原子对Rh-卡宾亚胺中间体A的亲核进攻开环得到中间体B, 接着发生[6+3]环加成反应产生了9元环中间体C.中间体C再通过分子内亲核加成和再次开环-闭环重排过程, 得到最终的产物83.该反应过程中只有一种副产物N2排出, 具有高原子经济性的绿色化学特征. C4-烷基的磺酰三唑底物1不能发生反应.多数C4-芳基底物1可以中等到好的收率完成转化.苯环上含Cl和Br的C4-芳基底物只能获得24%~35%的低收率产物, 但通过增加催化剂负载量(从5 mol%增加到9 mol%), 相应的反应产率可提高到45%~67%.含邻甲氧苯基、萘、噻吩等C4-芳基底物1也可获得中等产率. Ar上含有F和Cl等吸电子基的三嗪底物82可以获得好的产物收率, 含Br和Me、i-Pr以及t-Bu等供电子取代基团的底物也可获得60%以上中等收率.但是4-吡啶取代三嗪不能转化为预期的产物.
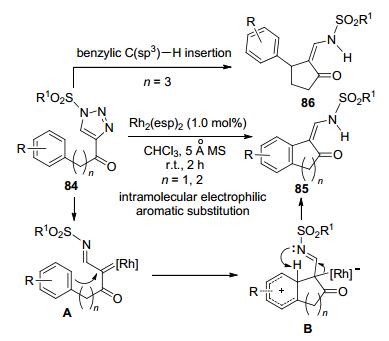
Miura和Murakami等[41a ]研究了含有苯环侧链的4-酰基-N-磺酰-1, 2, 3-三唑84的Rh-催化反应特性.这类连有苯环侧链的Rh-卡宾亚胺中间体可发生不同反应模式的分子内环化, 产物类型取主要决于苯环和羰基间的距离.当苯环和酰基相隔1~2个碳原子时, Rh-卡宾亚胺中间体A发生的是分子内芳环亲电取代反应, 得到苯并五元或六元环的产物85.苯环上的供电子基团有利于亲电取代的发生, 产物最高收率可达98%.苯环对位存在F、CF3和NO2等吸电子基时, 产率分别降低至77%、39%和12%.苯环3-Me取代底物发生反应时, 获得6-和2-位亲电取代成环的产物比例为65:35.苯环和酰基间隔3个碳原子时[41b ], 则发生苄基α-位的C(sp3)—H的插入反应, 得到苯基取得的环戊酮衍生物86 (Scheme 32).
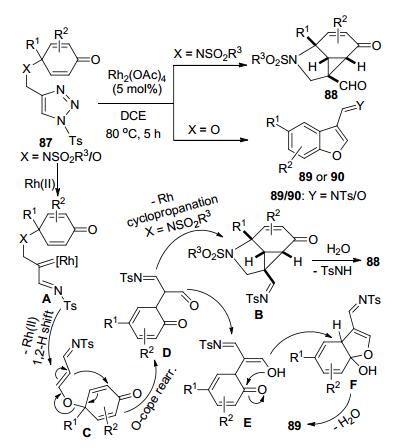
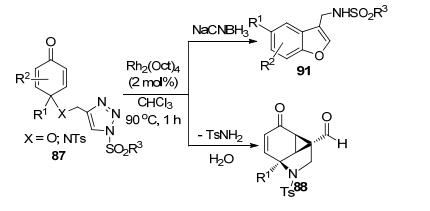
魏音和施敏等[42]研究了含有2, 5-环己二烯酮结构的磺酰三唑底物87在Rh2(OAc)4催化下发生的分子内环化反应(Scheme 33).当底物87的两个反应中心通过N连接时, 磺酰三唑开环产生的Rh-卡宾亚胺中间体A和环己烯二酮的缺电子双键之间通过脱去Rh-催化剂发生环丙烷化形成中间体B.该中间体水解后以最高可达99%的反应收率构建了一类环丙烷并[cd]吲哚类衍生物88.底物87的两个反应中心通过O连接时, 中间体A脱Rh会先后发生1, 2-H迁移和O-cope重排得到中间体D.中间体D通过烯醇化、分子内的O-亲核进攻羰基C和脱水芳构化三步串联反应, 以最高可达86%的收率得到取代苯并呋喃产物89.但是部分产物在柱分离时只能以较低收率获得亚胺水解的产物90. Volla等[43]以同样结构的底物87在Rh2(Oct)4催化下反应, 也获得了相似的结果(Scheme 34).当X=O时, 他们在分子内环化反应的基础上加入了NaCNBH3, 一锅法获得产物89的还原产物3-磺酰胺甲基苯并呋喃衍生物91 (Scheme 34), 总收率61%~84%.
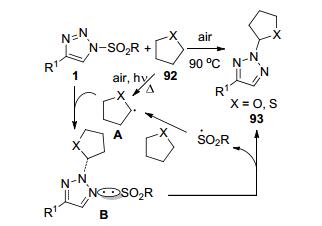
夏飞和刘顺英等[44]报道了一例基于N-磺酰-1, 2, 3-三唑1的自由基链转移反应.将底物1和四氢呋喃/四氢噻吩92简单加热, 经C(sp3)—H偶联反应可以得到N2-选择性-α-四氢呋喃/噻吩取代三唑衍生物93 (Scheme 35).根据对照实验提出的反应机理认为, 四氢呋喃/四氢噻吩92在加热和光照条件下产生的自由基A和N-磺酰-1, 2, 3-三唑发生SN2-类型的自由基取代反应, 得到中间体B.中间体B在C—N键形成的同时发生S—N键断裂得到相应的产物.新形成的磺酰基自由基进攻四氢呋喃/噻吩底物又获得新的自由基A, 从而完成自由基反应的链传递循环. R为Me或Ts基团, R1为取代苯基或噻吩环的底物1都可以中等到最高93%的收率转化为目标产物.其中R1为苯环上有F或CF3吸电子基团时, 反应收率相对较低(41%~47%).以2-甲基四氢呋喃为底物时, 得到四氢呋喃C2-取代和C5-取代两种异构产物的比例为3:1, 总收率仅有47%.该反应以空气中的氧气为氧化剂, 无需催化剂和反应溶剂, 仅在简单加热的条件下即可完成C(sp3)—H偶联反应, 拓展了N-磺酰- 1, 2, 3-三唑在参与自由基反应领域的新研究方向.
施敏等[45]以N-磺酰三唑1和双(N, N-二烷氨基)甲烷94为原料, 发展了一种碱促进下N, N-二烷氨甲基-2H- 1, 2, 3-三唑95的简便合成方法(Scheme 36).该方法适用的底物范围广, 各种N-磺酰三唑1均可获得满意的转化结果, 最高转化收率可达89%.底物94首先分解为中间体A和B.底物1在1, 4-二氮杂二环[2.2.2]辛烷(DABCO)促进下和中间体A反应, 转化为中间体C及其共振体D.中间体D和中间体B相结合得到目标产物95.
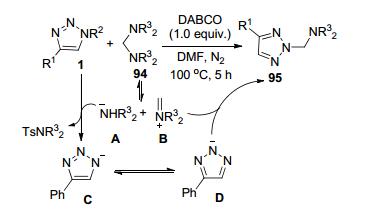
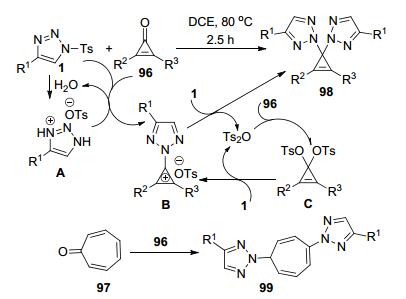
施敏等[46]报道了首例N-磺酰三唑1和环丙烯酮96或环庚烯酮98的自催化N2-选择性双三唑化反应, 实现了产物97和99的合成(Scheme 37).底物1在溶剂中残留的少量水中水解形成中间体A.中间体A作为酸催化剂促进了底物1和96之间的反应形成中间体B.该中间体再接受另一分子1的亲核进攻得到产物97和Ts2O. Ts2O继续参与底物1和96之间的反应循环, 通过中间体C转化为B, 直到所有的底物96消耗完全.该反应具有好的底物普适性、很高的转化效率和操作简便的反应条件.
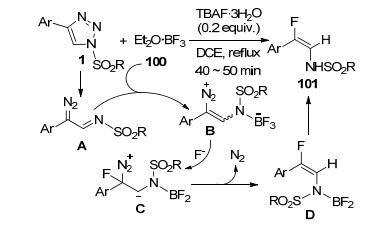
李传莹等[47]发现, N-磺酰基-4-芳基三唑1和Et2O• BF3 (100)可以在四丁基氟化铵三水化合物(TBAF)促进下反应, 立体选择性地合成了(E)-2-氟取代烯胺101 (Scheme 38).该反应无需金属催化, 首先是由BF3对来自底物1热分解产生的α-重氮亚胺中间体A的亲电进攻得到中间体B.中间体B再接受F-离子的亲核进攻形成中间体C.中间体C脱去N2, 再经中间体D异构化转化为最终产物101. TBAF的加入增加了F-的亲核进攻能力, 对反应有明显的促进作用.该反应具有很宽泛的底物普适性, R为烷基或芳基取代磺酰基的各种底物都可以顺利完成转化, 其中只有R为2-萘取代磺酰基时产物收率相对较低(48%). C4-芳取代基团的适用范围也很宽泛, 除了Ar为4-乙酰苯基、2-苯乙烯基苯基或2-萘基时只能得到较低的产率(28%~37%)外, 其余的邻、间或对位取代苯基都以中等或较好收率获得预期产物.
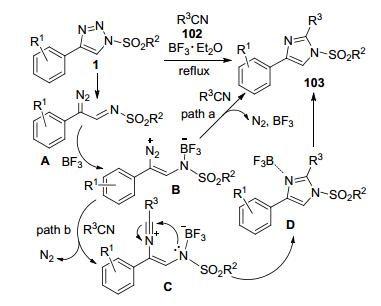
在上述工作的基础上, 作者又以1 equiv.的Et2O• BF3代替过渡金属催化剂, 促进N-磺酰基-4-芳基三唑1和乙腈或其它取代腈类衍生物102之间的环化反应, 高效的构建了多取代的磺酰基咪唑类化合物103 (Scheme 39)[48].该反应所需的最少反应时间仅为30 min, 最高收率可达99%. R2为Me、对甲基/溴-苯基或2-萘基磺酰基底物都可以完成转化, 其中2-萘磺酰基三唑获得高达71%的收率.更换C4-芳基中的各种取代基R1, 也可以中等到高收率得到预期产物, 其中只有R1为吸电子的p-CO2Me时, 相对收率较低(49%).除了乙腈外, 氯乙腈、丁腈、氯丙腈、苄腈和苯腈等都可以兼作反应底物和溶剂, 但是位阻效应会导致反应活性依次下降.苄腈和苯腈参与的反应收率分别低至39%和32%.根据作者提出的可能反应历程, 来自的底物1原位产生重氮亚胺中间体A和BF3先形成关键中间体B.中间体B可能和腈之间发生直接的环加成(path a), 一步脱去N2和BF3.或者中间体B接受腈的亲核进攻先脱去N2形成中间体C.后者再发生分子内亲核进攻构建C—N键, 在形成咪唑闭环的同时发生BF3迁移, 最终得到目标产物.
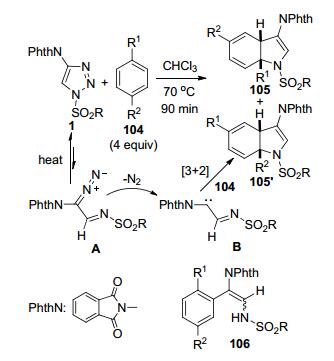
Davies等[49]以C4-邻苯二甲酰亚胺基取代的N-磺酰基-1, 2, 3-三唑1和非活化的芳烃104为原料, 通过加热条件下的[3+2]环加成实现了[3a, 7a]-二氢吲哚105/105'的区域选择性合成, 同时得到部分C—H插入反应产物106, 总收率最高达到82% (Scheme 40).作者还证实了产物106是由相应的产物105发生开环反应转化而来的.当选择CHCl3为反应溶剂时, 该反应可以在保证总收率的同时有利于提高产物中105的比例(105:106=2.7:1).芳烃底物104中的R1和R2为体积相差较大的取代基时, 更有利于提高产物105和105'的区域选择性比例.例如, 当R1为Me、R2为Et、i-Pr和t-Bu的底物时产物105/105'的rr值分别为1.6:1、4.5:1和>20:1.
近年来, N-磺酰基-1, 2, 3-三唑成为重要的有机合成原料.通过金属铑催化下N-磺酰基-1, 2, 3-三唑开环产生的Rh-卡宾亚胺中间体的反应, 可以构建很多结构新颖的杂环类或氨基取代芳环类化合物.近两年的报道以X—H (X=O, C等)插入和O-ylide的反应类型居多, 且主要通过将Rh-卡宾亚胺中间体的反应和[3, 3]-重排等多步骤反应串联, 构建更为复杂的N杂环类化合物, 进一步拓展了N-磺酰基-1, 2, 3-三唑在有机杂环多环化合物合成领域的应用范围和空间.由于N-磺酰基-1, 2, 3-三唑在合成领域的重要价值, 发现廉价易得的合成原料、开发新的合成方法和高效催化剂、拓展其在新型结构有机物合成以及在三氮唑的不对称控制反应领域的研究, 仍将是有机化学家们面临的新机遇和挑战.
(a) Horneff, T.; Chuprakov, S.; Chernyak, N.; Gevorgyan, V.; Fokin, V. V. J. Am. Chem. Soc. 2008, 130, 14972.
(b) Chattopadhyay, B.; Gevorgyan, V. Angew. Chem., Int. Ed. 2012, 51, 862.
(c) Gulevich, A. V.; Gevorgyan, V. Angew. Chem., Int. Ed. 2013, 52, 1371.
(d) Davies, H. M. L.; Alford, J. S. Chem. Soc. Rev. 2014, 43, 5151.
(e) Wang, Y.; Lei, X.; Tang, Y. Synlett 2015, 26, 2051.
(f) Anbarasan, P.; Yadagiri, D.; Rajasekar, S. Synthesis 2014, 46, 3004.
(g) Hockey, S. C.; Henderson, L. C. Aust. J. Chem. 2015, 68, 1796.
(h) Jiang, Y.; Sun, R.; Tang, X.-Y.; Shi, M. Chem.-Eur. J. 2016, 22, 17910.
(i) Volkova, Y. A.; Gorbatov, S. A. Chem. Heterocycl. Compd. 2016, 52, 216.
(j) Li, Y.; Yang, H.-J.; Zhai, H.-B. Chem.-Eur. J. 2018, 24, 12757.
(a) Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596.
(b) Wang, Q.; Chan, T. R.; Hilgraf, R.; Fokin, V. V.; Sharpless, K. B.; Finn, M. G. J. Am. Chem. Soc. 2003, 125, 3192.
(a) Bae, I.; Han, H.; Chang, S. J. Am. Chem. Soc. 2005, 127, 2038.
(b) Zhang, W.-S.; Xu, W.-J.; Kuang, C.-X. Chin. J. Org. Chem. 2015, 35, 2059(in Chinese). (张文生, 许文静, 匡春香, 有机化学, 2015, 35, 2059.)
Yoo, E. J.; Ahlquist, M.; Kim, S. H.; Bae, I.; Fokin, V. V.; Sharpless, K. B.; Chang, S. Angew. Chem., Int. Ed. 2007, 46, 1730.
Wang, F.; Fu, H.; Jiang, Y.-Y.; Zhao, Y.-F. Adv. Synth. Catal. 2008, 350, 1830.
Raushel, J.; Fokin, V. V. Org. Lett. 2010, 12, 4952.
Liu, Y.-T.; Wang, X.-Y.; Xu, J.-M.; Zhang, Q.; Zhao, Y.; Hu, Y.-F. Tetrahedron 2011, 67, 6294.
Zhang, W.-S.; Xu, W.-J. Chem. Heterocycl. Compd. 2016, 52, 192.
Zhang, W.-S.; Xu, W.-J.; Zhang, F. Heterocycl. Commun. 2016, 22, 165.
Rajasekar, S.; Anbarasan, P. Chem.-Asian J. 2019, 14, 4563.
Garlets Z. J.; Davies, H. M. L. Org. Lett. 2018, 20, 2168.
Kubiak, II, R. W.; Davies, H. M. L. Org. Lett. 2018, 20, 3771.
Tian, M.-M.; Liu, B.-X.; Sun, J.-A.; Li, X.-W. Org. Lett. 2018, 20, 4946.
Kahar, N. M.; Nabar, K. U.; Jadhav, P. P.; Dawande, S. G. Asian J. Org. Chem. 2019, 8, 79.
(a) Rajasekar, S.; Anbarasan, P. Org. Lett. 2019, 21, 3067.
(b) Rajasekar, S.; Anbarasan, P. J. Org. Chem. 2019, 84, 7747.
Mi, P.-B.; Wang, H.-N.; Zhao, R.; Song, J.-N. Eur. J. Org. Chem. 2018, 6, 759.
Jia, R.-M.; Meng, J.; Leng, J.-Y.; Yu, X.-X.; Deng, W.-P. Chem.-Asian J. 2018, 13, 2360.
Pal, K.; Hoque, A.; Volla, C. M. R. Chem.-Eur. J. 2018, 24, 2558
Pal, K.; Sontakke, G. S.; Volla, C. M. R. Org. Lett. 2019, 21, 3716.
Chen, Y.-H.; Dong, S.-X.; Xu, X.; Liu, X.-H.; Feng, X.-M. Angew. Chem., Int. Ed. 2018, 57, 16554.
Alcaide, B.; Almendros, P.; Fernández, I.; Martínez del Campo, T.; Palop, G.; Toledano-Pinedo, M.; Delgado-Martίnez, P. Adv. Synth. Catal. 2019, 361, 1160.
Kaladevi, S.; Kamalraj, M.; Altia, M.; Rajasekar, S.; Anbarasan, P. Chem. Commun. 2019, 55, 4507.
Xing, S.-Y.; Xia, H.-Y.; Wang, X.; Wu, D.; Xu, X.-R.; Wang, Y.-R.; Su, K.; Zhu, B.-L.; Guo, J.-S. Org. Chem. Front. 2019, 6, 3121.
Liu, Y.; Xie, P.; Li, J.-G.; Bai, W.-J.; Jiang, J. Org. Lett. 2019, 21, 4944.
Liu, S.-Y.; Yao, W.-F.; Liu, Y.; Wei, Q.-H.; Chen, J.-H.; Wu, X.; Xia, F.; Hu, W.-H. Sci. Adv. 2017, 3, e1602467.
Ma, X.-J.; Xie, X.-M.; Liu, L.; Xia, R.; Li, T.-Y.; Wang, H.-X. Chem. Commun. 2018, 54, 1595.
Yu, S.-S.; An, Y.-H.; Wang, W.-L.; Xu, Z.-F.; Li, C.-Y. Adv. Synth. Catal. 2018, 360, 2125.
Yadagiri, D.; Chaitanya, M.; Reddy, A. C. S.; Anbarasan, P. Org. Lett. 2018, 20, 3762.
Liu, Z.; Du, Q.-C.; Zhai, H.-B.; Li, Y. Org. Lett. 2018, 20, 7514.
Ma, X.-J.; Liu, L.; Wang, J.-Y.; Xi, X.-L.; Xie, X.-M.; Wang, H.-X. J. Org. Chem. 2018, 83, 14518.
Xu, Z.-F.; Shan, L.-H.; Zhang, W.; Cen, M.-J.; Li, C.-Y. Org. Chem. Front. 2019, 6, 1391.
Zhu, C.-Z.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2019, 361, 3430.
Xu, Z.-F.; An, Y.-H.; Chen, Y.-D.; Duan, S.-G. Tetrahedron Lett. 2019, 60, 1849.
Makarov, A. S.; Uchuskin, M. G.; Hashmi, A. S. K. Chem. Sci. 2019, 10, 8583.
Bosmani, A.; Guarnieri-Ibáñez, A.; Goudedranche, S.; Besnard, C.; Lacour, J. Angew. Chem., Int. Ed. 2018, 57, 7151.
Strelnikova, J. O.; Rostovskii, N. V.; Starova, G. L.; Khlebnikov, A. F.; Novikov, M. S. J. Org. Chem. 2018, 83, 11232.
Khaidarov, A. R.; Rostovskii, N. V.; Zolotarev, A. A.; Khlebnikov, A. F.; Novikov, M. S. J. Org. Chem. 2019, 84, 3743.
Khaidarov, A. R.; Rostovskii, N. V.; Starova1, G. L.; Khlebnikov, A. F.; Novikov, M. S. Chem. Heterocycl. Compd. 2018, 54, 946.
Furukawa, A.; Hata, T.; Shigeta, M.; Urabe, H. Tetrahedron Lett. 2019, 60, 815.
Ge, J.-M.; Wu, X.-L.; Bao, X.-G. Chem. Commun. 2019, 55, 6090.
(a) Zhao, Q.; Miura, T.; Murakami, M. Chem. Lett. 2019, 48, 510.
(b) Miura, T.; Zhao, Q.; Murakami, M. Angew. Chem., Int. Ed. 2017, 56, 16645.
Zhu, C.-Z.; Wei, Y.; Shi, M. Org. Chem. Front. 2019, 6, 2884.
Sontakke, G. S.; Pal, K.; Volla, C. M. R. J. Org. Chem. 2019, 84, 12198.
Li, Z.; Wei, Q.-H.; Song, L.-L.; Han, W. Y. J.; Wu, X.; Zhao, Y.; Xia, F.; Liu, S. Y. Org. Lett. 2019, 21, 6413.
Jiang, Y.; Wang, Q.; Sun, R.; Tang, X.-Y.; Shi, M. Org. Chem. Front. 2016, 3, 744.
Li, L.-H.; Jiang, Y.; Hao, J.; Wei, Y.; Shi, M. Adv. Synth. Catal. 2017, 359, 3304.
Xu, Z.-F.; Dai, H.-C.; Shan, L.-H.; Li, C.-Y. Org. Lett. 2018, 20, 1054.
Yang, D.-D.; Shan, L.-H.; Xu, Z.-F.; Li, C.-Y. Org. Biomol. Chem. 2018, 16, 1461.
Wilkerson-Hill, S. M.; Haines, B. E.; Musaev, D. G.; Davies, H. M. L. J. Org. Chem. 2018, 83, 7939.

图 1 N-磺酰三唑及其开环中间体结构
Figure 1 Structures of N-sulfonyltriazole and its ring opening intermediates

图式 2 硅烷和N-磺酰三唑的分子间C(sp3)—H功能化反应
Scheme 2 Intermolecular C(sp3)—H functionalization of silicon-substituted alkane with N-sulfonyl-triazoles

图式 4 Rh(II)-催化芳烃和4-酰基-1-磺酰基三唑C—C偶联
Scheme 4 Rh(II)-Catalyzed C—C coupling of arenes and 4-acyl-1-sulfonyltriazoles

图式 5 Rh(II)-催化吲哚嗪和N-磺酰基三唑的高立体选择性C3-功能化
Scheme 5 Rh(II)-Catalyzed highly stereoselective C3 functionalization of indolizines with N-sulfonyl-triazoles

图式 6 二氢-β-卡啉类和二氢异喹啉类化合物的合成
Scheme 6 Synthesis of dihydro-β-carbolines and dihydroiso-quinolines

图式 9 2-氨基苯并噁唑嗪酮和5-氨基噁二唑的合成
Scheme 9 Synthesis of 2-amino-benzoxazinones and 5-amino- oxadiazoles

图式 12 N-磺酰三唑和丙二烯基醇的Rh-催化反应
Scheme 12 Rh-catalyzed reactions of N-sulfonyl-triazoles with allenols

图式 21 2, 3-二氢吡咯衍生物的立体选择合成
Scheme 21 Stereoselective synthesis of 2, 3-dihydropyrrole derivatives

图式 23 1-N-螺[5.5]十一烷的立体选择合成
Scheme 23 Stereoselective synthesis of 1-azaspiro[5.5]undecanes

图式 26 多环吲哚啉-苯并二氮杂环庚烷类化合物的合成
Scheme 26 Synthesis of polycyclic indoline-benzodiazepines

图式 28 N-磺酰三唑和2, 2-二芳基-氮丙啶的Rh(II)-催化反应
Scheme 28 Rh(II)-catalyzed reaction of 2, 2-diaryl-2H-azirines with N-sulfonyl-triazoles

图式 30 2, 3-脱氢-3-甲磺酰基哌啶类化合物的合成
Scheme 30 Synthesis of 2, 3-didehydro-3-(methanesulfonyl)- piperidines

图式 32 含苯环侧链取代的4-酰基-N-磺酰-三唑的Rh-催化环化反应
Scheme 32 Rh-catalyzed cyclization of 4-acyl-N-sulfonyl- triazoles possessing a phenyl ring

图式 33 环丙烷并[cd]吲哚和苯并呋喃类衍生物的合成
Scheme 33 Synthesis of cyclopropa[cd]indole and benzofuran derivatives

图式 34 连有N-磺酰-三唑环己二烯酮的Rh-催化反应
Scheme 34 Rh-catalyzed reaction of N-sulfonyl-triazole- tethered cyclohexadienones

图式 35 N-磺酰-三唑的无催化自由基链转移反应
Scheme 35 Catalyst-free radical chain transformation of N- sulfonyl-triazoles

图式 36 N, N-二烷氨甲基-2H-1, 2, 3-三唑的合成
Scheme 36 Synthesis of N-dialkylaminomethyl-2H-1, 2, 3- triazoles

图式 37 环丙烯酮和环庚烯酮的自催化双N2-选择性三唑化反应
Scheme 37 N2-Selective autocatalytic ditriazolylation reactions of cyclopropenones and tropones

图式 38 (E)-单氟取代烯胺衍生物的非金属催化合成
Scheme 38 Metal-free synthesis of (E)-monofluoroenamine derivatives

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:





















