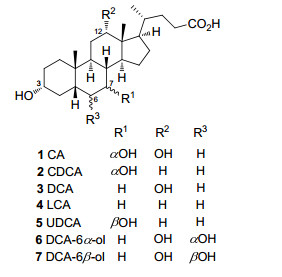
图 1.
选择性胆汁酸的结构
Figure 1.
Selective structures of bile acids

胆汁酸(BAs)是一类存在于人及其它脊椎动物胆汁中的具有重要生理功能的甾体化合物[1](图 1).人体中的胆汁酸根据代谢路径可以简单分为初级胆汁酸、次级胆汁酸和三级胆汁酸.其中初级胆汁酸包括胆酸(CA, 1)和鹅去氧胆酸(CDCA, 2), 它们是由胆固醇在肝脏中经多步酶反应合成[2].初级胆汁酸在肝脏中进一步与牛磺酸或甘氨酸形成水溶性偶联物并分泌至小肠, 在肠道细菌的作用下发生诸如去偶联、去羟基化或异构化生成次级胆汁酸包括脱氧胆酸(DCA, 3)、石胆酸(LCA, 4)和熊去氧胆酸(UDCA, 5)[3].由于上述胆汁酸具有促进脂肪和脂溶性维生素的乳化和吸收等重要生理功能, 分泌至肠道的胆汁酸约95%在回肠被再吸收, 并通过一种被称为“肠肝循环”的主动转运系统返回肝脏[4].被重吸收回肝脏的次级胆汁酸进一步在酶的作用下发生甾体骨架上多个位置的氧化(羟基化), 生成一系列复杂的三级胆汁酸代谢产物[5].探究清楚三级胆汁酸的代谢路径对于完善胆汁酸的协同代谢网络和机制非常关键.最近的胆汁酸代谢研究在超高效液相-串联质谱(UPLC-MS/MS)技术的支持下首次精准揭示了脱氧胆酸(DCA, 3)在细胞色素P450 3A代谢酶的作用下发生甾体骨架C-6位氧化是三级胆汁酸重要生成的路径之一[5a].为进一步深入研究该路径下特异性代谢产物6α和6β-羟基去氧胆酸[(DCA-6α-ol (6)和DCA-6β-ol (7)]在人体胆汁酸协同代谢网络机制中的具体作用以及作为表征P4503A代谢酶活性的潜在内源性探针, 合成更多的DCA-6α-ol和DCA-6β-ol具有显著需求.
DCA-6α-ol和DCA-6β-ol尽管早在20世纪50年代就有由胆酸CA (1)经氧化C-6位进行转化合成的报道[6], 然而这些早期的研究路线中涉及的关键的C-7位还原生成亚甲基的反应收率极低(约为5%), 不具备实际制备合成意义.随后Iida等[7]于1991年报道了由脱氧胆酸DCA (3)作为起始原料经C-6氧化进行转化合成的路线.路线由DCA出发经C-3酮基构建共轭烯酮, 进而生成△3, 5共轭烯醇醚, 再经选择性环氧△5, 6双键化实现C-6β羟基的引入, 总共经12步和19步分别实现了DCA-6β-ol (7)和DCA-6α-ol (6)的合成[7].该路线涉及多步的保护基引入和氧化还原操作, 从而导致步骤冗长及异构体难以分离.本研究拟将报道一条全新的以胆酸(1)为起始原料, 经在C-6位引入亚乙烯基和自由基还原脱氧等关键反应, 选择性地同时实现DCA-6α-ol (6)和DCA-6β-ol (7)的合成, 该合成路线完全有别于前述已知的合成研究.
合成研究如Scheme 1所示, 首先从商业易得的便宜的起始原料胆酸(1)出发, 经N-溴代琥珀酰亚胺(NBS)促进的C-7位羟基选择性氧化和甲酯化两步已知的反应顺利制备7-酮中间体8[8].随后8与叔丁基二甲硅烷基三氟甲磺酸酯(TBSOTf)在2, 6-二甲基吡啶的作用下反应, 以96%的收率制得烯醇硅醚化合物9, 在该反应该条件下同时实现了对C-3和C-12羟基的保护.尝试将烯醇硅醚9直接转化6-羟基酮10并试图经改良的Wollf-Kishner还原等多种条件脱除C-7羰基[9], 然而均无法得到期望的DCA-6-ol产物, 仅分离到少量C-6和C-7同时脱氧的副产物(即DCA 3).因此我们考虑在C-6位引入亚乙基再经氧化还原来间接实现C-6氧功能基的引入.参考文献条件, 烯醇硅醚9首先与乙醛在三氟化硼乙醚(BF3•Et2O)的作用下发生Mukaiyama羟醛缩合反应[10], 顺利制得引入亚乙基的共轭烯酮产物11.尝试在多种条件下[11](ZnI2/SiEt3或者BF3•Et2O/ NaCNBH3)直接还原共轭烯酮11中的羰基成亚甲基, 然而未获成功, 且反应十分复杂, 未分离到任何期望的脱氧产物.因此我们进一步将12中的C-7位羰基用硼氢化钠还原为醇, 醇随后与醋酐发生酰化制备得到烯丙醇乙酰酯化合物12.在尝试直接经靶催化还原脱除烯丙位乙酰氧基[12]失败后, 我们转而将12先经臭氧氧化裂解分子中的亚乙基, 以85%的收率制得酮产物13.接着13在四氢呋喃(THF)/甲醇的混合溶剂中进行碘化钐促进的羰基α位自由基还原脱氧反应[13], 以88%的收率最终顺利制得期望的C-7位乙酰氧基脱除的6-酮化合物14. 14随后经硼氢化钠还原羰基并进而在酸性条件下(HCl/MeOH)脱除TBS保护基, 以物质的量比为约1:5分别得到可以硅胶柱层析分离的6α-OH化合物15(收率15%)和6β-OH化合物16(收率75%).筛选羰基还原条件发现以硼烷叔丁基胺复合物(t-BuNH2•BH3)[7]为还原剂能够以物质的量比为1:1分别得到化合物15(收率46%)和16(收率46%).甲酯化合物15和16的1H NMR和13C NMR谱数据与1991年Iida等[7]首次报道的文献数据完全一致.最后, 15和16分别在碱性条件下水解甲酯, 以90%的收率分别制得最终的羧酸产物DCA-6α-ol (6)和DCA-6β-ol (7).
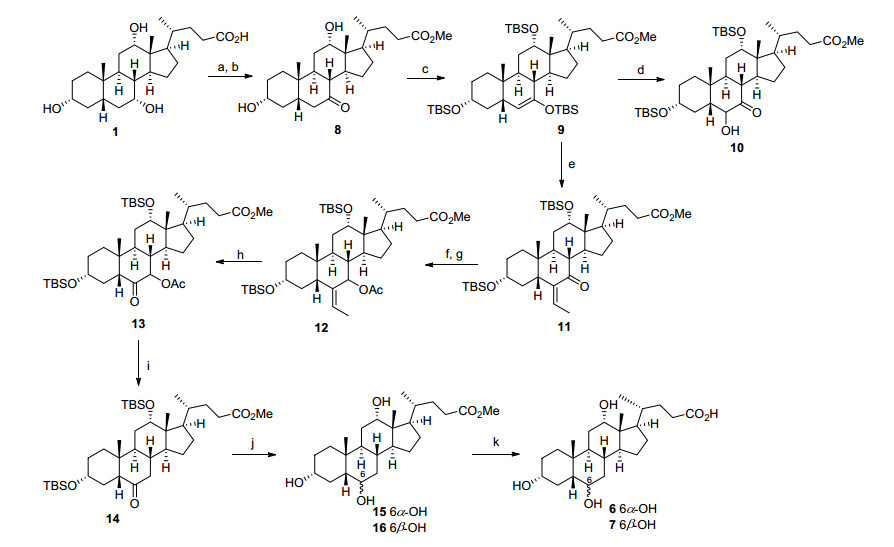
Reagents and conditions: (a) NBS, NaHCO3, acetone/H2O, r.t.~85 ℃, 95%; (b) MeOH, PTSA, reflux, 2 h, 100%; (c) TBSOTf, 2, 6-lutidine, DCM, r.t., 96%; (d) mCPBA, NaHCO3, DCM, 48%; (e) CH3CHO, BF3•Et2O, DCM, -78 ℃, 84%; (f) NaBH4, THF/MeOH (V:V=1:1), 0 ℃, 86%; (g) Ac2O, DMAP, pyridine, 80 ℃, 94%; (h) O3, DCM, -78 ℃, then Me2S, r.t., 85%; (i) SmI2, THF/MeOH (V:V=10:1), r.t., 88%; (j) t-BuNH2•BH3, DCM, r.t., then HCl/MeOH, 0 ℃, 46% for 15, 46% for 16; (k) LiOH, THF/MeOH/H2O (V:V:V=5:3:2), 90%.
本研究以便宜易得的胆酸为原料, 以总共10步反应19%的总收率发展了一条快捷实现三级胆酸代谢产物DCA-6α-ol (6)和DCA-6β-ol (7)高效合成的路线.路线利用Mukaiyama羟醛缩合和SmI2促进的羰基邻位还原脱氧为关键反应, 选择性地实现了胆酸甾体骨架中7-OH向邻位亚甲基(C-6)的转移.该新颖的羟基邻位位移的合成策略为更多胆酸衍生物的多样化修饰以及相关天然产物的合成提供了潜在的新途径, 相关研究仍在进行之中.
旋光光谱用Perkin Polarimeter 341型旋光光度测定仪测定; 核磁共振谱用Varian Unity NOVA-400/54核磁共振仪测定(TMS为内标). HRMS使用Bruker Daltonics Bio-TOF-Q质谱仪测定.未有特别说明反应均在干燥氩气保护下进行.所有实验药品均为市售分析纯试剂, 使用前未进行纯化处理.四氢呋喃、甲苯和乙醚使用前经过钠/二苯甲酮除水处理; 二氯甲烷使用前经过氢化钙除水处理; 薄层色谱(TLC)为GF254硅胶板, 烟台江友硅胶开发有限公司, 用10%磷钼酸/乙醇、I2、高锰酸钾溶液或荧光显色; 柱层析用硅胶为200~300目, 青岛海洋化工厂.
于0 ℃下, 向化合物8 (4.0 g, 9.5 mmol)的二氯甲烷(100 mL)溶液中, 依次滴加2, 6-二甲基吡啶(8.2 mL, 70.40 mmol)和TBSOTf (13.5 mL, 77.11 mmol).滴毕, 反应液升至室温, 并继反应3 h.加入饱和氯化铵水溶液(10 mL)淬灭反应, 二氯甲烷萃取(50 mL×3), 合并有机层, 饱和食盐水(15 mL)洗涤, 无水硫酸钠干燥, 抽滤, 减压浓缩所得粗品经闪式硅胶柱层析纯化[V(石油醚):V(乙酸乙酯)=30:1]即得3α, 7, 12α-三叔丁基二甲硅氧基-5β-胆酸-6-烯甲酯(9), 淡黄色固体, 7 g (9.19 mmol), 产率96%. m.p. 140~142 ℃; [α]D20+52 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.70 (d, J=5.2 Hz, 1H), 3.94 (s, 1H), 3.66 (s, 3H), 3.53~3.41 (m, 1H), 2.40~2.33 (m, 1H), 2.25~2.20 (m, 1H), 2.17~2.00 (m, 1H), 1.99~1.94 (m, 3H), 1.88~1.84 (m, 2H), 1.76~1.56 (m, 5H), 1.49~1.23 (m, 9H), 0.95 (s, 3H), 0.92 (s, 9H), 0.86 (s, 9H), 0.84 (s, 9H), 0.79 (s, 3H), 0.72 (s, 3H), 0.13 (s, 3H), 0.11 (s, 3H), 0.10 (s, 3H), 0.04 (s, 3H), 0.01 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 174.7, 152.0, 108.8, 72.8, 71.1, 51.4, 49.4, 45.7, 44.7, 42.6, 41.0, 40.9, 34.6, 34.3, 33.2, 32.6, 31.8, 31.1, 30.5, 28.6, 27.3, 26.5, 26.4, 26.4, 26.2, 26.2, 25.7, 25.7, 22.5, 18.9, 18.4, 18.3, 17.9, 13.2, -2.7, -2.9, -2.9, -3.6, -3.7, -3.9, -4.5, -4.6, -4.62; ESI-HRMS calcd for C43H83O5Si3 [M+H]+ 763.5543, found 763.5540.
于0 ℃下, 向烯醇硅醚9 (200 mg, 0.262 mmol)的二氯甲烷(4 mL)溶液中, 依次加入间氯过氧苯甲酸(70%, 96.1 mg, 0.39 mmol)和碳酸氢钠(88.2 mg, 1.05 mmol), 在该温度下继续搅拌反应30 min.之后, 加入饱和硫代硫酸钠水溶液(2 mL)淬灭反应, 乙酸乙酯萃取(10 mL×3), 合并有机层, 饱和食盐水(10 mL)洗涤, 无水硫酸钠干燥, 抽滤, 减压浓缩.所得粗品溶于THF (4 mL)中, 于0 ℃下, 加入四丁基氯化铵(99 mg, 0.36 mmol), 升温至室温, 并持续搅拌反应至TLC显示原料完全转化.加水(2 mL)淬灭反应, 乙酸乙酯萃取(10 mL×3), 合并有机层, 再分别以饱和碳酸氢钠水溶液(10 mL)和饱和食盐水(10 mL)洗涤有机层, 无水硫酸钠干燥, 抽滤, 减压浓缩, 粗品经闪式硅胶柱层析纯化[V(石油醚):V(二氯甲烷)=1:2]即得3α, 12α-二叔丁基二甲硅氧基-5-羟基-7-酮-5β-胆酸甲酯(10):白色固体, 83.6 mg (0.13 mmol), 产率48%. m.p. 150~152 ℃; [α]D20+18 (c 0.05, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.47 (d, J=6.4 Hz, 1H), 4.00 (s, 1H), 3.66 (s, 3H), 3.50~3.45 (m, 1H), 2.48~2.26 (m, 4H), 2.24~2.17 (m, 4H), 2.11~2.06 (m, 2H), 1.90~1.79 (m, 4H), 1.71~1.60 (m, 5H), 0.94 (d, J=6.2 Hz, 3H), 0.90 (s, 9H), 0.86 (s, 3H), 0.82 (s, 9H), 0.68 (s, 3H), 0.07 (s, 6H), 0.05 (s, 6H); ESI-HRMS calcd for C37H69O6Si2 [M+H]+ 665.4627, found 665.4635.
于-78 ℃下, 向化合物9 (6.4 g, 8.4 mmol)的二氯甲烷(70 mL)溶液中, 依次缓慢滴加乙醛(1.7 mL, 30.33 mmol)和三氟化硼乙醚溶液(2.1 mL, 17.0 mmol), 并于该温度下继续搅拌反应直至TLC检测显示反应结束.加入饱和碳酸氢钠水溶液(20 mL)淬灭反应, 二氯甲烷(30 mL×3)萃取, 合并有机层, 饱和食盐水(20 mL)洗涤, 无水硫酸钠干燥, 抽滤, 减压浓缩, 粗品经闪式硅胶柱层析纯化[V(石油醚):V(乙酸乙酯)=30:1]即得3α, 12α-三叔丁基二甲硅氧基-6-亚乙基-7-酮-5β-胆酸甲酯(11):白色固体, 4.76 g (7.06 mmol), 产率84%. m.p. 174~176 ℃; [α]D20+47 (c 0.05, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 6.09 (q, J=6.8 Hz, 1H), 4.00 (s, 1H), 3.66 (s, 3H), 3.51~3.59 (m, 1H), 2.53 (d, J=12.8 Hz, 1H), 2.44~2.09 (m, 7H), 1.79 (d, J=11.8 Hz, 4H), 1.67 (d, J=6.8 Hz, 3H), 1.57~1.42 (m, 4H), 1.39~1.20 (m, 6H), 0.98 (s, 3H), 0.94 (d, J=5.6 Hz, 3H), 0.91 (s, 9H), 0.82 (s, 9H), 0.65 (s, 3H), 0.08 (s, 3H), 0.05 (s, 3H), -0.01 (s, 3H), -0.02 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 205.1, 174.70, 143.9, 128.5, 72.6, 70.7, 51.5, 48.9, 48.3, 45.8, 44.9, 41.4, 38.0, 35.4, 34.5, 34.4, 32.5, 31.4, 30.8, 30.6, 28.9, 27.63, 26.2, 26.2, 25.7, 25.7, 25.7, 23.0, 18.2, 18.3, 17.9, 13.0, 12.6, -2.9, -2.9, -3.8, -3.8, -4.6, -4.6; ESI-HRMS calcd for C39H71O5Si2 [M+H]+ 675.4835, found 675.4828.
将化合物11 (2.8 g, 4.15 mmol)溶于甲醇(18 mL)和四氢呋喃(18 mL)的混合溶剂中, 于0 ℃加入硼氢化钠(302 mg, 7.98 mmol), 继续反应至TLC检测显示反应完全.加水(5 mL)淬灭, 乙酸乙酯(30 mL×3)萃取, 合并有机层, 饱和食盐水(20 mL)洗, 无水硫酸钠干燥, 抽滤, 减压浓缩粗品经闪式硅胶柱层析[V(石油醚):V(乙酸乙酯)=10:1]即得3α, 12α-三叔丁基二甲硅氧基-6-亚乙基-7-羟基-5β-胆酸甲酯(12s):白色固体, 2.42 g (3.57 mmol), 产率86%. m.p. 183~185 ℃; [α]D20+95 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.61 (q, J=6.8 Hz, 1H), 4.04~4.08 (m, 1H), 3.96 (d, J=3.2 Hz, 1H), 3.66 (s, 3H), 3.52~3.58 (m, 1H), 2.49~2.39 (m, 1H), 2.39~2.30 (m, 1H), 2.25~2.12 (m, 2H), 2.02~1.69 (m, 6H), 1.69~1.60 (m, 6H), 1.49~1.42 (m, 5H), 1.34~1.24 (m, 4H), 0.95 (s, 12H), 0.85 (s, 9H), 0.75 (s, 3H), 0.67 (s, 3H), 0.08 (s, 3H), 0.05 (s, 3H), 0.01 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 174.7, 142.2, 113.6, 73.4, 72.9, 71.5, 51.5, 48.9, 46.9, 45.7, 44.9, 44.7, 37.0, 35.2, 35.2, 34.7, 32.3, 31.5, 31.3, 30.8, 28.8, 27.5, 27.2, 26.3, 26.3, 25.7, 25.7, 23.0, 18.4, 18.4, 17.9, 13.0, 12.4, -2.7, -2.7, -3.9, -3.9, -4.5, -4.5; ESI-HRMS calcd for C39H73O5Si2 [M+H]+ 677.4991, found 677.5007.
室温下, 向化合物12s (1.8 g, 2.66 mmol)的吡啶(27 mL)溶液中依次加入4-二甲氨基吡啶(DMAP) (325 mg, 2.66 mmol)和醋酐(1.3 mL, 13.75 mmol).升温至80℃, 并继续反应至TLC检测显示反应转化完全.加入饱和氯化铵水溶液(10 mL)淬灭反应, 乙酸乙酯(30 mL×3)萃取, 合并有机层, 饱和食盐水(15 mL)洗涤, 无水硫酸钠干燥, 抽滤, 减压浓缩粗品经闪式硅胶柱层析纯化[V(石油醚):V(乙酸乙酯)=20:1]即得3α, 12α-三叔丁基二甲硅氧基-6-亚乙基-7-乙酰氧基-5β-胆酸甲酯(12):白色固体, 1.8 g (2.51 mmol), 产率94%. m.p. 136~138 ℃; [α]D20+110 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.27 (d, J=10.4 Hz, 1H), 5.12 (q, J=6.8 Hz, 1H), 3.96~3.94 (m, 1H), 3.66 (s, 3H), 3.57~3.52 (m, 1H), 2.45 (dd, J=13.2, 3.8 Hz, 1H), 2.37~2.32 (m, 1H), 2.25~2.10 (m, 3H), 2.10 (s, 3H), 1.94~1.67 (m, 11H), 1.55~1.47 (m, 6H), 1.43~1.36 (m, 3H), 0.90~0.98 (m, 12H), 0.85 (s, 9H), 0.77 (s, 3H), 0.68 (s, 3H), 0.09~0.01 (m, 12H); 13C NMR (101 MHz, CDCl3) δ: 174.6, 170.4, 137.5, 113.8, 75.1, 72.7, 71.4, 51.5, 48.8, 46.3, 45.0, 44.7, 42.2, 36.4, 35.16, 33.1, 34.6, 32.7, 31.5, 31.3, 30.6, 28.9, 27.2, 26.3, 26.3, 25.7, 25.7, 25.7, 22.8, 21.6, 18.4, 18.3, 17.9, 13.0, 12.5, -2.7, -2.7, -4.0, -4.0, -4.6, -4.6; ESI-HRMS calcd for C41H75O6Si2 [M+H]+ 719.5097, found 719.5091.
于-78 ℃下, 向化合物12 (1.8 g, 2.5 mmol)的二氯甲烷(200 mL)溶液中通入臭氧, 搅拌反应至薄层色谱检测显示反应结束.加入二甲硫醚(8.5 mL)淬灭反应, 升至室温并继续反应3 h, 减压浓缩所得粗品经闪式硅胶柱层析纯化[V(石油醚):V(乙酸乙酯)=10:1]即得3α, 12α-三叔丁基二甲硅氧基-6-羰基-7-乙酰氧基-5β-胆酸甲酯(13):白色固体, 1.5 g (2.1 mmol), 产率85%. m.p. 201~203 ℃; [α]D20+75 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.21 (d, J=10.4 Hz, 1H), 4.02~4.00 (s, 1H), 3.66 (s, 3H), 3.58~3.43 (m, 1H), 2.59~2.49 (m, 1H), 2.39~2.32 (m, 1H), 2.30~2.15 (m, 2H), 2.13 (s, 3H), 2.10~1.90 (m, 3H), 1.90~1.68 (m, 7H), 1.61~1.57 (m, 5H), 1.54~1.38 (m, 3H), 0.97 (s, 9H), 0.94 (d, J=6.4 Hz, 3H), 0.85 (s, 9H), 0.82 (s, 3H), 0.70 (s, 3H), 0.11 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H), 0.01 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 205.83, 174.59, 170.20, 72.46, 70.20, 58.88, 51.53, 48.81, 47.01, 44.93, 41.86, 37.43, 35.28, 35.28, 34.78, 34.30, 32.38, 31.46, 30.88, 30.70, 28.80, 27.23, 26.33, 26.33, 25.70, 25.70, 25.56, 23.08, 21.05, 18.39, 18.36, 17.87, 12.92, -2.78, -2.78, -3.88, -3.88, -4.67, -4.73; ESI-HRMS calcd for C39H71O7Si2 [M+H]+ 707.4733, found 707.4714.
将化合物13 (0.8 g, 1.1 mmol)的四氢呋喃和甲醇溶液(V:V=10:1, 11 mL)置换氩气三次, 于室温下, 加入碘化钐的四氢呋喃溶液(10 mL, 10 mmol), 并继续反应至TLC检测显示反应完全.加饱和碳酸氢钠水溶液(2 mL)淬灭反应, 乙酸乙酯(10 mL×3)萃取, 合并有机层, 饱和食盐水(10 mL)洗涤, 无水硫酸钠干燥, 抽滤, 减压浓缩所得粗品经闪式硅胶柱层析纯化[V(石油醚):V(乙酸乙酯)=10:1]即得3α, 12α-三叔丁基二甲硅氧基-6-羰基-5β-胆酸甲酯(14):白色固体, 650 mg (1.0 mmol), 产率88%. m.p. 176~178 ℃; [α]D20+36 (c 1.05, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.05 (s, 1H), 3.66 (s, 3H), 3.56~3.48 (m, 1H), 2.41~2.02 (m, 8H), 1.98~1.65 (m, 9H), 1.58~1.50 (m, 7H), 0.97 (s, 9H), 0.94 (d, J=6.4 Hz, 3H), 0.85 (s, 9H), 0.81 (s, 3H), 0.67 (s, 3H), 0.12 (s, 3H), 0.08 (s, 3H), 0.01 (s, 3H), -0.00 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 214.1, 174.5, 73.2, 70.5, 59.8, 51.4, 47.9, 47.7, 45.9, 42.8, 37.7, 37.2, 35.7, 35.6, 34.3, 33.2, 31.3, 30.9, 30.8, 28.6, 27.4, 26.2, 26.2, 25.6, 25.6, 23.8, 23.3, 18.3, 17.9, 17.8, 12.6, -2.7, -2.7, -4.0, -4.0, -4.6, -4.7; ESI-HRMS calcd for C37H69O5Si2 [M+H]+ 649.4678, found 649.4665.
于0 ℃下, 向化合物14 (500 mg, 0.77 mmol)的二氯甲烷(25 mL)溶液中, 加入硼烷-叔丁胺络合物(75 mg, 0.89 mmol), 升至室温, 并继续反应至TLC检测显示反应结束.加饱和氯化铵水溶液(10 mL)淬灭反应, 二氯甲烷(30 mL×3)萃取, 合并有机层, 饱和食盐水(10 mL)洗涤, 无水硫酸钠干燥, 抽滤.将减压浓缩所得粗品溶于甲醇(23 mL)中, 于0 ℃滴加浓盐酸的甲醇溶液(23 mL).滴毕, 升至室温, 并继续反应液至TLC检测显示反应完全.加入饱和碳酸氢钠水溶液(10 mL)淬灭反应, 乙酸乙酯(30 mL×3)萃取, 合并有机层, 饱和食盐水(20 mL)洗涤, 无水硫酸钠干燥, 减压浓缩所得粗品经闪式硅胶柱层析[V(石油醚):V(乙酸乙酯)=1:5]得到3α, 6α, 12α-三羟基-5β-胆酸甲酯(15)[7]和3α, 6β, 12α-三羟基-5β-胆酸甲酯(16)[7].
3α, 6α, 12α-三羟基-5β-胆酸甲酯(15)[7]:白色固体, 150 mg (0.35 mmol), 两步收率46%. m.p. 102~104 ℃; [α]D20+37 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.07~4.04 (m, 1H), 4.00~3.99 (m, 1H), 3.66 (s, 3H), 3.64~3.62 (m, 1H), 2.39~2.34 (m, 2H), 2.26~2.22 (m, 1H), 1.94~1.85 (m, 2H), 1.85~1.77 (m, 2H), 1.77~1.58 (m, 7H), 1.54~1.45 (m, 4H), 1.44~1.29 (m, 4H), 1.28~1.13 (m, 2H), 0.97 (d, J=6.2 Hz, 3H), 0.89 (s, 3H), 0.67 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 174.8, 72.9, 71.7, 68.0, 51.5, 48.3, 47.6, 47.1, 46.5, 35.5, 35.5, 35.1, 34.9, 34.4, 32.8, 31.1, 30.8, 29.7, 28.7, 28.4, 27.4, 23.6, 23.1, 17.2, 12.6; LC-MS m/z: 422.4 [M+ H]+.
3α, 6β, 12α-三羟基-5β-胆酸甲酯(16)[7]:白色固体, 149 mg (0.35 mmol), 两步收率46%. m.p. 91~92 ℃; [α]D20+45 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.01~4.00 (m, 1H), 3.79~3.77 (m, 1H), 3.66 (s, 3H), 3.63~3.57 (m, 1H), 2.41~2.33 (m, 1H), 2.20~2.27 (m, 1H), 1.86~1.77 (m, 4H), 1.61~1.65 (m, 13H), 1.43~1.35 (m, 4H), 1.10 (s, 3H), 0.97 (d, J=6.2 Hz, 3H), 0.89~0.82 (m, 1H), 0.71 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 174.7, 73.0, 72.9, 71.1, 51.5, 48.4, 47.8, 47.2, 46.4, 36.2, 35.6, 35.1, 34.2, 33.8, 33.8, 31.0, 30.8, 30.8, 29.8, 28.3, 27.4, 25.2, 23.6, 17.2, 12.7; MS m/z: 422.4 [M+H]+.
参考文献[7].室温下将氢氧化锂的一水合物(10 mg, 0.24 mmol)加入化合物15 (30 mg, 0.07 mmol)的四氢呋喃(5 mL)、甲醇(3 mL)和水(2 mL)的混合溶剂中, 并在该温度下搅拌反应至薄层色谱检测显示反应完全.向反应液中滴加2 mol•L-1盐酸, 调节反应液pH为3~4, 乙酸乙酯(10 mL×3)萃取, 合并有机相, 饱和食盐水(10 mL)洗涤, 无水硫酸钠干燥, 抽滤, 减压浓缩所得粗品经闪式硅胶柱层析纯化[V(二氯甲烷):V(丙酮)=1:2]得到3α, 6α, 12α-三羟基-5β-胆酸(6), 白色固体, 26 mg (0.064 mmol), 产率90%. m.p. 135~137 ℃ (lit.[7] m.p. 135~137 ℃); [α]D20+55 (c 0.5, 95% EtOH); MS (ESI) m/z: 409.4 [M+H]+.
以化合物16 (18 mg, 0.043 mmol)为原料经上述相同的方法即制备得到3α, 6β, 12α-三羟基-5β-胆酸(7):白色固体, 15.7 mg (0.038 mmol), 产率90%. m.p. 137~139 ℃ (lit.[7] m.p. 137~139 ℃); [α]D20+35 (c 0.41, 95% EtOH) [lit.[6c] [α]D20+36 (c 0.41, 95% EtOH)]; MS (ESI) m/z: 409.3 [M+H]+.
辅助材料(Supporting Information) 化合物9~16的1H NMR谱和13C NMR谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Monte, M. J.; Marin, J. J.; Antelo, A.; Vazqueztato, J. World J. Gastroenterol. 2009, 15, 804. doi: 10.3748/wjg.15.804
Chiang, J. Y. L. J. Lipid Res. 2009, 50, 1955. doi: 10.1194/jlr.R900010-JLR200
Ridlon, J. M.; Kang, D.; Hylemon, P. B. J. Lipid Res. 2006, 47, 241. doi: 10.1194/jlr.R500013-JLR200
Hofmann, A. F. Front. Biosci., Landmark Ed. 2009, 14, 2584.
(a) Zhang, J.; Gao, L.; Chen, Y.; Zhu, P.; Yin, S.; Su, M.; Ni, Y.; Miao, J.; Wu, W.; Chen, H.; Brouwer, K. L. R.; Liu, C.; Xu, L.; Jia, W.; Lan, K. Drug Metab. Dispos. 2019, 47, 283.
(b) Chen, Y. J.; Zhang, J.; Zhu, P.; Tan, X.; Lin, Q.; Wang, W.; Yin. S.; Gao. L.; Su, M.; Liu, C.; Xu, L.; Jia, W.; Sevrioukova, I. F.; Lan, K. Drug Metab. Dispos. 2019, 47, 574.
(c) Hayes, M. A.; Li, X. Q.; Gronberg, G.; Diczfalusy, U.; Andersson, T. B. Drug Metab. Dispos. 2016, 44, 1480.
(d) Bodin, K.; Lindbom, U.; Diczfalusy, U. Biochim. Biophys. Acta 2005, 1687, 84.
(e) Araya, Z.; Wikvall, K. Biochim. Biophys. Acta 1999, 47, 1438.
(f) Deo, A. K.; Bandiera, S. M. Drug Metab. Dispos. 2009, 37, 1938.
(g) Gustafsson, J.; Andersson, S.; Sjovall, J. Biol. Neonate 1985. 47, 26.
(a) Haslewood, G. A. D. Biochem. J. 1958, 70, 551.
(b) Takeda, K.; Igarashi, K. J. Biochem. 1959, 46, 1313.
(c) Ratliff, R. L.; Matschiner, J. T.; Doisy, E. A.; Hsia, J. S. L.; Thayer, S. A.; Elliott, W. H. J. Biol. Chem. 1961, 236, 685.
Iida, T.; Tamaru, T.; Chang, F.C.; Goto, J.; Nambara, T. J. Lipid Res. 1991, 32, 649.
(a) Fieser, T. L.; Rajagopalan, S. J. Am. Chem. Soc. 1949, 71, 3935.
(b) Ibrahim-Ouali, M.; Romero, E. Steroids 2013, 78, 651.
Qian, S.; Zhao, G. Synlett 2011, 722.
(b) Vasu, N.; Sinbababu, A. K. J. Org. Chem. 1978, 43, 1978.
(c) Zhou, W.-S.; Wang Z.-Q.; Jiang, B. J. Chem. Soc., Perkin Trans. 1 1990, 1.
(d) Iida, T.; Tamura, T.; Matsumoto, T. Synthesis 1984, 957.
Pellicciari, R.; Gioiello, A.; Macchiarulo, A.; Thomas, C.; Rosatelli, E.; Natalini, B.; Sardella, R.; Pruzanski, M.; Roda, A.; Pastorini, E.; Schoonjans, K.; Auwerx, J. J. Med. Chem. 2009, 52, 7958.
(a) Li, Z.; Deng, G.; Li, Y.-C. Synlett 2008, 3053.
(b) Srikrishna, A.; Viswajanani, R.; Sattigeri, J. A.; Yelamaggad, C. V. Tetrahedron Lett. 1995, 36, 2347.
(a) Tsuji, J.; Minami, I.; Shimizu, I. Synthesis 1986, 623.
(b) Tsuji, J.; Mandai, T. Synthesis 1996, 1.
(c) Waalboer, D. C. J.; Schaapman, M. C.; van Delft, F. L.; Rutjes, F. P. J. T. Angew. Chem., Int. Ed. 2008, 47, 6576.
Molander, G. A.; Hahn, G. J. Org. Chem. 1986, 51, 1135. doi: 10.1021/jo00357a040

图式 1 制备DCA-6α-ol (6)和DCA-6β-ol (7)
Scheme 1 Preparation of DCA-6α-ol (6) and DCA-6β-ol (7)
Reagents and conditions: (a) NBS, NaHCO3, acetone/H2O, r.t.~85 ℃, 95%; (b) MeOH, PTSA, reflux, 2 h, 100%; (c) TBSOTf, 2, 6-lutidine, DCM, r.t., 96%; (d) mCPBA, NaHCO3, DCM, 48%; (e) CH3CHO, BF3•Et2O, DCM, -78 ℃, 84%; (f) NaBH4, THF/MeOH (V:V=1:1), 0 ℃, 86%; (g) Ac2O, DMAP, pyridine, 80 ℃, 94%; (h) O3, DCM, -78 ℃, then Me2S, r.t., 85%; (i) SmI2, THF/MeOH (V:V=10:1), r.t., 88%; (j) t-BuNH2•BH3, DCM, r.t., then HCl/MeOH, 0 ℃, 46% for 15, 46% for 16; (k) LiOH, THF/MeOH/H2O (V:V:V=5:3:2), 90%.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载: