当有机化合物引入含磷基团时, 其物理状态、化学性质以及生物活性都将发生极大的变化.有机磷化合物在许多方面都显示出了它的重要性, 一些磷酸衍生物作为核酸和辅酶的组成部分, 成为维持生命所不可缺少的物质.芳基膦酸酯类化合物作为一类重要的有机磷化合物, 在作为药物中间体、功能材料、有机合成配体以及农药等方面均显示出优秀的性质并且被广泛应用[1 3 , 因此, 芳基膦酸酯的合成在近年来受到了有机合成科研人员的广泛关注[4 11 .
传统合成芳基膦化合物的方法, 通常是采用芳基格氏试剂或者芳基锂试剂与氯代磷(膦)酸酯反应制备得到.该方法能够有效地合成一系列含磷配体, 是目前合成该系列化合物的主要方法.但是该类方法所使用的氯代磷试剂怕水不稳定, 反应底物对这些活泼的有机金属试剂的官能团耐受性差.同时, 有机金属试剂对水异常敏感, 潮湿的环境中反应物均容易变质, 使得该类试剂在储存和反应时都必须严格保证无水, 限制了该方法的应用范围.另外, 芳基卤代物和三价磷化合物可以通过Arbuzov或类Arbuzov反应来实现芳基膦化合物的合成.然而, 该类反应通常都需要较高的反应温度(超过180 ℃), 而且在反应过程中生成的卤代烷烃容易继续与亚磷酸三酯反应得到烷基磷酸二酯副产物.
近年来很多化学研究者报道了以过渡金属催化和光诱导等方法构筑Ar—P键的多种芳基来源:卤代芳烃(Ar—X)、芳基硼(Ar—B)化合物、芳基氮(Ar—N)化合物、芳基醚(Ar—O, Ar—S)化合物、芳基铋、芳基锑和芳基硅(Ar—Bi, Ar—Sb, Ar—Si)化合物等, 甚至Ar—H和Ar—C化合物都可以通过C—H键活化或形成芳基自由基和C—C键断裂提供芳基来源.本文以各种构筑Ar—P键方法中芳基源的种类进行分类, 对近年来过渡金属催化、光诱导和电化学促进下合成芳基膦酸酯类化合物所取得的研究进展作简要综述.
1.
以Ar—X为芳基源构筑Ar—P键
芳基卤代物因具有活性高和商品化价格低等优点使得其成为最广泛使用的工业合成原料之一.在过渡金属催化构筑C—C键反应发展的热潮下, Hirao等在1981年[12 和1982年[13 分别报道了钯催化下卤代芳烃与亚磷酸酯通过偶联反应构筑Ar—P键的方法, 并合成了一系列芳基膦酸酯类化合物(Eq. 1).该方法为过渡金属催化合成芳基膦化合物的研究树立了风向标.在该工作的基础下, 芳基膦化合物的合成方法进入快速发展阶段.
2009年, Stawinski等[14 报道了钯催化卤代芳烃和亚磷酸酯作用得到芳基膦酸酯的反应(Eq. 2).与Hirao等报道的工作不同的是:该方法以二价的醋酸钯为催化剂, 反应过程中醋酸钯与双齿膦配体双(二苯基膦基)二茂铁(dppf)原位生成零价钯复合物, 进一步与卤代芳烃发生氧化加成, 与亚磷酸酯发生亲核取代和去质子化反应, 并进一步通过还原消除得到芳基膦产物.作者通过磷谱核磁共振比对方法, 发现该反应在转化率达到95%时, 醋酸根阴离子在反应中能够显著促进该反应的进行, 从而降低反应时间.
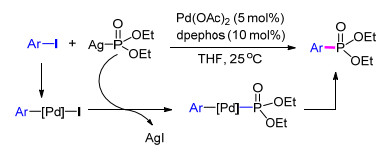
2009年, Stockland等[15 报道了室温条件下的Hirao型反应.该方法在5 mol%的醋酸钯催化下, 以10 mol%的双(2-二苯基磷苯基)醚(dpephos)为配体, 四氢呋喃为溶剂, 二乙氧基磷酸银作为磷酸脂来源与芳基碘化合物反应得到对应的芳基膦化合物.作者认为该反应首先由钯催化剂插入卤代芳烃, 再与二乙基磷酸银发生转金属, 最后经过还原消除得到芳基膦产物(Scheme 1
图式 1
我国的科学家对于温和条件下合成芳基膦化合物的方法也进行了探索. 2013年, 赵玉芬等[16 以二芳基碘鎓盐为原料, 在廉价金属铜催化下与亚磷酸酯反应得到对应的芳基膦酸酯产物.该反应同样需要在碱性条件下反应, 条件温和(25 ℃)且反应非常迅速(10 min).在底物拓展实验中, 芳环上不管是吸电子还是供电子官能团甚至溴代的二芳基碘鎓盐都能以优良的产率得到目标产物(Eq. 3).作者认为这些实验结果表明该反应与已有报道中生成Cu(Ⅲ)中间产物的机理不同, 其可能为自由基机理, 原因是具有大位阻的以及缺电子的芳基自由基更能够稳定存在, 从而得到更多的对应的芳基膦产物.
需要指出的是, 赵玉芬等[17 早在2006年就报道过铜催化的卤代芳烃与亚磷酸脂或者二膦氧化物发生偶联反应的方法(Eq. 4).该方法为典型的Ullmann类型反应, 氮气条件下以脯氨酸或2-哌啶甲酸为配体, 碳酸铯或者4-二甲氨基吡啶(DMAP)为碱.该方法对于芳基碘的反应效果比较好, 各种官能团条件下均能够得到产率中等至优秀的产物.芳基溴代物在该条件下只能得到30%~40%的产率.该方法的亮点在于反应所使用的配体为便宜易得的脯氨酸或2-哌啶甲酸, 这使得该方法具有一定的工业应用价值.
2014年, Taillefer等[18 开发了一种铜催化下的Mi- chaelis-Arbuzov反应, 并成功将其应用于合成芳基膦酸二乙酯、二芳基膦酸乙酯和三芳基膦氧化合物, 该方法不需要额外的膦配体(Scheme 2 Scheme 2
图式 2
2012年, 韩福社等[19 报道了镍催化下卤代芳烃与亚磷酸二甲酯的反应(Eq. 5).反应采用2 equiv.的K3 PO4 作碱, 以二氧六环为溶剂, 在100 ℃条件下反应6~12 h就能够高效地得到相对应的产物.反应无需添加其它还原剂和配体, 不管是溴代芳烃还是氯代芳烃都能以较高的产率得到目标产物.该方法对氯代芳烃底物具有良好的官能团耐受性, 同时对一些卤代杂环底物也能提供中等偏上的产率.
同样是采用镍催化剂, Yamagishi等[20 在2015年发展了芳基碘代物与次磷酸酯类化合物反应构建Ar—P键(Eq. 6).该反应以10 mol%的氯化镍为催化剂, 20 mol%的2, 2'-联吡啶作为配体, 添加2 equiv.的三乙胺碱和2 equiv.的Zn, 在N , N -二甲基甲酰胺(DMF)为溶剂和50 ℃下反应24 h, 得到芳基次膦酸酯类目标产物, 芳环上各种对位和间位取代位阻较大的芳基碘化物均得到较好的反应产率, 但是溴代芳基作为芳基源时反应效果较差.
Hirao类型的反应还可以在微波加热的条件下进行(Eq. 7). 2008年, Stawinski等[21 在前人的基础上, 以四三苯基膦钯作为催化剂, 采用微波加热的方法促使卤代芳烃与亚磷酸酯反应构筑Ar—P键.相对于传统的Hirao反应, 该方法大大提高了反应速度, 可以将反应时间缩短至10 min, 卤代芳烃与卤代烯烃均适合本方法.随后Keglevich等[22 发现, 微波法条件下Hirao反应可以在不需要膦配体的条件下进行, 以醋酸钯为催化剂, 三乙胺为碱, 甚至不需要溶剂, 就能够高效地合成芳基磷酸酯和三芳基膦氧化合物(Eq. 8).
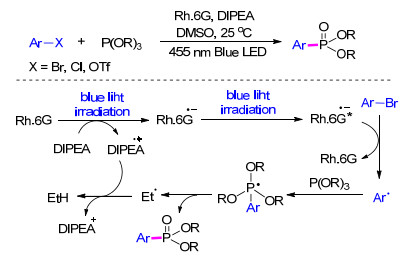
光催化/诱导技术作为一种高效、安全的环境友好型环境净化技术, 近年来得到了快速发展并被广泛用于有机合成中, 其中在Ar—P键的构筑方面也有了一些研究进展.光促进下的Arbuzov反应是经典Michaelis- Arbuzov反应的一种变体. 2016年, König等[23 报道了一种可见光促进下的Arbuzov反应来合成芳基膦酸酯的方法.机理研究认为:首先光催化剂有机染料罗丹明6G在二甲基亚砜(DMSO)溶剂中被蓝光照射生成罗丹明6G自由基阴离子, 而电子供体N , N -二异丙基乙胺(DIPEA)失去电子变成自由基阳离子, 罗丹明6G自由基阴离子继续在蓝光照射下跃迁到激发态.溴代芳烃在激发态的罗丹明6G自由基阴离子作用下生成芳基自由基, 芳基自由基和亚磷酸三烷基酯发生加成反应得到不稳定的磷自由基, 该磷自由基通过释放乙基自由基和重排得到芳基膦酸酯目标产物(Scheme 3
图式 3
紧接着在2017年, König等[24 又报道了一种敏化引发的双中心光氧化还原法合成芳基膦酸酯的方法.该策略是利用单电子转移类型的光敏剂Ru(bpy)3 Cl2 吸收可见光, 并将能量转移到能够进行氧化还原反应的多环芳烃中, 该方应同样能够使芳基卤代物产生芳基自由基从而用于Ar—P键的形成(Eq. 9).因为反应过程中存在芳基自由基从溶剂或DIPEA的自由基阳离子中获取氢原子的副反应, 作者选择了亲核试剂P(OR)3 作为磷酸酯化的来源, 利用P(OR)3 对芳基自由基的良好反应性能阻止副反应的竞争.同时, 该方法对于杂芳基卤化物和其他亚磷酸酯同样适用.
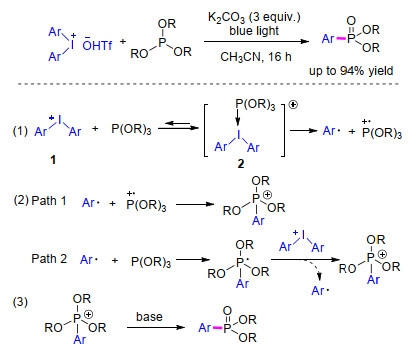
2018年, Lakhdar等[25 报道了可见光促进下二芳基碘鎓盐与亚磷酸酯通过偶联反应合成芳基磷酸酯类化合物的方法.该反应使用3 equiv.的K2 CO3 为碱, 以乙腈作为溶剂, 在蓝光照射下反应16 h就以良好收率得到芳基磷酸酯类化合物.作者通过实验和计算提出的可能机理包含两种途径(Scheme 4
图式 4
2019年, 李朝军和曾会应等[26 报道了在光诱导下的芳基卤化物与磷酸酯类化合物的交叉偶联反应.该方法中各种芳基和杂芳基卤化物与二芳基膦氧化物、二烷基膦酸酯或芳基膦酸酯均能通过交叉偶联得到相对应的产物.该方法对于各种官能团都具有良好的耐受性, 例如芳环上带有不同类型取代基的芳基卤代化合物杂环芳烃等均能以中等到良好的产率得到目标产物(Eq. 10).该方法最大的优点是无需添加过渡金属和外部光敏剂, 但是该反应需要在氩气氛围下进行.
有机电化学反应采用电能转化为化学能从而实现有机化合物合成, 被认为是一种典型的绿色合成方法.近年来, 有机电化学反应技术在Ar—P键的构筑方面也有了一些研究进展. 2018年, Léonel等[27 报道了首例电化学辅助下镍催化的(杂)芳基卤化物和亚磷酸二甲酯反应合成(杂)芳基膦酸酯的方法(Eq. 11).该方法以可牺牲的金属镍作为阳极, 在恒定电流下, 添加三乙胺为碱, 四丁基溴化铵为电解质, 以乙腈为溶剂在未分隔的电解池中反应2~4 h就能高效的得到(杂)芳基膦酸酯化合物.该方法同样也适用于芳基碘化物和其它类型的亚磷酸二烷基酯化合物.
2019年, 王剑波等[28 开发了一种无需催化剂的芳基卤化物与亚磷酸三烷基酯的电化学交叉偶联构筑Ar—P键的方法.该反应以镍作为阳极, 石墨作为阴极, 在不分隔电池中, 在温和中性的条件下高效进行得到目标产物.该方法具有良好的底物普适性和官能团耐受性, 并且在克级放大实验中能以非常高的产率得到目标产物.但是在亚磷酸三甲酯作为磷酸化试剂时该反应只能得到痕量的产物, 使用亚磷酸三苯酯作为磷酸化试剂时该反应并不能进行.机理推测认为:首先芳基卤化物被阴极还原生成的自由基阴离子进一步发生碎片化产生芳基自由基, 芳基自由基被亚磷酸三烷基酯捕获生成芳基膦酸自由基, 紧接着芳基膦酸自由基被阳极产生的Ni2+ 离子氧化芳基膦正离子, 该自由基与反应体系中的亲核试剂反应得到目标产物(Scheme 5
图式 5
2019年, 项金宝等[29 也报道了一种在镍催化下芳基溴化物与膦氧化合物的电化学交叉偶联反应(Eq. 12).该方法并没有采用传统牺牲阳极的电化学方法, 而是使用廉价的碳棒电极, 通过阳极和阴极相互协调作用, 在未分割的电池中制备芳基膦化合物.该反应条件温和, 在室温空气中就可以反应, 而且具有非常好的底物普适性和官能团耐受性.
2.
以Ar—B化合物为芳基源构筑Ar—P键
芳基硼酸具有无毒、对空气和湿度不敏感及官能团兼容性好等特点, 芳基硼酸被广泛用于偶联反应.近年来, 分别在Pd、Cu、Ni和Co等催化剂作用下, 以芳基硼酸作为芳基来源构筑Ar—P键方面也取得了不错的研究进展.
2009年, Larhed等[30 报道了微波条件下醋酸钯催化以硼酸为芳基源合成芳基膦酸酯的方法.与卤代芳烃相比, 该方法需要2, 9-二甲基-1, 10-菲啰啉为配体, 以对苯醌(p -BQ)作为氧化剂.反应只需要30 min就可以完成, 具有很好的底物耐受性, 芳环上带有Br, Ac和CO2 Me等取代基团时都能以中等到良好的产率得到目标产物, 烯烃类硼酸底物也能以较低的产率得到目标产物(Eq. 13).作者通过电喷雾质谱法(ESI-MS)分析了该反应的中间体, 从而推测该反应的机理与零价钯插入卤代芳烃的机理并不一样, 而是通过转金属化反应实现整个反应循环.
2011年, 赵玉芬等[31 发展了一种芳基硼酸和亚磷酸脂类化合物通过偶联反应生成含磷有机化合物的方法.该反应在铜催化体系下, 加入1, 10-菲罗啉(phen)作为氮配体, 加入(i -Pr)2 NEt营造一种碱性环境, 乙腈作为溶剂, 在室温下反应24 h.该反应为首次芳基硼酸在廉价金属Cu的催化下构建Ar—P键.该方法反应条件温和, 在空气中就可以反应, 不需要惰性气体保护, 对底物具有很好的普适性, 芳环上带有耐受性较差基团如NO2 , CN, CO2 Me和Br的芳基硼酸也能以中等到良好的产率得到目标产物(Eq. 14).
廉价金属镍作为催化剂在偶联反应中也有不错的表现, 近年来受到大家的广泛关注. 2013年, 赵玉芬等[32 报道了镍催化剂作用下芳基硼酸与膦氧化合物通过交叉偶联反应得到芳基膦类化合物(Eq. 15).该方法加入溴化镍作为催化剂, 吡啶为氮配体, 以二氯乙烷为溶剂, 加入碳酸钾调节反应体系至碱性, 在100 ℃下反应24 h.该反应需要氩气保护, 优点在于催化剂用量小, 膦氧底物具有很好的普适性, 采用廉价的溴化镍催化剂也有效地降低了生产成本, 具有商业价值.
廉价金属钴催化的氧化偶联反应一般通过单电子转移发生, 使得钴催化的反应与钯和镍催化相比更具特异性, 近年来钴催化剂广泛用作各种C—C和C—N交叉偶联反应. 2020年, Kaur等[33 发展了首例钴催化下芳基硼酸和亚磷酸脂类化合物的交叉偶联反应(Eq. 16).值得指出的是, 该反应以锌粉作为添加剂, 以三联吡啶作为配体.该方法具有良好的官能团耐受性和底物普适性, 对于各种类型的二烷氧基酯均能以良好产率得到相对应的芳基膦氧化合物.
芳基硼酸酯具有高化学稳定性、易溶于非质子溶剂中和易提纯等优点, 也被用于Ar—P的构筑反应. 2017年, Lee等[34 报道了首例Pd催化下芳基硼酸酯和磷酸二烷氧基酯的交叉偶联反应制备芳基膦化合物(Eq. 17).该方法不需要添加碱和配体, 相转移催化剂四甲基氯化铵(TMACl)的加入对反应有很好的促进作用.乙醇作为溶剂是该反应的关键, 作者认为乙醇在该反应体系中并不单纯作为溶剂, 还起到了与芳基硼酸酯进行配位的作用.
3.
以Ar—N类化合物为芳基源构筑Ar—P键
经典的Sandmeyer反应是一种重氮官能团在亚铜盐的催化下被卤素或氰基所取代的反应, 随着合成化学的发展, 该反应已能实现将重氮官能团转化为羟基、磺酸盐等一系列官能化反应. 2010年Cacchi等[35 成功将芳基重氮盐应用于Michaelis-Arbuzov类型反应芳制备基膦化合物(Eq. 18).该方法以醋酸钯为催化剂, 以KI活化三烷氧基磷化合物, 以碳酸铯为碱, 80 ℃乙腈为溶剂下平稳进行.该反应具有良好的底物普适性, 二芳基膦氧与二环己基膦氧化合物同样适用于该方法.另外, 直接以芳基氨为起始原料, 通过加入亚硝酸叔丁酯与三氟化硼乙醚原位生成重氮盐, 再投入到反应中也能得到中等的产率.值得指出的是, 该反应对含氯、溴、氰基和酮等官能团的芳基重氮盐具有良好的官能团耐性.
近年来, 光诱导的反应广泛用于芳基重氮盐的偶联反应中. 2015年, Toste等[36 报道了一种金催化剂与光催化剂协同作用下芳基重氮盐的磷酸酯化反应(Eq. 19).该反应可以在温和的反应条件下进行(室温、无碱或无添加剂), 并显示出非常优异的底物适用性.同时, 该反应与已知的金催化的偶联反应相比避免了强氧化剂的使用.
2016年, 王剑波等[37 报道了一种基于Sandmeyer型转化的有效无金属膦酸脂化方法(Eq. 20), 该方法以易得的芳基胺化合物作为底物来合成芳基膦酸酯.这种无金属转化的反应采用温和的t
芳基肼类化合物是易得的重要化工原料, 2014年, 赵玉芬等[38 报道了一种Pd催化下芳基肼的磷酸酯化反应(Eq. 21).该反应以Pd(OAc)2 为催化剂, 以1, 3-双(二苯基膦)丙烷(dppp)为膦配体, 加入三氟乙酸来营造酸性环境, 在氧气条件下反应24 h.值得一提的是, 该反应具有良好的底物普适性, 在氧气作为氧化剂前提下能以良好至优秀的产率得到目标产物.
2014年, 邹建平等[39 也以芳基肼类化合物为芳基来源, 在金属铜催化下实现了芳基肼与亚磷酸三烷基酯的偶联反应.该方法以10 mol%的氧化铜为催化剂, 碳酸铯为碱, 乙腈为溶剂.该反应不需要添加额外的氧化剂和配体且具有良好的底物普适性, 大部分底物都以中等至良好的产率得到所需目标化合物.然而, 芳环上带有F等吸电子基团会导致产率下降, 芳基肼类化合物带有邻位取代基会有明显抑制反应进行, 尤其当两个邻位被乙基占据时会阻断该反应的进行(Eq. 22).
2018年, 於兵等[40 报道了在可见光的介导下, 同样以芳基肼类化合物为原料与亚磷酸三烷基酯反应制备芳基磷酸酯的方法.该方法以价廉且无毒的有机染料曙红B作为催化剂, 空气作为氧化剂, 乙腈作为溶剂, 添加50 mol%的有机碱1, 4-二氮杂二环[2.2.2]辛烷(DABCO), 在45 ℃以白色LED灯照射6 h就能得到相应的芳基磷酸酯产物, 反应体系无需添加金属且反应条件温和.该方法具有良好的底物普适性, 例如亚磷酸三丁酯、亚磷酸三异丙酯和亚磷酸三苯酯均能在该条件下得到相对应的目标产物(Eq. 23).值得一提的是, 采用该方法在克级放大实验中通过日光照射能以55%的收率得到目标产物(Eq. 24).
2019年, Protti等[41 开发了一种在可见光促进下芳基偶氮砜与三烷基和三芳基亚磷酸酯的磷酸酯化反应.该方法只需以乙腈为溶剂, 在氮气氛围下用24 W蓝色LED灯在室温下照射24 h就可以得到目标化合物.反应具有高效、无需额外添加金属和光催化剂等优点.同时, 该方法具有出色的官能团耐受性和底物普适性, 芳环上带有给电子基团和吸电子基团的芳基偶氮砜甚至杂环芳烃偶氮砜均适用于该方法(Eq. 25).
苯并三唑具有独特的结构, 它能脱氮以构建新的键, 在金属催化反应中被发展成为有用的合成子. 2018年, 夏吾炯等[42 采用苯并三唑为芳基源, 在反应体系中加入N , N' -二异丙基乙胺(DIPEA)和过氧化苯甲酰, 在光催化剂作用下进行可见光脱氮磷酰化反应, 获得了带不同取代基的芳基膦酸酯(Eq. 26).反应对芳环上带有各种类型取代基的苯并三唑底物均具有良好的官能团耐受性, 同时该方法在克级放大实验中也能以良好的产率得到目标产物.作者对机理进行了研究, 表明反应经历自由基过程且芳基自由基通过苯并三氮唑还原脱氮形成.
4.
以Ar—O和Ar—S类衍生物为芳基源构筑Ar—P键
作为亲电试剂的芳基三氟甲烷磺酸酯的性质类似于芳基卤素, Michaelis-Arbuzov反应同样能在镍催化的条件下进行.早在2011年, 张万斌等[43 首次报道了以氯化镍为催化剂的条件下, 芳基三氟甲烷磺酸酯与亚磷酸三乙酯进行反应构筑Ar—P键的方法(Eq. 27).该反应中碱金属卤代盐的加入能够有效促进反应的进行, 但是需要在很高的温度下完成.作者认为反应能够顺利进行的关键在于阴离子交换和Ar—P键形成过程、EtOTf较高的沸点以及其能够在较高的温度下催化亚磷酸三乙酯发生重排反应, 从而生成乙基膦酸二乙酯副产物.
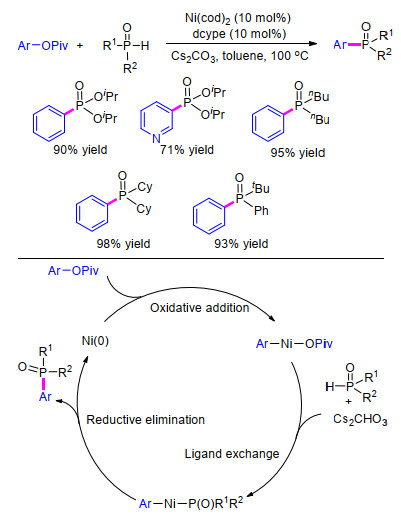
酚类化合物广泛存在于自然界中, 同时镍催化剂被广泛用于C—O键的活化断裂构筑新的化学键. 2015年, 韩立彪[44 等报道了首例在廉价金属镍催化下酚类衍生物通过C—O键的断裂来合成有机膦化合物的方法.作者选用新戊酸2-萘酯和二苯膦氧化物作为反应底物模型, 以Ni(COD)2 作为催化剂, 1, 2-双(二环己基磷基)-乙烷(dcype)为膦配体, 加入碳酸钾碱, 在二氧六环溶剂中80 ℃条件下反应18 h, 能以高产率得到目标化合物(Eq. 28).该方法反应条件温和, 具有良好的底物普适性和官能团耐受性, 二苯基氧膦和磷酸二烷氧基酯均可适用于该方应.
紧接着在2016年, 韩立彪等[45 进一步研究了镍催化下苯酚衍生物的磷酸脂化反应, 该方法通过Ar—O活化使用易得的苯酚衍生物作为芳基化试剂, 成功制备了各种芳基膦化合物, 各种类型的膦(磷)氧类化合物都能以良好至优秀的产率得到目标产物.在机理推测中, 作者认为首先Ni(0)对Ar—O进行氧化加成, 膦(磷)氧类化合物在碱性条件下进行配位交换形成中间体Ar—Ni— P(O)R1 R2 , 随后进行还原消除得到目标产物(Scheme 6
图式 6
2017年, 余达刚等[46 报道了室温下通过光催化/镍的催化体系使芳基C—O键发生断裂从而与各种类型磷氧化物发生磷酸化的反应(Eq. 29).该反应体系中邻菲罗啉作为配体, 1, 8-二氮杂二环十一碳-7-烯(DBU)作为有机碱, 在室温下光照反应12 h就能以良好至优异的产率得到芳基膦氧化物[51 .反应具有优异的官能团耐受性, 显示出广阔的底物范围, 各种类型的酚类衍生物均能与膦(磷)氧类化合物反应, 以良好至优秀的收率得到目标产物.
2014年, 王磊等[47 报道了一种Pd催化下芳基亚磺酸钠与亚磷酸脂类化合物的脱硫交叉氧化偶联反应, 该方法以Pd(PPh3 )2 Cl2 为催化剂, 以Ag2 CO3 为氧化剂, 以DMSO为溶剂, 添加四甲基氯化铵(TBAC)为相转移催化剂, 反应温度80 ℃下平稳进行.该反应具有高效性, 并有着良好的卤素官能团耐受性, 值得一提的是芳环上含溴代基团的芳基亚磺酸钠底物也能以优秀的产率得到目标产物(Eq. 30).不过, 当芳环上带有硝基时, 仅有痕量目标产物生成.
无独有偶, 在2014年肖军华等[48 也同样实现了Pd催化下芳基亚磺酸钠与亚磷酸脂类化合物脱硫交叉氧化偶联反应.不同的是, 该方法是以PdCl2 为催化剂, 以Ag2 CO3 为氧化剂, 在120 ℃温度下用微波加热10 min就可以高效地得到芳基膦化合物, 反应具有良好的底物普适性和官能团耐受性(Eq. 31).另外, 其它芳基磺酸金属盐, 例如芳基磺酸锌、芳基磺酸钾、芳基磺酸锂、芳基磺酸银和芳基磺酸铜等均适用于该方法得到目标产物.
2016年, 韩立彪等[49 报道了一种镍催化下C—S键断裂的磷酸脂化反应来构建Ar—P键的方法(Eq. 32), 该反应以Ni(cod)2 为催化剂, 加入叔丁醇钠提供较强的碱性环境, 以二氧六环为溶剂, 在100 ℃下反应18 h, 即以高产率得到芳基膦化合物.该反应只需要0.1 mol%的镍催化剂, 该方法具有良好的底物普适性并能提供良好至优秀的产率.推测的反应机理基本上和镍催化下以苯酚类衍生物构筑Ar—P键的反应机理类似:首先Ni(0)进行氧化加成, 接着Ar—S衍生物在碱性下进行配位交换得到反应的中间体, 中间体进行还原消除生成最终的目标产物.
5.
以Ar—H为芳基源构筑Ar—P键
从原子经济的角度来看, Ar—H键直接和膦酸类化合物通过偶联构筑Ar—P键是一种非常直接方便的策略.目前发展的主要有两种:过渡金属催化下C—H活化和自由基策略(包括磷自由基和芳基自由基阳离子).
5.1
过渡金属催化发生C—H活化形成Ar—P键
过渡金属催化下的C—H键直接官能团化法是有机合成化学的热点领域.近年来, 通过C—H键活化的策略合成芳基膦化合物的方法也有了一些进展. 2013年, 余金权等[50 首先报道了以氮杂环为导向基团, 钯催化的C—H键活化构筑Ar—P键的方法(Eq. 33).首先选取了两种含吡啶或噁唑啉的钯复合物与亚磷酸二异丙酯反应, 发现在对苯醌存在下, 100 ℃反应8 h就能得到目标产物.然而, 作者直接采用一锅法, 用二苯基吡啶与亚磷酸二异丙酯直接在上述条件下反应, 却不能得到产物.作者认为是亚磷酸酯与钯发生很强的螯合作用, 抑制了C—H活化过程.然后作者采用特殊的注射器, 使亚磷酸酯在100℃的条件下缓慢滴加至反应体系中, 就能够得到目标产物.采用优化的最佳反应条件, 各种类型的亚磷酸酯与二芳基膦氧化合物对该条件的兼容性均较好.在机理研究中, 分离出来的C—H活化的Pd中间体可以直接和亚磷酸酯在相应的反应条件下反应得到目标产物(Eq. 34).
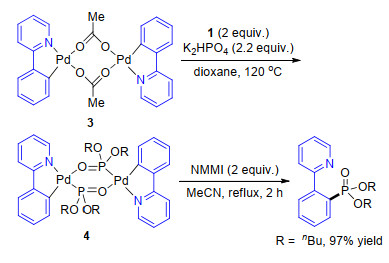
基于上述报道, 发现亚磷酸酯原料会与钯催化剂发生强烈的螯和作用抑制C—H活化过程, 同在2013年Murakami等[51 提出一种新的优化方案(Eq. 35), 即将亚磷酸酯与丙酮反应生成的磷酸脂化试剂1 , 再投入反应.磷酸脂化试剂1 在反应过程中能够缓慢释放出亚磷酸酯参与反应, 从而避免了其抑制C—H活化的过程.在对反应机理的探索过程中, 作者首先通过已知的文献合成了2-苯基吡啶与醋酸钯的络合物3 , 并将其投入到以K2 HPO4 为碱, 二氧六环为溶剂的反应中, 反应得到了2-苯基吡啶与亚磷酸酯的络合物4 , 并取得了其单晶结构.继续将络合物4 在2.0 equiv.的N -甲基马来酰亚胺(NMMI)乙腈溶液中回流2 h, 以97%的产率得到目标产物(Scheme 7
图式 7
2016年, Hong等[52 同样以亚磷酸酯与丙酮反应生成磷酸脂化试剂, 实现了铑催化的C—H键磷酸酯化反应.该反应以[Cp*RhCl2 ]2 与AgNTf2 共同作为催化体系, 碳酸银作为氧化剂, 在120 ℃下反应8 h即可得到目标产物.反应中导向基团可以为亚胺、吡啶、嘧啶、酰胺或其他氮杂环(Eq. 36).反应对反应底物具有较好的官能团耐受性, 同时各种亚磷酸酯衍生物在该反应中都能够顺利反应得到对应芳基膦酸酯类产物.
偶氮基团作为非常有效的导向基团常用于金属催化的邻位C—H活化反应中. 2014年, 王利民等[53 实现了钯催化下C—H活化的邻位膦酰化反应.该方法以醋酸钯作为催化剂, 加入醋酸锰作为氧化剂, 在醋酸为溶剂的体系下室温反应12 h能以中等至极好的收率得到邻二烷基膦酰基二芳基偶氮化合物(Eq. 37).该方法具有良好的底物普适性, 对于不对称取代的偶氮芳烃同样适用, 并且显示出了良好的区域选择性, 膦酰化反应位点主要发生在未取代的苯环上, 而不是发生在具有吸电子取代基的苯环上, 即富电子芳环比缺电子芳环更具反应活性.
很多钯催化的反应同样适用于廉价金属铜催化剂, 2014年, 余孝其等[54 以双氮的8-氨基喹啉为导向基团, 实现了铜催化的C—H键磷酸酯化反应(Eq. 38).以二价铜为催化剂具有非常突出的优越性, 不必考虑亚磷酸酯抑制C—H活化的过程, 所以该反应直接以亚磷酸酯为原料, 碳酸银与N -甲基吗啉-N -氧化物(NMO)共同为氧化剂, 在55 ℃下就能够得到对应的磷酸酯化产物.作者尝试了各种双配位原子导向基团, 该反应对于导向基团有非常严格的要求, 经过筛选发现只有8-氨基喹啉作为导向基团可以兼容该反应, 其他均不适用.
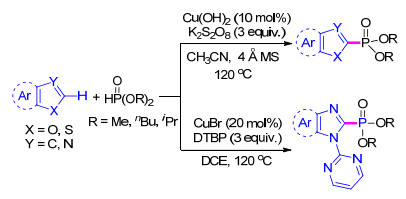
同样采用铜催化剂, Singh等[55 在2019年报道了亚磷酸二烷基酯分别与吡咯、噁唑、咪唑、苯并咪唑和吲哚的C(sp2 )—H脱氢交叉偶联制备相应磷酸化产物的方法.当以苯并噻唑、噁唑和苯并噁唑为底物时, 该方法以Cu(OH)2 为催化剂, 加入K2 S2 O8 作为氧化剂, 在乙腈为溶剂的120 ℃反应体系下反应可高产率地得到相应的产物(Scheme 8 N -嘧啶保护的苯并咪唑为底物时, 则需要在以溴化铜作为催化剂, 二叔丁基过氧化物(DTBP)作为氧化剂, 二氯乙烷为溶剂的体系下反应(Scheme 8
图式 8
5.2
磷自由基和芳基自由基阳离子策略形成Ar—P键
早在1985年, Effenberger等[56 报道了常温下, 硝酸银与过硫酸钠可以将亚磷酸酯氧化为亚磷酸酯自由基, 继而直接与芳香烃发生自由基加成, 得到芳基膦酸酯产物(Eq. 39).然而, 该方法存在着化学选择性不好、且只兼容带供电子基团底物等缺点.
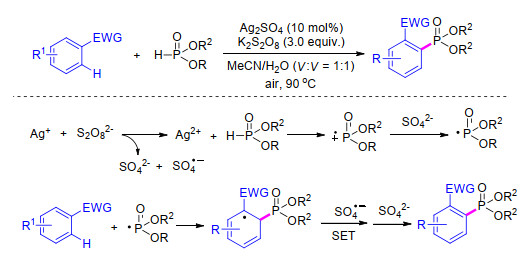
2013年, 成义祥等[57 在该工作的基础上, 发展了带有吸电子基团的芳基化合物的磷酸酯化反应.该反应以Ag2 SO4 为催化剂, 过硫酸钾为氧化剂, 在MeCN/H2 O (V :V =1:1)为溶剂的条件下, 实现了带吸电子基团芳基化合物的磷酸酯化.作者认为该反应中, 首先以过硫酸根将一价银离子氧化为二价银离子, 二价银离子能够将亚磷酸酯氧化为亚磷酸酯自由基, 该自由基与带有吸电子基团的芳香化合物发生自由基亲电加成反应, 最后通过单电子转移过程得到目标产物(Scheme 9
图式 9
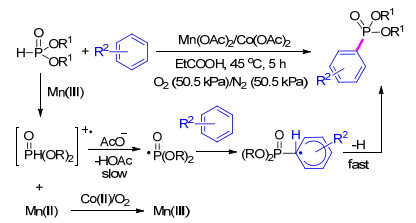
2006年, Ishii等[58 报道了一种直接以芳香烃为原料通过芳基自由基策略制备芳基膦酸酯的方法.该方法采用Mn(OAc)2 /Co(OAc)2 /O2 氧化还原偶联体系, 将亚磷酸酯氧化为亚磷酸酯自由基, 再与芳烃类化合物进行加成得到了芳基磷酸酯目标产物(Scheme 10
图式 10
2019年, Montchamp等[59 报道了锰催化下简单芳烃与H-磷酸盐的磷酸化反应.该方法以醋酸锰作为催化剂, 二氧化锰为氧化剂, 醋酸钠为碱, 醋酸为溶剂, 在70 ℃氮气氛围下反应即可得到所需目标产物(Eq. 40).值得注意的是, 该方法需要分批且缓慢加入次磷酸酯反应物.该反应具有良好的底物普适性, 各种芳族化合物和杂芳族化合物大多能得到目标产物, 但是该反应不适用于苯胺、硝基苯和苯酚等底物.反应机理推断中, 作者认为是自由基类型的亚磷酰基化过程.
光催化或光诱导方法是产生自由基非常有效的手段之一. 2017年, 雷爱文等[60 报道了一种二甲苯和亚磷酸三乙酯反应构筑C(sp2 )—P键的方法(Eq. 41), 该反应采用钴催化剂和光诱导下的光催化剂相结合的双催化体系, 在外部无氧化剂的条件下, 各种类型的C(sp2 )—H键, 包括芳烃、杂化芳烃和烯烃等化合物等都可以被有效地膦酸脂化, 反应产率良好及较高, 产物具有较好的化学选择性.作者认为芳基或烯烃自由基阳离子是Ar—H键膦酸脂化的关键中间体, 而亚磷酸三乙酯作为亲核试剂捕获芳基自由基阳离子, 从而得到目标产物.
同样采用芳基自由基阳离子策略, 雷爱文等[61 在2019报道了电化学合成法构筑Ar—P键的方法(Eq. 42).作者选择了2-苯基咪唑并[1, 2-a ]吡啶和亚磷酸三乙酯为底物, 碳棒和铂板分别作为阳极和阴极, n 4 NBF4 为电解质, 乙腈为溶剂, 反应在一个无隔膜电解槽内以4 mA电流下进行, 能够以70%的收率得到目标产物.作者认为2-苯基咪唑并[1, 2-a ]吡啶在阳极失去一个电子形成自由基阳离子, 紧接着与亚磷酸三乙酯进一步在阳极失去电子氧化脱氢, 最后脱烷基化形成目标化合物.值得一提的是该反应不需要任何外加氧化剂和金属催化剂, 而且底物具有普适性, 不仅对C(sp2 )—P键的构筑适用, 对于C(sp3 )—P键的构筑同样也适用.
6.
Ar—C类化合物通过C—C键断裂构筑Ar—P键
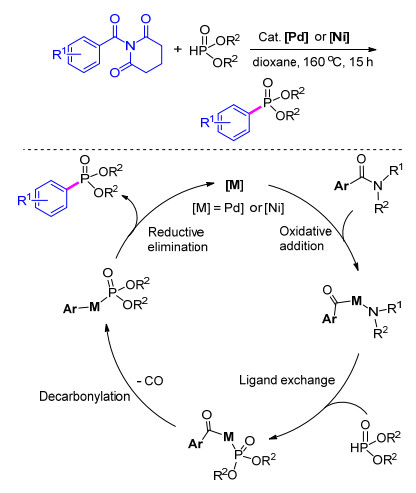
在过渡金属作用下对C—C键进行活化断裂从而形成新的化学键, 在近年来得到了广泛关注.其中, 芳基酰胺类化合物的脱羰后进行交叉偶联在构建新的化学键方面也有了较好的研究进展. 2017年, Szostak等[62 首次报道了使用钯或镍催化剂催化的芳基酰胺类化合物的脱酰胺膦酸脂化反应, 从而构筑Ar—P键得到芳基膦酸酯, 该方法具有良好的底物官能团适用性, 大多数产物的产率都很高.机理推测其主要经历了氧化加成、配位交换、脱羰和还原消除等过程, 最终得到目标产物(Scheme 11
图式 11
紧接着在2018年, Yamaguchi等[63 报道了一种镍催化下芳基酯类化合物与HP(O)R1 R2 化合物通过脱羰偶联反应.该反应仅需使用5~10 mol%的醋酸镍作为催化剂, 在使用3, 4-双(二环己基膦基)噻吩(dcypt)作为配体时能够得到较高的产率.反应具有良好的底物普适性, 各种类型的亚磷酸酯与二芳基膦氧化合物对该条件的兼容性均较好.特别值得指出的是, 许多杂环类芳香脂类化合物也能以中等偏上的产率得到目标产物(Eq. 43).
2019年, 韩立彪等[64 以工业上易得的芳基酰肼类化合物为原料, 通过脱羰与亚磷酸酯或二芳基磷氧发生偶联反应来构建Ar—P键(Eq. 44).该方法以醋酸钯为催化剂, 以双(二苯基膦)甲烷(dppm)作双膦配体, 以过硫酸钠为氧化剂, 苯磺酸作为添加剂, 100 ℃在二甲基亚砜和二氯乙烷混合溶剂下进行反应, 各种类型的芳基酰肼和磷酸类反应底物取得中等偏上的产率.反应体系中添加强质子酸的作用是使H-膦氧化合物形成膦酸盐, 从而抑制H-膦氧化合物的氧化与配位, 同时在双膦配体的共同作用下可以选择性活化碳碳键, 从而得到芳基膦化合物.
在各种脱碳偶联的策略中, 采用价廉且可广泛获得的芳基羧酸作为芳基源已引起广泛的关注. 2014年, 肖军华等[65 以邻硝基苯甲酸为原料, 通过脱羧与亚磷酸酯发生偶联反应来构建Ar—P键.该反应采用微波加热的方法, 反应时间短, 只需要10 min就可以得到目标产物.同时, 许多常见的官能团, 如硝基、甲基、氟和氯等基团在该方法中有着较好的相容性(Eq. 45).然而, 该反应产率普遍偏低, 而且底物局限于邻硝基芳基甲酸类化合物, 普通类型的芳基羧酸并不适用于该方法.
2019年, Szostak等[66 报道了Pd催化下直接使用普遍存在的芳基羧酸通过脱羧偶联反应构筑Ar—P键的方法(Eq. 46).该方法在5 mol% Pd(OAc)2 催化下, 以1, 4-双(二苯基膦)丁烷(dppb)为配体, 加入Et3 N和特戊酸酐(piv2 O)作为添加剂, 反应以良好的产率得到所需的芳基磷酸酯产物.机理研究中, 作者认为该方法基于羧酸与酸酐piv2 O酯化后原位活化并选择性将金属插入C(O)—Opiv并进行脱羰反应, 副产物为温和有机酸pivOH和一氧化碳.该方法具有非常好的官能团耐受性和底物普适性, 底物不再限制于特定的芳基羧酸, 甚至对于乙烯基羧酸同样适用.
7.
其他芳基源构筑Ar—P键
除了采用以上常见的芳基源来构建Ar—P键, 其他一些如苯炔、芳基铋、芳基硅和芳基锑等化合物也被发展作为芳基源来合成芳基膦酸酯类化合物.
2013年, Mhaske等[67 实现了苯炔参与的Michaelis Arbuzov类型反应(Eq. 47).该方法在常温条件下, 以邻(三甲基硅基)三氟甲磺酸芳基酯为苯炔的前驱体, 氟化铯为引发剂, 乙腈为溶剂, 在温和的反应条件下得到芳基膦酸酯、二芳基膦酸酯与三芳基氧膦化合物.反应具有良好的化学选择性, 膦酸酯基团主要出现在原三氟甲烷磺酸基的位点处. 2016年, 同样以邻(三甲基硅基)三氟甲磺酸芳基酯为苯炔的前驱体, Chen[68 和张万斌等[69 报道了苯炔与亚磷酸酯或二芳基膦氧作用得到芳基膦酸酯的方法(Eq. 48).反应在碳酸铯为碱, 氟化铯为引发剂的条件下, 均能够以中等以上的产率得到目标产物, 且具有良好的化学选择性.
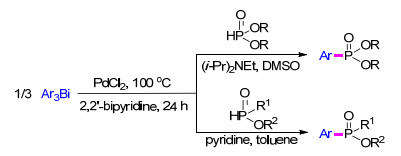
有机铋化合物具有无毒、易获得等特点, 并在有机合成中占有特殊地位. 2014年, 赵玉芬等[70 报道了首例以三芳基铋为芳基源与膦氧化合物通过交叉偶联形成芳基膦化合物的方法.该方法以二氯化钯作催化剂, 2, 2-联吡啶作双氮配体, 以二异丙基乙胺或吡啶提供碱性环境, 100 ℃在甲苯或二甲基亚砜溶剂下平稳进行(Scheme 12
图式 12
芳基硅烷因具有无毒、高稳定性、环境友好及易于合成等优点而被广泛应用于交叉偶联反应中.近年来, 以芳基硅烷作为芳基来源在C—C键构筑方面也取得了很多不错的研究进展. 2017年, 我们课题组[71 报道了首例芳基硅烷与亚磷酸脂通过氧化交叉偶联形成芳基膦化合物的方法.该反应在钯催化体系下, 加入碳酸银作为氧化剂, 以氟化钾作为氟源, 80 ℃二甲基甲酰胺溶剂下平稳反应12 h.该反应条件温和, 在空气中就可以反应, 而且对底物具有较好的普适性和官能团耐受性.比较遗憾的是, 当把二芳基膦氧化合物作为偶联底物时, 基本上得不到目标产物(Eq. 49).
2015年, Yasuike等[72 报道了Pd催化下五价有机锑化合物三芳基锑二乙酸酯与亚磷酸酯的磷酸酯化反应.该方法条件温和, 无需使用任何碱就可以使三芳基锑二乙酸酯与亚磷酸二烷基氢酯反应得到相对应的芳基磷酸酯产物.该反应在亚磷酸酯类底物的拓展方面显示出良好的普适性, 对于亚磷酸二异丙酯、亚磷酸二甲酯、亚磷酸二乙酯、亚磷酸二异丁酯都能高收率地得到所需目标产物(Eq. 50).
另外, 一些特定底物通过环化反应也可构建Ar—P键. 2016年, 江焕峰等[73 报道了一种Cs2 CO3 促进的烯-炔-酮与亚磷酸脂的串联反应构建呋喃类膦化合物(Eq. 51).该反应条件温和, 无需添加金属催化剂且反应速度较快, 具有良好的底物普适性.当Cs2 CO3 的添加量提高到1.5 equiv.时, 二芳基膦氧化合物也适合该环化反应, 并能以46%的产率得到目标产物.
8.
结论与展望
近年来, 各种通过构筑Ar—P键来合成芳基膦酸酯类化合物的方法得到了快速发展, 越来越多的芳基源被开发出来.反应条件从传统的高温、怕水怕氧和添加复杂配体等发展到简单快速、高效环保, 实现了巨大的飞跃, 也为有机膦化合物在光电、阻燃材料以及药物化学等研究领域的广泛应用奠定了良好基础.近几十年来过渡金属催化的Ar—P键构筑已经被广泛深入研究, 也取得了很多不错的成绩.最近, 环境友好的光化学和电化学合成方法在芳基膦酸酯类化合物的合成上开始得到快速发展, 相信这方面的研究会带给我们意想不到的收获.



 下载:
下载:




















































 下载:
下载:













