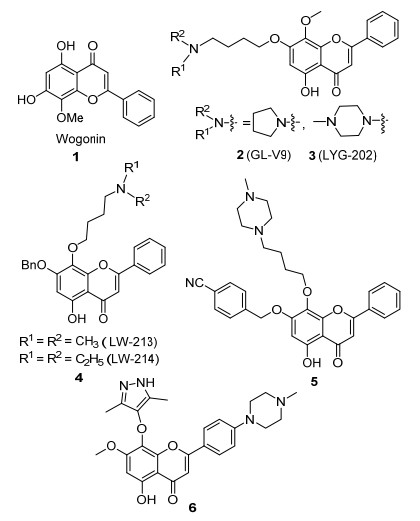
图 1.
汉黄芩素及代表性的衍生物
Figure 1.
Structure of Wogonin and representative derivatives

黄酮是一类具有广泛药理活性的重要天然产物[1], 汉黄芩素(Wogonin, 1)是从黄芩根中分离得到的一种黄酮类化合物(图 1)[2], 具有抗氧化[3]、抗病毒[4]、抗炎[5]及神经保护[6]等多种药理活性.尤其是近年来研究发现, 汉黄芩素及其衍生物对肿瘤细胞具有独特的杀伤机制, 可以在不影响正常细胞的情况下, 直接诱导癌细胞凋亡[7].目前, 汉黄芩素作为化学I类抗肿瘤新药即进入临床研究阶段[8].同时, 对于汉黄芩素的结构优化工作也开始受到重视.李志裕课题组[9]在汉黄芩素的7-位引入取代基得到了化合物2 (GL-V9)和3 (LYG-202) (图 1), 它们对乳腺癌、肝癌和胃癌的抑制活性均优于汉黄芩素, 其中化合物3非常值得作为抗肿瘤候选药物进行深入研究[10].在此研究基础上, 该研究组[11~13]又通过在8位引入含氨基的侧链合成了具有较好水溶性和成药性的LW-系列化合物4; Wang等[14]对汉黄芩素A环的7-和8-位同时进行结构修饰, 得到的化合物5对多种肿瘤细胞株都具有良好的抗肿瘤活性, 是潜在的CDK9抑制剂; 对化合物5的8位和B环进一步优化, 得到了成药性更好的CDK9抑制剂6[15].
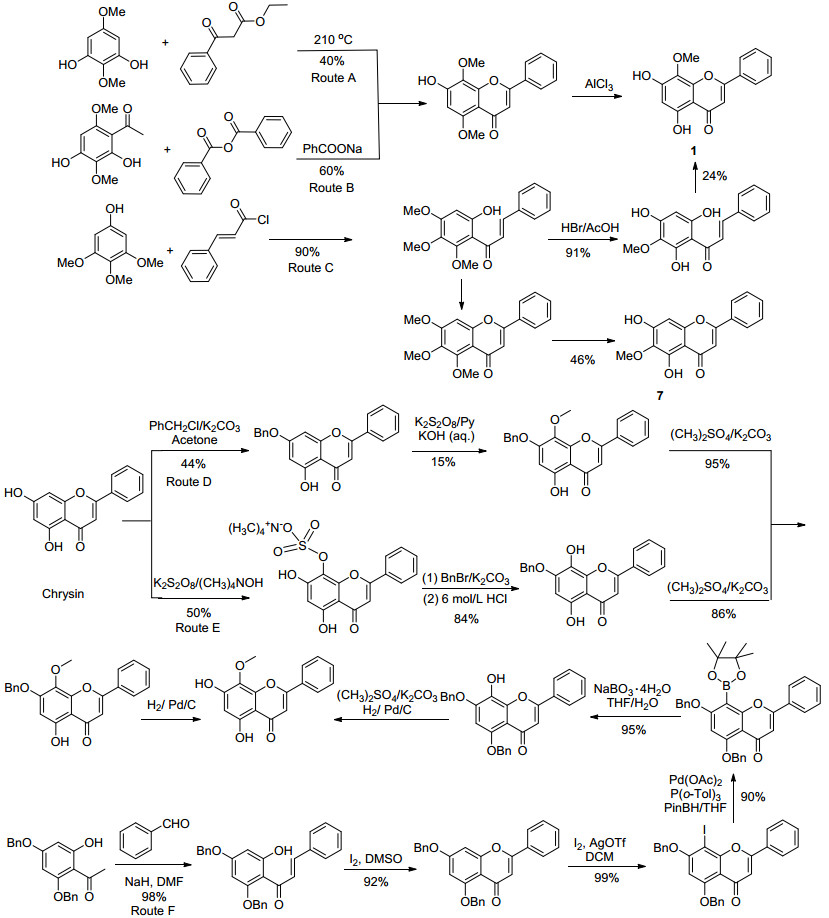
汉黄芩素的大量制备对于其结构优化至关重要.然而, 目前汉黄芩素主要还是依靠从黄芩中提取得到, 由于其含量很低(0.01%~0.3%)[16], 存在提取工艺繁琐、质量不稳定等问题, 远满足不了需求[17].汉黄芩素的全合成研究已取得了显著进展, 目前, 主要有两类合成方法(Scheme 1).第一类合成方法是从简单的原料出发, 在构建黄酮骨架的同时引入官能团或潜在的官能团.如路线A[18]和B[19], 分别以2, 5-二甲氧基间苯二酚或2, 4-二羟基-3, 6-二甲氧基苯乙酮为原料, 只需两步反应即可得到汉黄芩素, 但是存在原料不易得到、反应条件苛刻及收率低等问题. 2003年, Huang等[20]报道了路线C, 以3, 4, 5-三甲氧基苯酚为原料, 通过Claisen-Schmidt缩合反应生成查尔酮, 进而关环得到汉黄芩素.该路线虽然起始原料便宜易得, 但是关环反应有两种方向, 生成的是汉黄芩素(1)和千层纸素(7)的混合物, 分离纯化困难, 收率低(总收率19%).另一类合成方法是选择黄酮类化合物白杨素为起始原料, 通过不同的方法在8位引入甲氧基得到汉黄芩素.路线D[21]和E[22]是以Elbs氧化为关键反应, 该反应需要使用大量的过硫酸钾, 反应条件苛刻, 操作复杂, 收率很低; 袁虎等[23]借鉴路线D和E的思路, 由5, 7-二苄氧基白杨素经8位硼酸酯化、氧化引入8-位羟基的方法合成了汉黄芩素(路线F), 5, 7-二苄氧基白杨素是由2, 4-二苄氧基-6-羟基苯乙酮经两步反应制得.路线F避免了路线D、E采用Elbs氧化反应的不足, 每步反应收率都比在90%以上, 但是存在原料不易得到、路线长、试剂昂贵, 如钯试剂和银试剂的使用等问题.因此, 仍然很有必要研发出一种适合于大量制备汉黄芩素的有效方法.
比较已有的合成路线, 路线F最值得借鉴, 如果能够解决5, 7-二苄氧基白杨素的来源问题, 避免或减少贵重试剂的使用, 将有可能成为一条大量制备汉黄芩素的高效合成路线.路线D中, 在由白杨素制备7-苄基保护的白杨素时[21], 收率低的主要原因是易形成5, 7-二苄氧基白杨素, 那么, 若提高苄基化试剂的用量, 改变反应条件有可能使5, 7-二苄氧基白杨素成为主产物; 另外, 查阅文献发现, 2005年Seijas等[24]报道, 在无溶剂微波条件下, 间三苯酚与苯甲酰乙酸乙酯经缩合、合环“两步一锅”法反应, 可高收率地合成白杨素类衍生物, 尽管微波条件不适合于放大制备, 但是通过优化反应条件, 有可能避免微波的使用.基于上述分析, 我们设计了如Scheme 2所示的新合成路线.
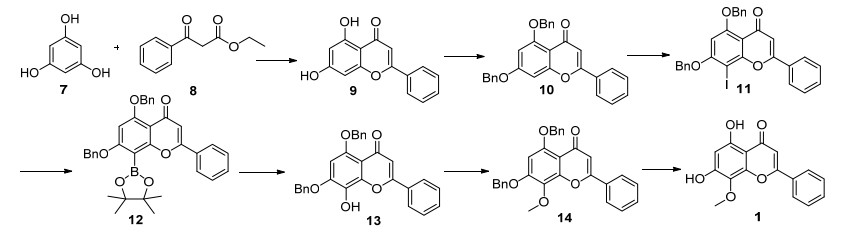
白杨素(9)的合成主要是参考Seijas[24]的方法.为了避免微波的使用, 以均三苯酚和苯甲酰乙酸乙酯为原料, 筛选了不同的碱、溶剂和温度对反应的影响.结果发现, 以4-二甲氨基吡啶(DMAP)为催化剂, 在无溶剂条件下, 200 ℃反应4 h, 可以87%的收率得到白杨素(9).
参照路线D由白杨素制备7-苄基保护的白杨素的方法, 当溴苄与白杨素的物质的量之比提高到1:2.4, 以碳酸钾为碱, 丙酮为溶剂, 在Ar保护下, 60 ℃反应15 h, 可以96%的收率得到5, 7-二苄氧基白杨素.
在路线F中, 袁虎等[23]是通过AgOTf/I2体系进行碘代反应完成中间体10的8-碘代反应的.为降低成本和减少污染, 希望能避免三氟甲磺酸银和碘的使用.为解决这一问题, 考察了不同催化剂和碘化剂对反应的影响.最终发现在不使用任何催化剂的情况下, 以N-碘代丁二酰亚胺(NIS)为碘化试剂[25], 在Ar保护下, 70 ℃反应18 h, 可以95%的收率得到5, 7-二苄氧基-8-碘-2-苯基-4H-1-苯并呋喃-4-酮(11).
曾尝试由中间体11直接水解得到5, 7-二苄氧基-8-羟基-2-苯基-4H-1-苯并呋喃-4-酮(13)[26], 但是尝试多种反应条件反应, 结果并不理想.最后, 借鉴路线F的方法, 先经硼酸酯化反应, 得到5, 7-二苄氧基-8-频哪醇硼酸酯-2-苯基-4H-1-苯并呋喃-4-酮(12), 然后, 氧化水解得到5, 7-二苄氧基-8-羟基-2-苯基-4H-1-苯并呋喃-4-酮(13).值得注意的是, 路线F中由中间体硼酸酯12制备13时, 使用了大量氧化剂过硼酸钠, 反应不易控制, 不适合于大规模生产.我们参照文献[27], 以乙醇-水为溶剂, NaOH为碱, 12和盐酸羟胺室温反应24 h, 可以94%收率得到关键中间体13.
根据文献[22]方法, 中间体13经8-甲基化和脱苄基, 顺利得到目标化合物汉黄芩素(1), 经核磁和HRMS确证, 其结构与天然产物图谱数据一致[15].
以便宜易得的间苯三酚为起始原料, 经过七步反应, 以58%的总收率成功地合成了汉黄芩素.解决了已有合成工艺中原料难得、试剂昂贵、分离纯化困难等问题.每步反应的产物均不需要柱层析纯化, 具有操作简单、反应条件温和、收率高等优点, 为汉黄芩素的大量制备奠定了基础.
核磁共振仪为Brucker Avance-400型, 四甲基硅烷(TMS)为内参; 质谱仪为LTQ-Orbitrap XL型(Thermo Fisher), 离子源采用电喷雾离子化(ESI); 薄层色谱(TLC)使用Silica Gel GF254(购自青岛海洋化工厂).所有试剂均为国产或进口分析纯, 不经处理直接使用.无水溶剂用常规方法干燥处理.
取间苯三酚(1.26 g, 10.0 mmol)置于烧瓶中, 依次加入苯甲酰乙酸乙酯(5.0 mL, 29.0 mmol)、DMAP (0.036 g, 0.3 mmol), 在Ar保护下, 升温至200 ℃搅拌反应4 h, TLC(石油醚/乙酸乙酯, V:V=2:1)检测, 原料反应完全, 停止反应.反应冷却至室温, 缓慢加水(40 mL)淬灭反应, 静置30 min, 析出固体, 抽虑, 滤饼依次用30 mL水和10 mL乙酸乙酯洗.滤饼干燥得粗品, 用四氢呋喃(THF)重结晶, 得到2.21 g黄色固体, 收率87%. m.p. 165~167 ℃(文献值[23]: 159~162 ℃); 1H NMR (400 MHz, DMSO-d6) δ: 12.83 (s, 1H), 10.9 (s, 1H), 8.08~8.05 (m, 2H), 7.60~7.57 (m, 3H), 6.91 (d, J=4.0 Hz, 1H), 6.53 (d, J=4.0 Hz, 1H), 6.22 (d, J=4.0 Hz, 1H).
将中间体9 (2.16 g, 8.5 mmol)溶于40 mL丙酮, 加入碳酸钾(5.7 g, 40.8 mmol), 再缓慢加入溴苄(3.49 g, 20.4 mmol), Ar保护, 升温至60 ℃, 反应15 h, TLC(石油醚/乙酸乙酯, V:V=1:1)检测, 原料反应完全停止反应.反应冷却至室温, 蒸除反应液中大部分丙酮, 加入水60 mL, 搅拌30 min析出固体, 固体过滤, 干燥后的粗品用THF重结晶, 得3.54 g (8.16 mmol)黄色固体, 收率96%. m.p. 163~164 ℃(文献值[25]: 165~167 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.91~7.89 (m, 2H), 7.65~7.64 (m, 2H), 7.54~7.50 (m, 3H), 7.46~7.44 (m, 4H), 7.43~7.39 (m, 3H), 7.34~7.32 (m, 1H), 6.70 (d, J=4.0 Hz, 2H), 6.54 (d, J=4.0 Hz, 1H), 5.21 (s, 2H), 5.16 (s, 2H).
将中间体10 (3.52 g, 8.10 mmol)溶于20 mL N, N-二甲基甲酰胺(DMF), 加入NIS(2.19 g, 9.72 mmol), Ar保护, 升温至70 ℃反应18 h, TLC(石油醚/乙酸乙酯, V:V=2:1)检测, 原料反应完全停止反应.反应冷却至室温, 向反应液加入饱和碳酸氢钠溶液20 mL, 析出固体, 固体过滤, 干燥后的粗品用二氯甲烷(DCM)重结晶, 得4.35 g (7.77 mmol)类白色固体, 收率95%. m.p. 178~180 ℃(文献值[23]: 174~177 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.10~8.01 (m, 2H), 7.59~7.55 (m, 4H), 7.54~7.39 (m, 9H), 6.75 (s, 1H), 6.50 (s, 1H), 5.26 (s, 2H), 5.21 (s, 2H).
将中间体11 (4.31 g, 7.70 mmol)溶于无水THF (30 mL), 依次加入Pd(OAc)2 (87 mg, 0.385 mmol)、P(o-Tol)3 (235 mg, 0.77 mmol)和三乙胺(TEA) (3.2 mL, 23.0 mmol), 再缓慢加入PinBH (1.08 g, 8.47 mmol), Ar保护, 升温至60 ℃搅拌6 h, TLC(石油醚/乙酸乙酯, V:V=2:1)检测, 原料反应完全停止反应.反应冷却至室温, 加水20 mL淬灭反应, 加入乙酸乙酯萃取(50 mL×3), 合并有机相, 用饱和食盐水洗, 无水硫酸钠干燥, 蒸除溶剂, 用丙酮重结晶, 得3.75 g (6.70 mmol)白色固体产物, 收率87%. m.p. 200~201 ℃(文献值[23]: 201~203 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.06~8.02 (m, 2H), 7.62~7.60 (m, 2H), 7.51~7.49 (m, 5H), 7.43~7.32 (m, 6H), 6.68 (s, 1H), 6.41 (s, 1H), 5.32 (s, 2H), 5.12 (s, 2H), 1.41 (s, 12H).
将中间体12 (3.64 g, 6.50 mmol)溶于乙醇(30 mL), 加入NH2OH·HCl (904 mg, 13.0 mmol)、3 mol/L NaOH水溶液6 mL, 室温搅拌反应24 h, TLC(石油醚/乙酸乙酯, V:V=2:1)检测, 原料反应完全停止反应.用2 mol/L HCl调pH于中性, 析出白色固体, 抽虑, 固体干燥, 粗品用THF重结晶, 得2.75 g (6.11 mmol)白色固体, 收率94%. m.p. 221~222 ℃(文献值[23]: 223~225 ℃); 1H NMR (400 MHz, CDCl3) δ: 8.00~7.95 (m, 2H), 7.52~7.54 (m, 4H), 7.48~7.28 (m, 9H), 6.71 (s, 1H), 6.69 (s, 1H), 6.09 (s, 1H), 5.19 (s, 2H), 5.16 (s, 2H).
将中间体12 (2.74 g, 6.10 mmol)溶于丙酮(30 mL), 加入碳酸钾(2.11 g, 15.25 mmol), 再缓慢加入硫酸二甲酯(1.54 g, 12.2 mmol), 升温至60 ℃反应约8 h. TLC(石油醚/乙酸乙酯, V:V=2:1)检测, 原料反应完全停止反应.反应冷却至室温, 蒸除反应液丙酮, 加20 mL水稀释, 搅拌30 min, 析出固体, 粗品用乙酸乙酯重结晶得到2.69 g (5.80 mmol)白色固体, 收率95%.将甲基化中间体(2.52 g, 5.50 mmol)溶于30 mL甲醇中, 加入甲醇溶解的10%钯碳(250 mg), 进行氢化反应, 室温搅拌至原料消耗完全, 过滤, 浓缩, 粗品用乙醇重结晶得1.48 g (5.23 mmol)浅黄色固体产物, 收率95%, m.p. 202~204 ℃(文献值[23]: 205~207 ℃); 1H NMR (400 MHz, DMSO-d6) δ: 12.83 (s, 1H), 10.9 (s, 1H), 8.01 (t, J=4.0 Hz, 2H), 7.62 (d, J=4.0 Hz, 2H), 7.00 (s, 1H), 6.33 (s, 1H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 182.4,163.4,160.8,157.7,155.2,132.0,130.9,129.2,126.8,125.9,105.9,105.4, 98.7, 62.0; HRMS (ESI) calcd for C16H13O5 [M+H]+ 285.0685, found 285.0671.
辅助材料(Supporting Information) 白杨素(9)和中间体11的合成条件、化合物1、9~13的1H NMR和13C NMR谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
王哲元, 张璐, 张有成, 西部中医药, 2018, 31, 138. http://www.cnki.com.cn/Article/CJFDTotal-GSZY201805043.htmWang, Y. Z.; Zhang, L.; Zhang, Y. C. Western J. Tradit. Chin. Med. 2018, 31, 138(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-GSZY201805043.htm
任晓东, 符伟, 张晓芸, 胡珀, 汪嵘, 李志裕, 中国新药杂志, 2011, 20, 777. http://www.cnki.com.cn/Article/CJFDTotal-ZXYZ201109005.htmRen, X. D.; Fu, W.; Zhang, X. Y.; Hu, P.; Wang, Z.; Li, Z. Y. J. New Drugs 2011, 20, 777(in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-ZXYZ201109005.htm
(a) Cho, J.; Lee, H. K. Eur. J. Pharmacol. 2004, 485, 105.
(b) Pang, Y. Y.; Qin, X. M.; Du, G. H. Zhou, Y. Z. Chin. Tradit. Herb. Drugs 2019, 50, 3207(in Chinese).
(庞溢媛, 秦雪梅, 杜冠华, 周玉枝, 中草药, 2019, 50, 3207.)
(a) Wu, Y.; Jin, Y. Z.; Zhang, S.; Yan, Y. J.; Zhang, J.; Hao, J. Chin. J. Exp. Tradit. Med. Formulae 2012, 18, 161(in Chinese).
(吴莹, 金叶智, 张舒, 闫玥捷, 张津, 郝钰, 中国实验方剂学杂志, 2012, 18, 161.)
(b) Wu, Y.; Jin, Y. Z.; Wu, J.; Yu, X. F.; Hao, J. J. Beijing Univ. Trad. Chin. Med. 2010, 33, 541(in Chinese).
(吴莹, 金叶智, 吴珺, 于雪飞, 郝钰, 北京中医药大学学报, 2010, 33, 541.)
(a) Lee, H.; Kim, H.; Kim, S. Y.; Noh, H. S.; Kang, S. S.; Cho, G. J.; Choi, W. S.; Suk, A. K. FASEB J. 2003, 17, 1943.
(b) Wang, P. M.; Chen, W.; Meng, X. L. Chin. J. Exp. Tradit. Med. Formulae 2016, 22, 193(in Chinese).
(王沛明, 陈文, 孟宪丽, 中国实验方剂学杂志, 2016, 22, 193.)
(a) Ling, H.; Hua, M. Lishizhen Med. Mater. Med. Res. 2014, 25, 168(in Chinese).
(凌芳, 华梅, 时珍国医国药, 2014, 25, 168.)
(b) Lee, S. O.; Jeong, Y. J.; Yu, M. H.; Lee, J. W.; Hwangbo, M. H.; Kim, C. H.; Lee, I. S. Biochem. Biophys. Res. Commun. 2006, 351, 122.
(c) Chang, Y. L.; Shen, J. J.; Wung, B. S. Mol. Pharmacol. 2001, 60, 512.
(d) Huang, H. C.; Wang, H. R.; Hsieh, L. M. Eur. J. Pharmacol. 1994, 251, 93.
(e) Huang, Z. D.; Liu, Y. M.; Kong, C. H.; Xie, X. R.; Fu, B. B. Int. J. Cerebrovasc. Dis. 2014, 22, 660(in Chinese).
(黄正德, 刘煜敏, 孔朝红, 解晓睿, 付蓓蓓, 国际脑血管病杂志, 2014, 22, 660.)
(a) Yan, J. Y.; Chen, B. A. Lishizhen Med. Mater. Med. Res. 2012, 23, 2210(in Chinese).
(严静怡, 陈宝安, 时珍国医国药, 2012, 23, 2210.)
(b) Zhong, N.; Li, Q.; Feng, Y. H.; Gai, X.; Hao, L. Clin. Misdiagn. Misther. 2019, 32, 112(in Chinese).
(种楠, 李勤, 冯艳红, 晁旭, 郝蕾, 临床误诊误治, 2019, 32, 112.)
(c) Polier, G.; Ding, J.; Konkimalla, B. V.; Eick, D.; Ribeiro, N.; Kohler, R.; Giaisi, M.; Efferth, T.; Desaubry, L.; Krammer, P. H.; Liweber, M. Cell Death Discovery 2011, 2,182.
(a) Xing, H.; Ning, C.; Kong, D. X.; Cai, H.; Zhou, S. Y.; Ren, C.; Kong, Y.; Cheng, J. X.; Chen, X. J. Chin. J. Clin. Pharmacol. Ther. 2019, 24, 889(in Chinese).
(邢晗, 宁晨, 孔德璇, 蔡卉, 周时雨, 任畅, 孔颖, 程钰洁, 陈西敬, 中国临床药理学与治疗学, 2019, 24, 889.)
(b) Talbi, A.; Zhao, D.; Li, Q. W.; Li, J. X.; Fan, A. L.; Yang, W.; Han, X.; Chen, X. J. Molecules 2014, 19, 5538.
(a) Li, G. J.; Zhao, K.; Yao, Y. Y.; Shen, L.; Guo, Q. L. J Chin Pharm Univ. 2015, 46, 464(in Chinese).
(李国君, 赵凯, 姚彧远, 申乐, 郭青龙, 中国药科大学学报, 2015, 46, 464.)
(b) Li, L. W.; Lu, N.; Dai, Q. S.; Wei, L. B.; Zhao, Q.; Li, Z. Y.; He, Q. H.; Dai, Y.; Guo, Q. L. J. Ethnopharmacol. 2011, 13, 670.
Zhao, L, ; Miao, H. C.; Li, W. J.; Sun, Y.; Huang, S. L.; Li, Z. Y.; Guo, Q. L. Mol. Carcinog. 2016, 55, 778. doi: 10.1002/mc.22321
Pan, D.; Li, W.; Miao, H.; Yao, J.; Li, Z. Y.; Wei, L. B.; Zhao, L.; Guo, Q. L. Biochem. Pharmacol. 2014, 87, 598. doi: 10.1016/j.bcp.2013.12.010
Zeng, S.; Liu, W.; Nie, F. F.; Zhao, Q.; Rong, J. J.; Wang, J.; Tao, L.; Qi, Q.; Lu, N.; Li, Z. Y.; Guo, Q. L. Biochem. Biophys. Res. Commun. 2009, 385, 551. doi: 10.1016/j.bbrc.2009.05.099
Chen, Y.; Lu, N.; Ling, Y.; Wang, L.; You, Q. D.; Li, Z. Y.; Guo, Q. L. J. Pharmacol. Sci. 2010, 112, 37. doi: 10.1254/jphs.09213FP
Wang, J. B.; Ge, R. L.; Qiu, X. Q.; Xu, X.; Wei, L. B.; Li, Z. Y.; Bian, J. L. Eur. J. Med. Chem. 2017, 140, 421. doi: 10.1016/j.ejmech.2017.09.046
Wang, J. B.; Li, T. H.; Zhao, T. T.; Wu, T. Z.; Ding, H.; Li, Z. Y. Eur. J. Med. Chem. 2019, 178,782. doi: 10.1016/j.ejmech.2019.06.024
柳文媛, 林力行, 张正行, 中国药科大学学报, 1997, 28, 162. doi: 10.3321/j.issn:1000-5048.1997.03.009Liu, W. Y.; Lin, L. X.; Zhang, Z. X. J. Chin. Pharm. Univ. 1997, 28, 162(in Chinese). doi: 10.3321/j.issn:1000-5048.1997.03.009
李文娟, 潘馨, 陈晓兰, 陈维中, 廖联明, 亚太传统医药, 2014, 10, 33.Li, W. J.; Pan, X.; Chen, X. L.; Chen, W. Z.; Liao, L. M. Asia-Pacific Tradit. Med. 2014, 10, 33(in Chinese).
Shah, R. C.; Mehta, C. R.; Wheeler, T. S. J. Chem. Soc. 1938, 1555. doi: 10.1039/jr9380001555
Rivailie, P.; Mentzer, C. J. Chem. Soc. 1969, 268, 2213.
Huang, W. H.; Chien, P. Y.; Yang, C. H.; Lee, A. R. Chem. Pharm. Bull. 2003, 51, 339. doi: 10.1248/cpb.51.339
Rao, K. V.; Seshadri, T. R.; Sc, F. A. Proc.-Indian Acad. Sci., Sect. A 1947, 26, 13. doi: 10.1007/BF03170943
Li, T. H.; Weng, T. W.; Wang, J. B.; Wei, J. H.; Li, Z. Y. Org. Process Res. Dev. 2017, 21, 171. doi: 10.1021/acs.oprd.6b00249
袁虎, 叶霁, 陈浩, 赵增, 李玉柱, 张卫东, 孙青龑, 有机化学, 2016, 36, 2960. doi: 10.6023/cjoc201604057Yuan, H.; Ye, J.; Chen, H.; Zhao, Z.; Li, Y. Z.; Zhang, W. D.; Sun, Q. Y. Chin. J. Org. Chem. 2016, 36, 2960(in Chinese). doi: 10.6023/cjoc201604057
Seijas, J. A.; Vazquez-Tato, M. P.; Carballido-Reboredo, R. J. Org. Chem. 2005, 70, 2855. doi: 10.1021/jo048685z
Caldwel, S. T.; Petersson, H. M.; Farrugia, L. J.; Mullen, W.; Crozier, A.; Hartley, R. C. Tetrahedron 2006, 62, 7257. doi: 10.1016/j.tet.2006.05.046
(a) Kabalka, G. W.; Mereddy, A. R. Tetrahedron Lett. 2005, 46, 6315. Miao(b) Vedejs, E.; Diver, S. T. J. Am. Chem. Soc. 1993, 115, 3358.
Lu, K.; Chu, J.; Wang, H. M.; Fu, X. L.; Quan, D. W.; Ding, H. X.; Yao, Q. W.; Yu, P. Tetrahedron Lett. 2013, 54, 6345. doi: 10.1016/j.tetlet.2013.09.051
Liger, F.; Pellet-Rostaing, S.; Popowycz, F.; Lemaire, M. Tetrahedron Lett. 2011, 52, 3736. doi: 10.1016/j.tetlet.2011.05.043
Li, Z. Y.; Cao, X.; Wang, X.; Guo, Q. L.; You, Q. D. Org. Prep. Proced. Int. 2009, 41, 327. doi: 10.1080/00304940903102739

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:

