Figure 1.
Chemical structures of thiosemicarbazone-containing anticancer agents
Camphor-Based Thiosemicarbazone Analogues Induced G2 Cell Cycle Arrest and Apoptosis via Reactive Oxygen Species (ROS)-Mediated Mitochondrial Pathway in Human Breast Cancer Cells
Yan Zhang , Yunyun Wang , Yuxun Zhao , Chenglong Zhang , Wen Gu , Zhonglong Wang , Yongqiang Zhu , Shifa Wang

Cancer is one of the greatest threatens to human health all over the world, accounting for 9.75 million deaths in 2015.[1] Lung cancer is the world's leading cause of death, and breast cancer ranks first in the incidence of malignant tumors among women.[2] There are many chemotherapeutic drugs for treating a variety of cancers, such as cisplatin, etoposide, chlorambucil, doxorubicin and so on, but these anticancer drugs failed to inhibit the growth of cancer cells completely and are still facing the serious side effects. Hence, there is an urgent need to seek for effective anticancer agents with low cytotoxicity.
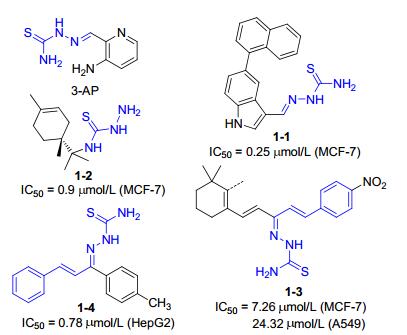
Thiosemicarbazones possess a remarkable pharmacological profile, and occupy a prominent position in pharmaceutical chemistry on account of their broad biological activity such as antiparasitic, [3-5] antibacterial, [6-7] antiviral[8-9] and anticancer.[10-12] Currently, the application of thiosemicarbazones in cancer therapy is being most extensively investigated. In 2007, de Oliveira et al.[13] reported a series of thiosemicarbazone derivatives, as well as their in vitro antiproliferative activity against eight human tumor cell lines, and compound 1-1 (Figure 1) was the most promising against michigan cancer foundation-7 (MCF-7) cell (IC50=0.25 µmol•L-1). In addition, R-(+)-limonene- based thiosemicarbazones were designed to be evaluated for antitumor activity, and compound 1-2 showed low value of IC50 (0.9 µmol•L-1) against MCF-7 cell.[14] Among the thiosemicarbazone-based anticancer agents, triapine (3-AP) is the most promising molecule that has entered Phase II clinical trial.[15] In view of the previously reported studies, those anticancer candidates based on thiosemicarbazone are associated with several factors including cell cycle arrest, [16] the disruption of the cellular redox homeostasis, [17] the chelation with important metal ions[18] and so on. In spite of this, there is still big gap in completely understanding the anticancer mechanism of thiosemicarbazone analogues, and it worth putting more effort into figuring out them.
Natural products, as raw materials, have widespreadly applied in the field of medicine on ground of their nice biocompatibility, green sourcing and low toxicity. Terpenoid, a large and diverse class of natural products, is the most promising source for the synthesis of a variety of structurally diverse compounds.[19] In recent years, our research group has been devoting to the synthesis of the derivatives from the natural terpenoids, and many designed compounds demonstrated a wide spectrum of biological potential including antibacterial, [20] insecticidal, [21] anti-inflammatory[22] and anti-tumor.[23-24] Camphor, an abundant monoterpenoid with a bicyclic structure, has been used in medicine since ancient times, [25] and it has good effect of insect repellent while has very low toxicity to human body. In addition to that, compounds modified by camphor exhibit various biological activities. For instance, camphane-based amides presented promising scaffolds for antimycobacterial agents.[26] Derivatives of camphor were considered as prospective inhibitors of influenza viruses with significant activity.[27] Camphor-based cyanopyrrolidines were identified as dipeptidyl peptidase IV (DPPIV) inhibitors for treating type 2 diabetes mellitus.[28]
In continuing our search for new compounds endowed with anticancer activity, camphor was selected as a building block to construct thiosemicarbazones. Besides, in biological and medical fields, the α, β-unsaturated carbonyl scaffold has been deemed as one of prepotent structures.[29] Chalcone, as a representative of α, β-unsaturated ketone, the molecular structure of which is relatively flexible and contains multiple reaction centers, which can bind to different receptors, thus possessing diverse pharmacological activities.[30-31] In past, it was reported that chalcone-based thiosemicarbazones 1-3 and 1-4 were described as antitumor analogs.[32-33] Thus, it was envisaged that novel molecule constructed by introducing α, β-unsaturated ketone structure into camphor-based thiosemicarbazones would show more potent biological properties. Encouraged by the above facts, a set of the camphor-based thiosemi-carbazones derivatives were designed, and their anticancer activities against three human tumor cell lines (A549, MDA-MB-231, RPMI- 8226) were evaluated. Furthermore, the preliminary anticancer mechanism of synthesized compounds was explored.
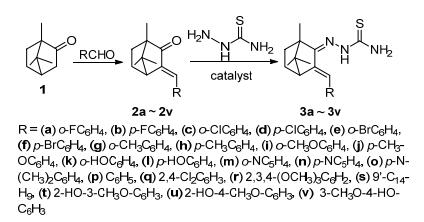
A series of thiosemicarbazone derivatives were synthesized according to the route depicted in Scheme 1, using camphor (1) as the starting point. Camphor 1 reacted with different substituted aromatic aldehydes by aldol condensation to generate compounds 2a~2v, and the target compounds 3a~3v were prepared by a condensation of intermediates 2a~2v and thiosemicarbazide in the presence of hydrochloric acid and absolute ethyl alcohol. All synthesized thiosemicarbazone analogs were purified by silica column chromatography and their structures were confirmed by 1H NMR, 13C NMR and HRMS analysis. As exemplified with the 1H NMR analysis of 2-(3-(2'-meth- oxybenzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]heptan-2- ylidene)hydrazinecarbothioamide (3i), 1H NMR spectra revealed appearance of NH2 signal at δ 7.38 and NH signal at δ 9.32, for olefinic bond, a singlet at δ 6.36 was found. The characteristic signals of different aromatic protons were observed at δ 6.89~7.33. Additionally, 13C NMR spectrum provided further structural evidence. The peak at δ 179.71 corresponded to C=S carbon and the presence of a single peak at δ 157.38 confirmed the imine structure. The 13C NMR spectra displayed two signals at δ 140.76 and 129.69, assigned to two C=C carbons, respectively. Besides, compound 3n was characterized by X-ray single crystal diffraction. Crystallographic data of the compound 3n was deposited with the Cambridge Crystallographic Data Centre (CCDC No 1838876).
The MDA-MB-231 (breast cancer cell line) and A549 (lung adenocarcinoma cell line) both are solid tumor cells with high morbidity and mortality, so they were selected to examined for the anticancer activities of the target compounds 3a~3v. Besides, multiple myeloma cell line (RPMI-8226), which is a kind of blood tumor cell, was added into test for comparison with solid tumor cells. And the cytotoxicity of compounds 3a~3v was evaluated using normal human gastric epithelial cell line (GES-1), and etoposide was used as a positive control. The IC50 values were summarized in Table 1.
 下载:
导出CSV
下载:
导出CSV
| Compound | IC50/(µmol•L-1) | |||
| MBA-MD-231 | RPMI-8226 | A549 | GES-1 | |
| 3a | 16.55±0.05 | 19.03±0.40 | 24.88±0.60 | > 50 |
| 3b | 48.91±0.22 | > 50 | > 50 | > 50 |
| 3c | 10.22±0.18 | 12.04±0.31 | 20.35±0.24 | > 50 |
| 3d | 37.40±0.14 | 46.88±0.28 | > 50 | > 50 |
| 3e | 15.96±0.50 | 14.33±0.22 | 29.63±0.02 | > 50 |
| 3f | 38.33±0.30 | > 50 | > 50 | > 50 |
| 3g | 11.22±0.21 | 11.90±0.18 | 16.46±0.35 | > 50 |
| 3h | > 50 | > 50 | > 50 | > 50 |
| 3i | 36.33±0.28 | > 50 | > 50 | > 50 |
| 3j | 39.46±0.24 | 48.46±0.33 | > 50 | > 50 |
| 3k | 24.62±0.22 | 10.64±0.29 | 17.65±0.25 | > 50 |
| 3l | 42.39±0.18 | > 50 | 48.39±0.40 | > 50 |
| 3m | 20.11±0.24 | 25.20±0.09 | 28.32±0.33 | > 50 |
| 3n | > 50 | > 50 | > 50 | > 50 |
| 3o | 46.57±0.14 | > 50 | > 50 | > 50 |
| 3p | > 50 | > 50 | > 50 | > 50 |
| 3q | > 50 | > 50 | > 50 | > 50 |
| 3r | 8.66±0.17 | 10.94±0.52 | 16.41±0.34 | > 50 |
| 3s | 3.90±0.04 | 6.56±0.27 | 10.64±0.19 | > 50 |
| 3t | 37.72±0.25 | 32.72±0.20 | 38.72±0.05 | > 50 |
| 3u | 12.00±0.09 | 11.00±0.23 | 18.97±0.18 | > 50 |
| 3v | 24.43±0.60 | 30.58±0.35 | 29.45±0.16 | > 50 |
| 1 | > 50 | > 50 | > 50 | > 50 |
| 2s | > 50 | > 50 | > 50 | > 50 |
| Etoposide | 3.48±0.08 | 1.49±0.36 | 15.22±0.56 | 6.86±0.06 |
It was found that majority of the tested analogs showed moderate to significant antitumor activity against selected cancer cell lines. Compound 3s showed the highest activities with IC50 values of 3.90 µmol•L-1 (MDA-MB-231 cell line), 6.56 µmol•L-1 (RPMI-8226 cell line) and 10.64 µmol•L-1 (A549 cell line), comparable to the reference drug (IC50 3.48±0.08, 1.49±0.36, 15.22±0.56 µmol•L-1 respectively). Compounds 3a, 3c, 3e, 3g, 3r and 3u exhibited the promising anticancer activity against MDA-MB- 231 and RPMI-8226 cell lines. Four compounds 3g, 3k, 3r and 3u had similar anticancer activity against A549 cell line to etoposide. Compared to compound 3p without any substituents on the benzene ring, almost all substituted derivatives showed improved anticancer activity. Furthermore, it was obvious that substitution position on the phenyl ring had a remarkable influence on the inhibitory activity. A significant boost in potency came from the ortho- substitution: compounds with group at o-position were more potent p-substituted analogs (3a vs 3b, 3c vs 3d, 3e vs 3f, 3g vs 3h, 3i vs 3j and 3k vs 3l). Another persuasive evidence was that compounds 3t~3v with different position of hydroxy and methoxy group had distinct difference in activity. In addition, a slight difference in inhibition of growth was observed for compounds 3a~3l with different substituting groups. Interestingly, introduction of two chlorine groups at ortho- and para-position (3q) caused a loss dramatically in the anticancer activity, but compound 3r bearing three methoxy groups presented a potent inhibitory against selected cancer cell lines. On top of this, the replacement of phenyl ring with 2-pyridine ring or anthracene ring also was beneficial to increasing the anticancer potency. And it worth noting that none of target compounds exhibited obvious toxicity toward normal human cells (IC50 > 50 µmol•L-1), whereas the reference drug had high toxicity (IC50=6.86 µmol•L-1). Our findings indicated that compound 3s has good selectivity between cancer and normal cells.
The cytotoxicity results suggested compound 3s display the most promising activity, especially in MDA-MB-231 cells, so compound 3s was selected to explore the possible mechanism of the observed cytotoxicity of these compounds. After being treated with various doses of compound 3s, the MDA-MB-231 cells were stained with propidium iodide (PI) and analyzed by flow cytometry. As shown in Figure 2, it could be observed that 3s treatment (doses from 0, 2, 4, 6 µmol•L-1, 48 h) clearly increased the proportion of cells in G2 phase from 11.41%, 16.52% and 26.97% to 43.45%, whereas the G0/G1 and S phase population decreased from 66.17% and 22.42% in the control to 38.56% and 17.99% (6.0 µmol•L-1), respectively. These results indicated that 3s caused G2 arrest in a dose-dependent manner.
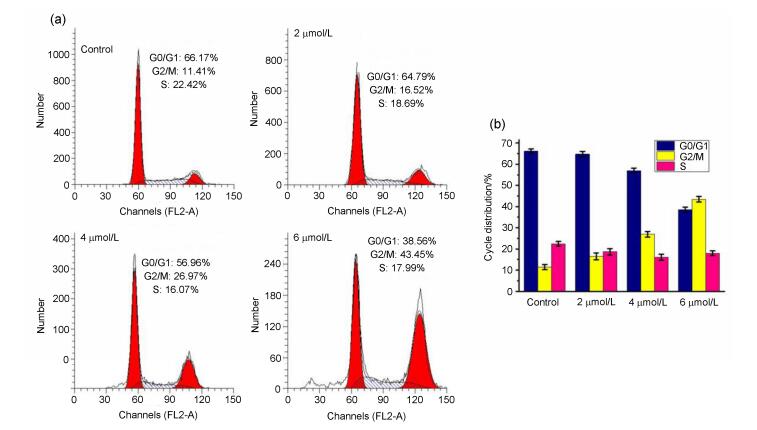
(a) Cells were treated with different concentrations (2, 4 and 6 µmol•L-1) of compound 3s for 48 h, which were then stained with propidium iodide (PI), and the DNA content was analyzed by flow cytometry. (b) Percentages of cells in various phases of the cell cycle
Next, we further examined whether the death of cancer cells treated with compound 3s was due to induction of apoptosis, the Annexin V-APC/7-AAD dual staining assay was conducted by flow cytometry. The results were presented in Figure 3, after MDA-MB-231 cells were exposed to different concentrations of compound 3s, the total rate of apoptosis rose from 10.15% (2 µmol•L-1) and 35.07% (4 µmol•L-1) to 58.7% (6 µmol•L-1), in which early and late apoptotic cells accounted for 32.7%, 26.0%, respecttively. The data indicated that compound 3s could effectively induce apoptosis of MDA-MB-231 cells, and the growth inhibitory of it might be related to triggering apoptosis.
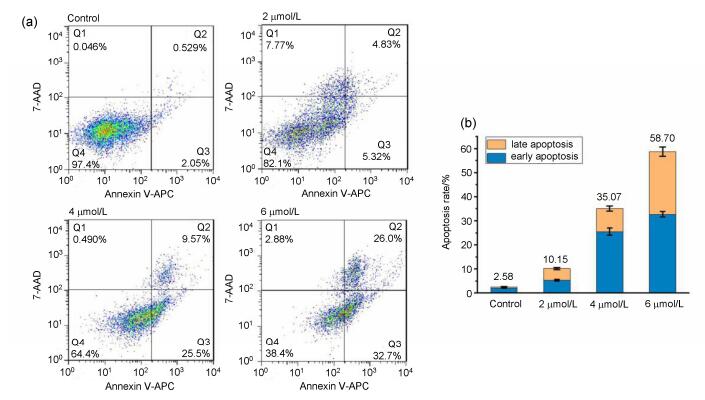
(a) Flow cytometry analysis of MDA-MB-231 cells treated with compound 3s (2, 4 and 6 µmol•L-1) for 48 h, respectively, and stained with Annexin V-APC/7-AAD. (b) Quantitative analysis of apoptotic cells in histogram
It's well-known that apoptosis can be initiated by extrinsic and intrinsic pathway, and both of them depend on reactive oxygen species (ROS).[34] ROS, metabolic byproducts of aerobic respiration, is recognized as a pivotal factor in maintaining redox homeostasis and participates in numerous intracellular signaling pathways.[35] When inducing cancer cells to produce excess ROS, oxidative stress- induced apoptosis could be triggered, which promotes cancer cell death.[36] To identify the impact of ROS in the course of 3s-induced apoptosis, MDA-MB-231 cells reacted with 3s (0, 2, 4 and 6 µmol•L-1) in the presence of 2', 7'-dichlorofluorescein diacetate (DCFH-DA), which can be converted into the fluorescent 2', 7'-dichlorofluoresceinn (DCF) upon the exposure to ROS. The results of flow cytometry analysis were shown in Figure 4, the level of ROS in the treated cells were found to positively correlate with the concentrations of compound 3s. Cells incubated with compound 3s at 2 µmol•L-1 concentration produced 2-fold increase of ROS in comparison to the control group (8.13%). After cells were incubated with compound 3s (4 and 6 μmol•L-1), the level of ROS production elevated to 28.8% (4 μmol•L-1) and 72.5% (6 μmol•L-1), respectively. Therefore, there is a rapid dose-dependent formation of ROS after compound 3s treatment, which suggested that overproduction of intracellular ROS might contribute to the apoptosis of MDA- MB-231 cells treated with compound 3s.
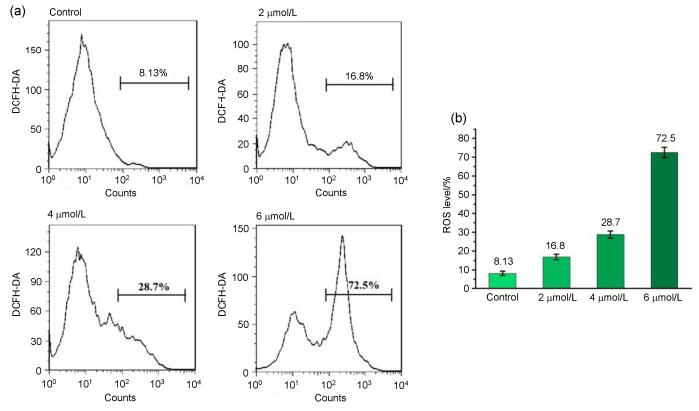
(a) Analysis of the ROS levels by flow cytometry after the MDA-MB-231 cells were treated with compound 3s at indicated concentrations or in presence of NAC for 48 h and stained with DCFH-DA. (b) Quantification of the flow cytometric results
As mentioned above, there are two major routes contributing to apoptosis. The extrinsic apoptotic pathway is initiated by binding between death receptors and ligand, and the intrinsic apoptotic pathway is involved in mitochondrial damage, including loss of mitochondrial membrane potential (ΔΨm) that plays a key role in maintaining mitochondrial integrity.[37-38] In this work, 5, 5', 6, 6'-tetra- chloro-1, 1', 3, 3'-tetraethyl-imidacarbocyanine (JC-1) was used as a fluorescent probe to determine whether mitochondrial membrane potential changes in MDA-MB-231 cells treated with compound 3s. The measured results are presented in Figure 5, in the control, there are 94.8% of red fluorescent cells indicating intact mitochondrial membrane potential. However, on addition of compound 3s (2, 4, 6 µmol• L-1), the proportion of green fluorescent cells increased gradually from 8.9% and 25.8% to 40.3%, demonstrating that compound 3s caused collapse of ΔΨm by concentration-dependent manner. Based on such study, it could be concluded that the compound 3s-induced apoptosis involved in intrinsic mitochondrial pathway.
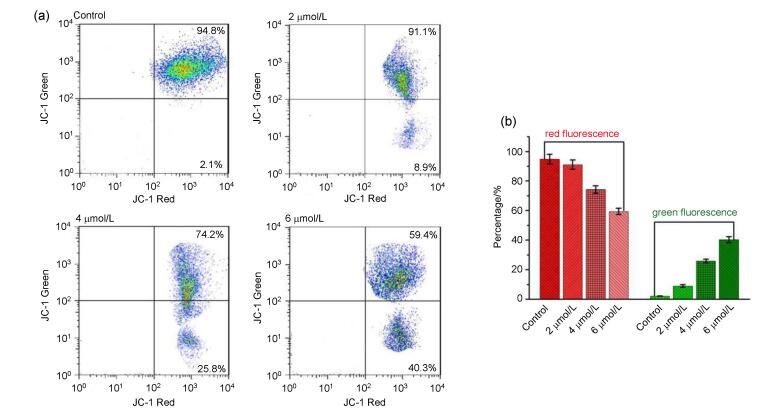
(a) Cells were treated with compound 3s (2, 4 and 6 µmol•L-1) for 48 h and observed the changes in mitochondrial membrane potential by flow cytometry. (b) Bar graph represents the percentage distribution of cells with red fluorescence or green fluorescence
According to aforementioned findings, compound 3s gave rise to the dissipation of mitochondrial membrane potential. Stimulation of mitochondrial damage promotes mitochondrial outer membrane permeabilization, causing release of apoptgenic factors such as cytochrome c (cyt c) from mitochondria to cytosol.[39] Binding to another mitochondrial protein, cytosolic cytochrome c triggers a cascade of caspase-dependent signaling events, and eventually, results in the execution of cell death.[40] To get insight into the molecular basis of the antitumor activity of compound 3s, the expression of several apoptosis-related proteins was explored by Western blot analysis. As illustrated in Figure 6, compound 3s up-regulated the level of pro-apoptotic protein Bax, whereas reduced the expression of anti-apoptosis protein Bcl-2 in the dose-dependent manner. These Bcl-2 superfamily of proteins are the vital members of modulating apoptotic susceptibility and mitochondrial outer membrane permeabilization.[41] Further-more, compound 3s promoted the release of cytochrome c into the cytoplasm. Executioner caspase-3 acts as a key signalling molecule of apoptosis in the downstream of the mitochondrial apoptotic pathway, in this experiment, the upregulation of caspase-3 was observed compared to the control. Hence, our results confirmed that compound 3s induced mitochondria related apoptosis.
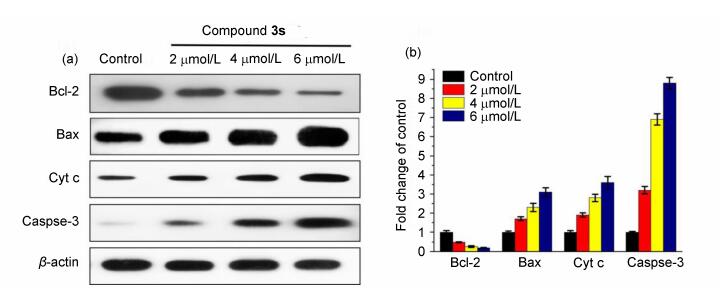
(a) Western blot analysis of Bcl-2, Bax, Cytochrome C, Caspase-3 in MDA-MB-231 cells treated with compound 3s (2, 4 and 6 µmol•L-1) for 48 h, β-actin was used as the internal reference. (b) Statistical analysis of protein expression levels
In this study, 22 novel camphor-based thiosemicarbazone derivatives were designed and synthesized and their antiproliferative activity against three human cancer cell lines were evaluated. The results of MTS assay showed that majority of the tested analogs showed good anti-tumor activity against selected cancer cell lines. particularly, compound 3s appeared to be the most active with IC50 values of 3.90 (MDA-MB-231), 6.56 (RPMI-8226) and 10.64 (A549) µmol•L-1, comparable to the reference drug and had low toxicity (IC50 > 50 µmol•L-1) to human normal gastric epithelial cells (GES-1). In terms of structural relationships, substitution position on the phenyl ring had a remarkable influence on the inhibitory activity. To explore the underlying mechanism of 3s, the impact of it on cell cycle and apoptosis in MDA-MB-231 cells was investigated. It was observed that 3s could arrest the cell cycle in the G2 phase and induce apoptosis in a dose-dependent manner. Furthermore, we found there was a concentration- dependent enhancement of reactive oxygen species (ROS) and the membrane potential (ΔΨm) decreased in MDA- MB-231 cells after 3s treatment, which contributing to destroying the balance of expression of anti-apoptosis and pro-apoptotic proteins. Above studies indicated that 3s exhibited potent anticancer efficiency via inducing G2 cell cycle arrest and apoptosis via ROS-mediated mitochondrial pathway.
Melting points were recorded using Optimelt-MPA100 melting point apparatus. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker AV 400 spectrometer in CDCl3 or DMSO-d6 solution. High resolution mass spectra (HRMS) were measured using an America Agilent 6540 mass spectrometer. X-ray diffraction data were collected on a Bruker D8 Venture single crystal diffractometer. All chemical reagents (AR grade) and solvents were provided by common commercial suppliers. Three cell lines were obtained from the Cell Bank of the Chinese Academy of Sciences. The chemical reactions were monitored by thin-layer chromatography (TLC) and gas chromatography (GC). All target compounds were purified by column chromatography (200~300 mesh silica gel), and their purity was determined by high performance liquid chromatography (HPLC) (America Agilent 1260).
Camphor (8 mmol), aromatic aldehyde (10 mmol), tert-butanol (35 mL) and potassium tert-butoxide (8 mmol) was added successively into a 50 mL dried flask. The reaction mixture was stirred and refluxed until the conversion ratio of camphor exceeded 90%. Then, tert-butanol was removed under vacuum and the concentrated reaction mixture was extracted with ethyl acetate and saturated salt water. Next, the organic layer were dried with sodium sulfate, followed by filtration and concentration, giving the yellow solid. Finally, the crude product was recrystallized from methanol and filtered to obtain intermediates 2a~2v.
(E)-3-(2-Fluorobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2a): Yield 90%; 1H NMR (400 MHz, CDCl3) δ: 7.45 (t, J=7.6 Hz, 1H), 7.38 (s, 1H), 7.26~7.36 (m, 1H), 7.16 (t, J=6.9 Hz, 1H), 2.99 (d, J=4.3 Hz, 1H), 2.13~2.21 (m, 1H), 1.73~1.84 (m, 1H), 1.47~1.65 (m, 2H), 1.03 (s, 3H), 1.00 (s, 3H), 0.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.6, 162.5, 160.0, 144.0, 130.1, 124.0, 119.7, 116.0, 115.8, 57.3, 49.4, 46.5, 30.6, 26.0, 20.6, 18.2, 9.2; HR-MS (ESI) calcd for C17H19FO [M+H]+ 259.1498, found 259.1499.
(E)-3-(4-Fluorobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2b): Yield 91%; 1H NMR (400 MHz, CDCl3) δ: 7.44~7.48 (m, 2H), 7.19 (s, 1H), 7.08 (t, J=8.7 Hz, 2H), 3.06 (d, J=4.3 Hz, 1H), 2.13~2.22 (m, 1H), 1.82~1.73 (m, 1H), 1.46~1.62 (m, 2H), 1.03 (s, 3H), 1.00 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.0, 164.0, 161.6, 141.8, 131.6, 126.3, 115.9, 115.6, 57.1, 49.1, 46.7, 30.7, 25.9, 20.6, 18.3, 9.3; HR-MS (ESI) calcd for C17H19FO [M+H]+ 259.1498, found 259.1502.
(E)-3-(2-Chlorobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2c): Yield 92%; 1H NMR (400 MHz, CDCl3) δ: 7.50 (s, 1H), 7.39~7.43 (m, 2H), 7.20~7.32 (m, 2H), 2.93 (d, J=4.3 Hz, 1H), 2.10~2.20 (m, 1H), 1.76~1.84 (m, 1H), 1.50~1.62 (m, 2H), 1.04 (s, 3H), 0.98 (s, 3H), 0.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.5, 144.1, 135.3, 134.1, 129.9, 129.6, 126.6, 124.0, 57.4, 48.9, 46.5, 30.5, 26.1, 20.6, 18.2, 9.3; HR-MS (ESI) calcd for C17H19ClO [M+H]+ 275.1203, found 275.1207.
(E)-3-(4-Chlorobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2d): Yield 91%; 1H NMR (400 MHz, CDCl3) δ: 7.34~7.42 (m, 4H), 7.18 (s, 1H), 3.05 (d, J=4.3 Hz, 1H), 2.13~2.22 (m, 1H), 1.74~1.84 (m, 1H), 1.46~1.62 (m, 2H), 1.03 (s, 3H), 1.00 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.9, 142.6, 134.6, 134.2, 130.9, 128.9, 126.2, 57.1, 49.2, 46.7, 30.7, 25.9, 20.6, 18.3, 9.3; HR-MS (ESI) calcd for C17H19ClO [M+H]+ 275.1203, found 275.1202.
(E)-3-(2-Bromobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2e): Yield 87%; 1H NMR (400 MHz, CDCl3) δ: 7.61 (d, J=8.0, 1.2 Hz, 1H), 7.43 (s, 1H), 7.38 (d, J=7.7, 1.8 Hz, 1H), 7.30~7.34 (m, 1H), 7.16~7.20 (m, 1H), 2.89 (d, J=4.3 Hz, 1H), 2.09~2.19 (m, 1H), 1.74~1.84 (m, 1H), 1.50~1.62 (m, 2H), 1.04 (s, 3H), 0.98 (s, 3H), 0.83 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.5, 143.9, 135.9, 133.1, 129.9, 129.7, 127.2, 126.5, 125.4, 57.5, 48.7, 46.5, 30.5, 26.1, 20.6, 18.2, 9.3; HR-MS (ESI) calcd for C17H19BrO [M+H]+ 319.0698, found 319.0698.
(E)-3-(4-bromobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2f): Yield 88%; 1H NMR (400 MHz, CDCl3) δ: 7.48~7.55 (m, 2H), 7.29~7.36 (m, 2H), 7.16 (s, 1H), 3.05 (d, J=4.3 Hz, 1H), 2.13~2.22 (m, 1H), 1.74~1.84 (m, 1H), 1.46~1.62 (m, 2H), 1.03 (s, 3H), 1.00 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.9, 142.8, 134.6, 131.9, 131.1, 126.2, 122.8, 57.1, 49.2, 46.7, 30.6, 25.9, 20.6, 18.3, 9.3; HR-MS (ESI) calcd for C17H19BrO [M+H]+ 319.0698, found 319.0701.
(E)-1, 7, 7-Trimethyl-3-(2-methylbenzylidene)bicycle- [2.2.1]heptan-2-one (2g): Yield 94%; 1H NMR (400 MHz, CDCl3) δ: 7.41 (s, 1H), 7.32~7.34 (m, 1H), 7.16~7.27 (m, 3H), 2.94 (d, J=4.3 Hz, 1H), 2.35 (s, 3H), 2.11~2.19 (m, 1H), 1.72~1.84 (m, 1H), 1.48~1.65 (m, 2H), 1.03 (s, 3H), 0.97 (s, 3H), 0.81 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 208.1, 142.8, 138.1, 134.7, 130.4, 128.6, 128.5, 125.8, 125.6, 57.5, 48.8, 46.6, 30.6, 26.3, 20.6, 20.0, 18.3, 9.3; HR-MS (ESI) calcd for C18H22O [M+H]+ 255.1749, found 255.1752.
(E)-1, 7, 7-Trimethyl-3-(4-methylbenzylidene)bicycle- [2.2.1]heptan-2-one (2h): Yield 95%; 1H NMR (400 MHz, CDCl3) δ: 7.35~7.42 (m, 2H), 7.19~7.21 (m, 3H), 3.10 (d, J=4.2 Hz, 1H), 2.37 (s, 3H), 2.13~2.21 (m, 1H), 1.74~1.83 (m, 1H), 1.48~1.64 (m, 2H), 1.03 (s, 3H), 0.99 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.3, 141.3, 138.9, 132.9, 129.8, 129.4, 127.6, 57.1, 49.3, 46.7, 30.8, 25.9, 21.4, 20.6, 18.4, 9.3; HR-MS (ESI) calcd for C18H22O [M+H]+ 255.1749, found 255.1748.
(E)-3-(2-Methoxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2i): Yield 95%; 1H NMR (400 MHz, CDCl3) δ: 7.57 (s, 1H), 7.39~7.42 (m, 1H), 7.28~7.33 (m, 1H), 6.96 (t, J=7.5, 1.1 Hz, 1H), 6.89~6.98 (m, 1H), 3.84 (s, 3H), 3.01 (d, J=4.3 Hz, 1H), 2.09~2.22 (m, 1H), 1.70~1.83 (m, 1H), 1.46~1.63 (m, 2H), 1.02 (s, 3H), 0.98 (s, 3H), 0.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.2, 158.5, 141.9, 130.1, 129.6, 124.8, 122.7, 120.7, 110.9, 57.3, 55.5, 49.2, 46.6, 30.7, 26.1, 20.6, 18.7, 9.3; HR-MS (ESI) calcd for C18H22O2 [M+H]+ 271.1698, found 271.1695.
(E)-3-(4-Methoxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2j): Yield 93%; 1H NMR (400 MHz, CDCl3) δ: 7.40~7.48 (m, 2H), 7.19 (s, 1H), 6.88~6.96 (m, 2H), 3.83 (s, 3H), 3.09 (d, J=4.2 Hz, 1H), 2.11~2.23 (m, 1H), 1.72~1.83 (m, 1H), 1.45~1.62 (m, 2H), 1.02 (s, 3H), 0.99 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.3, 160.1, 140.1, 131.4, 128.3, 127.3, 114.2, 57.0, 55.3, 49.2, 46.8, 30.9, 25.9, 20.5, 18.4, 9.3; HR-MS (ESI) calcd for C18H22O2 [M+H]+ 271.1698, found 271.1696.
(E)-3-(2-Hydroxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2k): Yield 89%; 1H NMR (400 MHz, CDCl3) δ: 7.68 (s, 1H), 7.37~7.40 (m, 1H), 7.20~7.26 (m, 1H), 6.90~6.95 (m, 2H), 3.05 (d, J=4.2 Hz, 1H), 2.12~2.21 (m, 1H), 1.76~1.82 (m, 1H), 1.49~1.63 (m, 2H), 1.04 (s, 3H), 0.99 (s, 3H), 0.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 155.5, 142.2, 130.4, 129.5, 122.8, 122.7, 120.3, 116.2, 57.5, 49.1, 46.8, 30.7, 26.1, 20.6, 18.3, 9.3; HR-MS (ESI) calcd for C17H20O2 [M+H]+ 257.1542, found 257.1544.
(E)-3-(4-Hydroxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2l): Yield 90%; 1H NMR (400 MHz, CDCl3) δ: 7.37~7.41 (m, 2H), 7.19 (s, 1H), 6.86~6.90 (m, 2H), 3.09 (d, J=4.2 Hz, 1H), 2.13~2.21 (m, 1H), 1.75~1.79 (m, 1H), 1.44~1.62 (m, 2H), 1.03 (s, 3H), 1.00 (s, 3H), 0.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.9, 156.6, 139.8, 131.7, 128.2, 127.7, 115.7, 57.2, 49.2, 46.9, 30.9, 25.9, 20.6, 18.4, 9.3; HR-MS (ESI) calcd for C17H20O2 [M+H]+ 257.1542, found 257.1543.
(E)-1, 7, 7-Trimethyl-3-(pyridin-2-ylmethylene)bicycle- [2.2.1]heptan-2-one (2m): Yield 82%; 1H NMR (400 MHz, CDCl3) δ: 8.73 (d, J=2.2 Hz, 1H), 8.54~8.56 (m, 1H), 7.75~7.78 (m, 1H), 7.30~7.38 (m, 1H), 7.19 (s, 1H), 3.08 (d, J=4.3 Hz, 1H), 2.14~2.27 (m, 1H), 1.72~1.87 (m, 1H), 1.48~1.65 (m, 2H), 1.04 (s, 3H), 1.02 (s, 3H), 0.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.4, 150.6, 149.3, 144.2, 136.3, 131.6, 123.6, 123.5, 57.1, 49.2, 46.7, 30.5, 25.9, 20.6, 18.2, 9.2; HR-MS (ESI) calcd for C16H19NO [M+H]+ 242.1545, found 242.1546.
(E)-1, 7, 7-Trimethyl-3-(pyridin-4-ylmethylene)bicycle- [2.2.1]heptan-2-one (2n): Yield 84%; 1H NMR (600 MHz, CDCl3) δ: 8.62 (d, J=5.2 Hz, 2H), 7.32 (d, J=5.3 Hz, 2H), 7.10 (s, 1H), 3.07 (d, J=4.4 Hz, 1H), 2.16~2.22 (m, 1H), 1.75~1.81 (m, 1H), 1.48~1.58 (m, 2H), 1.01 (s, 3H), 0.99 (s, 3H), 0.77 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 207.3, 149.6, 146.5, 143.8, 124.1, 123.8, 57.1, 49.3, 46.5, 30.4, 25.9, 20.6, 18.1, 9.2; HR-MS (ESI) calcd for C16H19NO [M+H]+ 242.1545, found 242.1549.
(E)-3-(4-(Dimethylamino)benzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]heptan-2-one (2o): Yield 90%; 1H NMR (400 MHz, CDCl3) δ: 7.42 (d, J=6.8 Hz, 2H), 7.18 (s, 1H), 6.70 (d, J=6.7 Hz, 2H), 3.10 (s, 1H), 3.00 (s, 6H), 2.12~2.17 (m, 1H), 1.72~1.78 (m, 1H), 1.46~1.59 (m, 2H), 1.01 (s, 3H), 0.98 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.4, 150.7, 137.8, 131.5, 128.4, 123.4, 111.9, 77.4, 77.1, 76.7, 56.9, 49.4, 46.9, 40.2, 31.0, 26.0, 20.5, 18.5, 9.4; HR-MS (ESI) calcd for C19H26NO [M+H]+ 284.2014, found 284.2016.
(E)-3-Benzylidene-1, 7, 7-trimethylbicyclo[2.2.1]heptan-2-one (2p): Yield 87%; 1H NMR (400 MHz, CDCl3) δ: 7.44~7.51 (m, 2H), 7.36~7.44 (m, 2H), 7.25~7.36 (m, 1H), 7.24 (s, 1H), 3.11 (d, J=4.3 Hz, 1H), 2.14~2.22 (m, 1H), 1.71~1.84 (m, 2H), 1.47~1.65 (m, 2H), 1.03 (s, 3H), 1.00 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 208.2, 142.1, 135.7, 129.8, 128.7, 128.7, 127.5, 57.1, 49.2, 46.7, 30.7, 25.9, 20.6, 18.3, 9.3; HR-MS (ESI) calcd for C17H20O [M+H]+ 241.1592, found 241.1590.
(E)-3-(2, 4-Dichlorobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2q): Yield 86%; 1H NMR (600 MHz, CDCl3) δ: 7.46 (d, J=2.1 Hz, 1H), 7.36 (d, J=8.4 Hz, 1H), 7.27~7.29 (m, 1H), 2.90 (d, J=4.3 Hz, 1H), 2.14~2.20 (m, 1H), 1.79~1.85 (m, 1H), 1.53~1.60 (m, 2H), 1.05 (s, 3H), 1.01 (s, 3H), 0.83 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 207.3, 144.5, 135.9, 134.7, 132.7, 130.5, 129.8, 127.0, 122.8, 57.4, 48.9, 46.6, 30.5, 26.0, 20.6, 18.2, 9.2; HR-MS (ESI) calcd for C17H18Cl2O [M+H]+ 309.0813, found 309.0816.
(E)-1, 7, 7-Trimethyl-3-(3, 4, 5-trimethoxybenzylidene)- bicyclo[2.2.1]heptan-2-one (2r): Yield 92%; 1H NMR (600 MHz, CDCl3) δ: 7.18 (s, 1H), 6.74 (s, 2H), 3.91 (s, 9H), 3.10 (d, J=4.2 Hz, 1H), 2.19~2.24 (m, 1H), 1.79~1.84 (m, 1H), 1.61~1.64 (m, 1H), 1.53~1.58 (m, 1H), 1.05 (s, 3H), 1.03 (s, 3H), 0.84 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 208.1, 153.3, 141.3, 138.9, 131.2, 127.8, 107.1, 77.2, 77.0, 76.8, 61.0, 57.0, 56.2, 49.4, 46.8, 30.7, 25.8, 20.6, 18.4, 9.3; HR-MS (ESI) calcd for C20H26O4 [M+H]+ 331.1909, found 331.1904.
(E)-3-(Anthracen-9-ylmethylene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-one (2s): Yield 83%; 1H NMR (400 MHz, CDCl3) δ: 8.38 (d, J=7.6 Hz, 1H), 7.97~8.00 (m, 5H), 7.42~7.48 (m, 4H), 2.16 (s, 3H), 1.66~1.71 (m, 2H), 1.61 (d, J=7.7 Hz, 1H), 1.34 (t, J=8.2 Hz, 1H), 1.09 (s, 3H), 1.00 (s, 3H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 207.3, 147.9, 131.3, 130.3, 129.3, 128.8, 128.2, 127.2, 127.0, 125.9, 125.7, 125.4, 125.3, 58.2, 48.7, 46.1, 30.6, 25.9, 20.8, 18.1, 9.4; HR-MS (ESI) calcd for C25H24O [M+H]+ 341.1905, found 341.1898.
(E)-3-(2-Hydroxy-3-methoxybenzylidene)-1, 7, 7-tri- methylbicyclo[2.2.1]heptan-2-one (2t): Yield 89%; 1H NMR (600 MHz, DMSO-d6) δ: 9.13 (s, 1H), 7.41 (s, 1H), 6.98~7.01 (m, 2H), 6.83 (t, J=7.9 Hz, 1H), 3.82 (s, 3H), 3.04 (d, J=4.1 Hz, 1H), 2.11~2.17 (m, 1H), 1.74~1.79 (m, 1H), 1.43~1.48 (m, 1H), 1.34~1.38 (m, 1H), 0.96 (s, 3H), 0.92 (s, 3H), 0.72 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ: 207.3, 148.3, 146.5, 141.3, 122.7, 122.2, 121.2, 119.4, 112.9, 56.9, 56.4, 49.0, 46.6, 30.6, 26.2, 20.7, 18.4, 9.8; HR-MS (ESI) calcd for C18H22O3 [M+H]+ 287.1647, found 287.1649.
(E)-3-(2-Hydroxy-4-methoxybenzylidene)-1, 7, 7-tri- methylbicyclo[2.2.1]heptan-2-one (2u): Yield 87%; 1H NMR (600 MHz, DMSO-d6) δ: 10.07 (s, 1H), 7.35 (t, J=4.2 Hz, 2H), 6.49 (d, J=2.5 Hz, 1H), 6.47 (s, 1H), 3.73 (s, 3H), 3.02 (d, J=4.1 Hz, 1H), 2.10~2.15 (m, 1H), 1.72~1.75 (m, 1H), 1.41~1.45 (m, 1H), 1.31~1.36 (m, 1H), 0.95 (s, 3H), 0.91 (s, 3H), 0.72 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ: 207.2, 161.7, 158.8, 138.7, 130.5, 122.1, 115.4, 106.1, 101.6, 56.8, 55.5, 49.0, 46.8, 30.7, 26.1, 20.6, 18.5, 9.8; HR-MS (ESI) calcd for C18H22O3 [M+H]+ 287.1647, found 287.1644.
(E)-3-(4-Hydroxy-3-methoxybenzylidene)-1, 7, 7-tri- methylbicyclo[2.2.1]heptan-2-one (2v): Yield 86%; 1H NMR (600 MHz, DMSO-d6) δ: 10.07 (s, 1H), 7.35 (t, J=4.2 Hz, 2H), 6.49 (d, J = 2.5 Hz, 1H), 6.47 (s, 1H), 3.73 (s, 3H), 3.02 (d, J=4.1 Hz, 1H), 2.11~2.16 (m, 1H), 1.74~1.79 (m, 1H), 1.43~1.48 (m, 1H), 1.34~1.38 (m, 1H), 0.95 (s, 3H), 0.91 (s, 3H), 0.72 (s, 3H); 13C NMR (150 MHz, DMSO-d6) δ: 207.2, 161.7, 158.8, 138.7, 130.5, 122.1, 115.4, 106.1, 101.6, 56.8, 55.5, 49.0, 46.8, 30.7, 26.1, 20.6, 18.5, 9.8; HR-MS (ESI) calcd for C18H22O3 [M+H]+ 287.1647, found 287.1646.
The intermediates 2a~2v (2 mmol) was refluxed with an equimolar amount of thiosemicarbazide in the presence of a few drops of concentrated HCl. After 12 h, the mixed solution turned from colorless to a yellow color and the new spot of product almost did not deepen on TLC plate. Then, the mix solution was concentrated and purified by column chromatography [ethyl acetate/petroleum ether, V:V=1:6, 1% triethylamine (TEA)] to afford the target compounds 3a~3v.
2-(3-(2-Fluorobenzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]- heptan-2-ylidene)hydrazinecarbothioamide (3a): Yield 78%. m.p. 156.1~156.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.22 (s, 1H), 7.36 (s, 1H), 7.25~7.32 (m, 2H), 7.23 (s, 1H), 7.07~7.18 (m, 2H), 6.40 (s, 1H), 2.72 (d, J=4.2 Hz, 1H), 1.99~2.05 (m, 1H), 1.80~1.88 (m, 1H), 1.47~1.56 (m, 2H), 1.05 (s, 3H), 0.91 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 180.0, 161.3, 158.3, 143.3, 130.2, 129.9, 129.1, 124.0, 122.4, 115.7, 54.4, 52.2, 47.8, 33.5, 26.1, 20.0, 18.1, 11.3; HR-MS (ESI) calcd for C18H23FN3S [M+H]+ 332.1597, found 332.1595.
2-(3-(4-Fluorobenzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]- heptan-2-ylidene)hydrazinecarbothioamide (3b): Yield 79%. m.p. 192.5~192.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.18 (s, 1H), 7.30 (d, J=6.3 Hz, 2H), 7.27 (s, 1H), 7.21 (s, 1H), 7.05~7.10 (m, 2H), 6.40 (s, 1H), 2.86 (d, J=4.2 Hz, 1H), 2.05~2.10 (m, 1H), 1.77~1.87 (m, 1H), 1.53 (d, J=11.3 Hz, 1H), 1.04 (s, 3H), 0.91 (s, 3H), 0.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.9, 161.2, 158.9, 141.2, 132.2, 130.9, 128.7, 115.72, 54.2, 51.7, 48.0, 33.4, 25.8, 20.0, 18.2, 11.3; HR-MS (ESI) calcd for C18H23FN3S [M+H]+ 332.1597, found 332.1591.
2-(3-(2-Chlorobenzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]- heptan-2-ylidene)hydrazinecarbothioamide (3c): Yield 82%. m.p. 199.7~200.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.24 (s, 1H), 7.41~7.43 (m, 1H), 7.39 (s, 1H), 7.32 (s, 1H), 7.27 (d, J=1.5 Hz, 2H), 7.21~7.24 (m, 1H), 6.46 (s, 1H), 2.66 (d, J=4.3 Hz, 1H), 1.97~2.03 (m, 1H), 1.81~1.86 (m, 1H), 1.51 (d, J=8.9 Hz, 2H), 1.05 (s, 1H), 0.89 (s, 1H), 0.80 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 180.0, 158.1, 142.8, 134.7, 133.9, 130.2, 129.6, 129.3, 126.8, 117.6, 54.4, 51.7, 47.8, 33.4, 25.9, 20.1, 18.1, 11.3; HR-MS (ESI) calcd for C18H23ClN3S [M+H]+ 348.1301, found 348.1297.
2-(3-(4-Chlorobenzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]- heptan-2-ylidene)hydrazinecarbothioamide (3d): Yield 82%. m.p. 157.1~157.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.20 (s, 1H), 7.35 (t, J=6.3 Hz, 3H), 7.31 (s, 1H), 7.25 (s, 1H), 7.20 (s, 1H), 6.51 (s, 1H), 2.86 (d, J=4.3 Hz, 1H), 2.06~2.09 (m, 1H), 1.81~1.85 (m, 1H), 1.48~1.53 (m, 2H), 1.04 (s, 3H), 0.91 (s, 3H), 0.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.9, 158.8, 141.9, 134.6, 134.1, 130.4, 128.7, 119.5, 54.2, 51.8, 48.0, 33.4, 25.7, 20.0, 18.2, 11.3; HR-MS (ESI) calcd for C18H23ClN3S [M+H]+ 348.1301, found 348.1295.
2-(3-(2-Bromobenzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]- heptan-2-ylidene)hydrazinecarbothioamide (3e): Yield 81%. m.p. 203.2~203.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.25 (s, 1H), 7.61 (d, J=8.0 Hz, 1H), 7.37 (d, J=7.9 Hz, 1H), 7.29~7.35 (m, 1H), 7.27 (s, 2H), 7.19 (t, J=7.6 Hz, 1H), 6.46 (s, 1H), 2.64 (d, J=4.3 Hz, 1H), 1.96~2.02 (m, 1H), 1.78~1.85 (m, 1H), 1.51 (d, J=8.5 Hz, 2H), 1.05 (s, 3H), 0.89 (s, 3H), 0.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 180.0, 158.1, 142.4, 136.5, 132.8, 130.3, 129.0, 127.2, 124.1, 120.1, 54.4, 51.5, 47.8, 33.3, 25.9, 20.1, 18.1, 11.3; HR-MS (ESI) calcd for C18H23BrN3S [M+H]+ 392.0796, found 392.0795.
2-(3-(4-Bromobenzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]- heptan-2-ylidene)hydrazinecarbothioamide (3f): Yield 80%. m.p. 159.1~159.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.20 (s, 1H), 7.51 (d, J=8.4 Hz, 2H), 7.37 (s, 1H), 7.18 (d, J=4.4 Hz, 2H), 6.48 (s, 1H), 2.85 (d, J=4.3 Hz, 1H), 2.05~2.10 (m, 1H), 1.79~1.85 (m, 1H), 1.48~1.52 (m, 2H), 1.04 (s, 3H), 0.91 (s, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.9, 158.7, 142.0, 135.0, 131.7, 130.6, 128.6, 122.3, 54.2, 51.8, 48.0, 33.4, 25.7, 20.0, 18.2, 11.3; HR-MS (ESI) calcd for C18H23BrN3S [M+H]+ 392.0796, found 392.0788.
2-(3-(2-Methylbenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3g): Yield 78%. m.p. 186.2~186.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.21 (s, 1H), 7.35 (s, 1H), 7.29 (s, 1H), 7.20~7.22 (m, 2H), 7.16~7.19 (m, 2H), 6.41 (s, 1H), 2.66 (d, J=4.3 Hz, 1H), 2.29 (s, 3H), 1.96~2.02 (m, 1H), 1.80~1.85 (m, 1H), 1.50 (d, J=8.8 Hz, 2H), 1.05 (s, 3H), 0.88 (s, 3H), 0.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.8, 158.8, 141.8, 136.5, 135.4, 130.1, 128.9, 128.2, 125.8, 119.4, 54.4, 51.6, 47.6, 33.5, 26.0, 20.1, 20.1, 18.1, 11.3; HR-MS (ESI) calcd for C19H26N3S [M+H]+ 328.1847, found 328.1843.
2-(3-(4-Methylbenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3h): Yield 80%. m.p. 164.1~164.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.21 (s, 1H), 7.24 (s, 1H), 7.22 (s, 2H), 7.18 (d, J=8.4 Hz, 3H), 6.43 (s, 1H), 2.92 (d, J=4.4 Hz, 1H), 2.37 (s, 3H), 2.04~2.09 (m, 1H), 1.80~1.86 (m, 1H), 1.57~1.60 (m, 1H), 1.47~1.52 (m, 1H), 1.04 (s, 3H), 0.91 (s, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.8, 159.3, 140.6, 138.3, 133.3, 129.2, 129.2, 120.8, 54.2, 51.9, 48.0, 33.5, 25.8, 21.3, 20.0, 18.2, 11.3; HR-MS (ESI) calcd for C19H26N3S [M+H]+ 328.1847, found 328.1841.
2-(3-(2-Methoxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3i): Yield 84%. m.p. 181.4~181.7 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.32 (s, 1H), 7.38 (s, 2H), 7.29~7.33 (m, 1H), 7.24 (d, J=7.5 Hz, 1H), 6.96 (t, J=7.5 Hz, 1H), 6.90 (d, J=8.3 Hz, 1H), 6.36 (s, 1H), 3.85 (s, 3H), 2.80 (d, J=4.2 Hz, 1H), 2.00~2.06 (m, 1H), 1.79~1.85 (m, 1H), 1.48~1.55 (m, 2H), 1.04 (s, 3H), 0.90 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.7, 159.1, 157.4, 140.8, 129.7, 128.9, 125.7, 120.2, 116.1, 110.6, 55.5, 54.3, 51.9, 47.8, 33.5, 26.0, 20.1, 18.2, 11.3; HR-MS (ESI) calcd for C19H26N3OS [M+H]+ 344.1797, found 344.1791.
2-(3-(4-Methoxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3j): Yield 83%. m.p. 174.6~174.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.13 (s, 1H), 7.28 (s, 1H), 7.23 (s, 1H), 7.21 (s, 1H), 7.13 (s, 1H), 6.85 (s, 1H), 6.83 (s, 1H), 6.35 (s, 1H), 3.77 (s, 3H), 2.85 (d, J=4.2 Hz, 1H), 1.97~2.02 (m, 1H), 1.72~1.79 (m, 1H), 1.38~1.45 (m, 2H), 0.96 (s, 3H), 0.84 (s, 3H), 0.67 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.7, 159.5, 157.7, 139.5, 130.7, 129.7, 128.7, 114.0, 55.3, 54.1, 51.8, 48.0, 33.5, 25.8, 20.0, 18.3, 11.3; HR-MS (ESI) calcd for C19H26N3OS [M+H]+ 344.1797, found 344.1797.
2-(3-(2-Hydroxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3k): Yield 82%. m.p. 178.7~189.1 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.63 (s, 1H), 7.36 (s, 2H), 7.18 (t, J=7.1 Hz, 1H), 7.11 (d, J=7.5 Hz, 1H), 6.87~6.92 (m, 2H), 6.43 (s, 1H), 2.75 (d, J=4.1 Hz, 1H), 1.97~2.03 (m, 1H), 1.78~1.85 (m, 1H), 1.49 (d, J=7.2 Hz, 2H), 1.03 (s, 3H), 0.89 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.3, 159.9, 153.9, 141.9, 129.6, 129.1, 125.1, 123.5, 120.3, 116.1, 54.5, 51.7, 47.7, 33.3, 26.1, 20.1, 18.2, 11.4; HR-MS (ESI) calcd for C18H24N3OS [M+H]+ 330.1640, found 330.1461.
2-(3-(4-Hydroxybenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3l): Yield 81%. m.p. 157.5~157.8 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.22 (s, 1H), 7.36 (s, 1H), 7.25~7.32 (m, 2H), 7.23 (s, 1H), 7.07~7.18 (m, 2H), 6.40 (s, 1H), 2.7 (d, J=4.2 Hz, 1H), 1.99~2.05 (m, 1H), 1.80~1.88 (m, 1H), 1.47~1.56 (m, 2H), 1.05 (s, 3H), 0.91 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 179.2, 160.9, 157.4, 137.3, 130.7, 129.7, 127.3, 115.5, 53.6, 50.7, 47.4, 32.8, 25.6, 19.8, 18.1, 11.5; HR-MS (ESI) calcd for C18H24N3OS [M+H]+ 330.1640, found 330.1638.
2-(3-(Pyridin-2-ylmethylene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3m): Yield 82%. m.p. 181.1~181.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.36 (s, 1H), 8.63 (s, 1H), 8.50 (d, J=4.2 Hz, 1H), 7.65 (d, J=7.9 Hz, 1H), 7.29~7.32 (m, 2H), 7.06 (s, 1H), 6.39 (s, 1H), 2.91 (d, J=4.1 Hz, 1H), 2.13~2.19 (m, 1H), 1.81~1.88 (m, 1H), 1.75~1.78 (m, 1H), 1.59~1.62 (m, 1H), 1.27 (s, 1H), 1.07 (s, 1H), 0.94 (s, 3H), 0.88 (s, 3H), 0.78 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 178.7, 156.4, 150.0, 148.2, 145.1, 135.5, 132.6, 123.4, 116.9, 54.7, 49.7, 49.4, 32.6, 26.1, 20.7, 17.5, 13.86; HR-MS (ESI) calcd for C17H23N4S [M+H]+ 315.1643, found 357.1641.
2-(3-(Pyridin-4-ylmethylene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3n): Yield 83%. m.p. 162.3~162.5 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.36 (s, 1H), 8.58~8.59 (m, 2H), 7.20~7.21 (m, 3H), 6.99 (s, 1H), 6.51 (s, 1H), 2.93 (d, J=4.3 Hz, 1H), 2.12~2.20 (m, 1H), 1.78~1.82 (m, 1H), 1.70~1.74 (m, 1H), 1.58~1.61 (m, 1H), 1.06 (s, 1H), 0.94 (s, 4H), 0.86 (s, 3H), 0.78 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 180.1, 158.0, 150.1, 147.2, 144.8, 126.7, 124.0, 117.9, 54.2, 51.9, 48.0, 33.2, 25.7, 20.1, 18.1, 11.2; HR-MS (ESI) calcd for C17H23N4S [M+H]+ 315.1643, found 357.1642.
2-(3-(4-Dimethylaminobenzylidene)-1, 7, 7-trimethyl- bicyclo[2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3o): Yield 84%. m.p. 135.3~135.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.19 (s, 1H), 7.28 (d, J=8.7 Hz, 3H), 7.16 (s, 1H), 6.70 (d, J=8.8 Hz, 2H), 6.28 (s, 1H), 3.01 (s, 3H), 2.97 (d, J=4.2 Hz, 1H), 2.07~2.10 (m, 1H), 1.78~1.85 (m, 1H), 1.56~1.59 (m, 1H), 1.45~1.59 (m, 1H), 1.02 (s, 3H), 0.91 (s, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.5, 160.2, 150.2, 137.1, 130.8, 130.6, 123.8, 111.8, 54.1, 52.0, 48.1, 40.2, 33.6, 25.9, 20.0, 18.4, 11.4; HR-MS (ESI) calcd for C20H29N4S [M+H]+ 357.2113, found 357.2106.
2-(3-Benzylidene)-1, 7, 7-trimethylbicyclo[2.2.1]heptan- 2-ylidene)hydrazinecarbothioamide (3p): Yield 83%. m.p. 126.5~126.8 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.24 (s, 1H), 7.36~7.42 (m, 3H), 7.34 (s, 2H), 7.30~7.32 (m, 2H), 6.45 (s, 1H), 2.92 (d, J=4.3 Hz, 1H), 2.04~2.10 (m, 1H), 1.81~1.87 (m, 1H), 1.47~1.59 (m, 2H), 1.04 (s, 3H), 0.91 (s, 3H), 0.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.9, 159.1, 141.3, 136.1, 129.9, 129.2, 128.5, 120.8, 54.2, 51.8, 47.9, 33.5, 25.8, 20.0, 18.2, 11.3; HR-MS (ESI) calcd for C18H24N3S [M+H]+ 314.1691, found 314.1685.
2-(3-(2, 4-Dichlorobenzylidene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3q): Yield 79%. m.p. 149.7~150.6 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.33 (s, 1H), 7.42 (s, 1H), 7.23 (s, 2H), 7.18~7.22 (m, 1H), 7.17 (s, 1H), 6.28 (s, 1H), 2.70 (d, J=4.3 Hz, 1H), 2.05~2.13 (m, 1H), 1.78~1.87 (m, 1H), 1.70~1.76 (m, 1H), 1.46~1.52 (m, 1H), 1.05 (s, 1H), 0.90 (s, 4H), 0.88 (d, J=10.2 Hz, 7H), 0.79 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 178.7, 156.1, 145.1, 135.0, 133.7, 130.5, 129.5, 127.0, 125.5, 116.5, 54.8, 51.7, 49.5, 32.6, 26.2, 20.7, 17.5, 13.9; HR-MS (ESI) calcd for C18H22Cl2N3S [M+H]+ 382.0911, found 382.0903.
2-(3-(2, 3, 4-Trimethoxybenzylidene)-1, 7, 7-trimethyl- bicyclo[2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3r): Yield 79%. m.p. 155.7~155.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.19 (s, 1H), 7.20 (s, 1H), 6.58 (s, 1H), 6.56 (s, 2H), 6.42 (s, 1H), 3.88 (d, J=2.6 Hz, 9H), 2.95 (d, J=4.4 Hz, 1H), 2.05~2.14 (m, 1H), 1.82~1.89 (m, 1H), 1.58~1.63 (m, 1H), 1.48~1.51 (m, 1H), 1.05 (s, 3H), 0.93 (s, 3H), 0.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 179.85, 158.96, 153.17, 140.75, 131.59, 130.01, 120.93, 106.65, 60.95, 56.20, 54.17, 52.20, 48.00, 33.43, 25.70, 20.04, 18.25, 11.32; HR-MS (ESI) calcd for C21H30N3O3S [M+H]+ 404.2008, found 404.1998.
2-(3-(Anthracen-9-ylmethylene)-1, 7, 7-trimethylbicyclo- [2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3s): Yield 78%. m.p. 218.5~218.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.50 (s, 1H), 8.46 (s, 1H), 8.02~8.05 (m, 4H), 7.86 (s, 1H), 7.48~7.51 (m, 5H), 6.45 (s, 1H), 1.98 (d, J=4.3 Hz, 1H), 1.71~1.78 (m, 1H), 1.59~1.63 (m, 1H), 1.31~1.37 (m, 1H), 1.09 (s, 3H), 0.91 (s, 3H), 0.71 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 180.1, 157.5, 145.4, 131.4, 129.9, 128.8, 127.3, 126.6, 125.9, 125.7, 125.4, 117.7, 54.8, 52.0, 47.5, 33.0, 25.6, 20.3, 18.0, 14.2; HR- MS (ESI) calcd for C26H28N3S [M+H]+ 414.2004, found 414.1995.
2-(3-(2-Hydroxy-3-methoxybenzylidene)-1, 7, 7-trimeth- ylbicyclo[2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3t): Yield 80%. m.p. 120.5~120.9 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.83 (s, 1H), 8.94 (s, 1H), 8.35 (s, 1H), 7.63 (s, 1H), 7.36 (s, 1H), 6.91~6.95 (m, 1H), 6.81~6.83 (m, 2H), 3.81 (s, 3H), 2.74 (d, J=4.1 Hz, 1H), 1.95~2.02 (m, 1H), 1.74~1.80 (m, 1H), 1.37~1.48 (m, 2H), 1.02 (s, 3H), 0.87 (s, 3H), 0.72 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 179.8, 160.0, 148.2, 145.0, 140.2, 125.5, 124.3, 121.6, 119.1, 112.2, 60.2, 56.3, 54.2, 47.6, 33.3, 26.2, 20.2, 18.5, 11.8; HR-MS (ESI) calcd for C19H26N3- O2S [M+H]+ 360.1746, found 360.1738.
2-(3-(2-Hydroxy-4-methoxybenzylidene)-1, 7, 7-trimeth- ylbicyclo[2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3u): Yield 80%. m.p. 180.6~181.8 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.74 (s, 2H), 8.31 (s, 1H), 7.59 (s, 1H), 7.31 (s, 1H), 7.15 (d, J=8.6 Hz, 1H), 6.41~6.49 (m, 2H), 3.72 (s, 3H), 2.75 (d, J=4.3 Hz, 1H), 1.99~2.03 (m, 1H), 1.73~1.79 (m, 1H), 1.38~1.48 (m, 2H), 1.01 (s, 3H), 0.87 (s, 3H), 0.71 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 179.1, 160.4, 159.9, 156.7, 137.7, 130.0, 124.8, 116.2, 105.1, 101.1, 55.0, 53.7, 50.8, 47.2, 32.9, 25.7, 19.7, 18.0, 11.4; HR-MS (ESI) calcd for C19H26N3O2S [M+H]+ 360.1746, found 360.1738.
2-(3-(3-Methoxy-4-hydroxybenzylidene)-1, 7, 7-trimeth- ylbicyclo[2.2.1]heptan-2-ylidene)hydrazinecarbothioamide (3v): Yield 81%. m.p. 207.1~207.6 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.99 (s, 1H), 9.32 (s, 1H), 8.26 (s, 1H), 7.54 (s, 1H), 7.28 (s, 1H), 6.93 (s, 1H), 6.78~6.88 (m, 2H), 3.79 (s, 3H), 2.89 (d, J=4.0 Hz, 1H), 1.99~2.07 (m, 1H), 1.69~1.81 (m, 1H), 1.39~1.50 (m, 2H), 1.01 (s, 3H), 0.89 (s, 3H), 0.72 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 179.8, 161.1, 147.9, 147.3, 138.1, 130.5 128.3, 122.8, 116.1 113.8, 56.0, 54.1, 51.4, 47.9, 33.3, 26.1, 20.3, 18.6, 12.0; HR-MS (ESI) calcd for C19H26N3O2S [M+H]+ 360.1746, found 360.1738.
Three human cells including breast cancer (MDA- MB-231), myeloma cancer (RPMI-8226), lung cancer (A549) and normal gastric epithelial (GES-1) cells were grown in RPMI-1640 medium [φ(FBS)=5%] under standard conditions [a humidified atmosphere, φ(CO2)=5%, 37 ℃]. Then, the cells (70%~80% confluence) were seeded in 96-well plates (5×103 cells/well) and incubated overnight. Then, the stock solutions of synthesized compounds (5 mmol•L-1) and etoposide (5 mmol•L-1) were prepared with dimethyl sulfoxide (DMSO) and diluted to the required concentrations (25.00, 12.50, 6.25, 3.12, 1.55, 0.75, 0.37 µmol•L-1) with serum free medium. Subsequently, the cultured cells were treated with different candidate solutions. After 48 h incubation, 10 µL of solution of MTS was added to each well and cells were treated for 0.5 h. The absorbance (OD) of the samples were measured at 490 nm on a microplate reader. The results (IC50) were calculated using GraphPad Prism 5.
MDA-MB-231 cells (2×105 cell/well) in the logarithmic growth were seeded in a 6-well plate. After overnight adherence to the plate at 37 ℃, cells were exposed to different concentrations (2, 4 and 6 μmol•L-1) of compound 3s for 48 h. Later, the cultured cells were harvested and fixed in ice-cold 70% ethanol at -20 ℃ for 24 h. After being washed with phosphate buffered saline (PBS), the cells were labeled with RNase A (1 mg/mL) and stained with propidium iodide (10 mg/mL) solution in the dark for 30 min. Finally, the DNA content in samples was measured by flow cytometry, and was analyzed using Modfit software.
MDA-MB-231 cells were seeded into a 6-well plates at 2×105 cell/well under usual conditions. After incubation overnight, cells were reacted with various concentrations (2, 4 and 6 µmol•L-1) of the selected compound for 48 h. Next, cells were collected, washed (PBS, twice) and resuspended in the binding buffer. Subsequently, the cells in suspension were stained with Annexin V-APC/7-AAD solution in the dark and incubated for 15 min. Lastly, the stained cells were examined by a flow cytometry system.
MDA-MB-231 cells were incubated in 6-well plates at a density of 1×106 cell/well overnight at 37 ℃, φ(CO2)=5%, and treated with compound 3s at different concentrations (2, 4 or 6 µmol•L-1). After 48 h incubation, the collected cells were incubated in PBS containing 10 µmol•L-1 of DCFH-DA (ROS indicator) for 20~30 min in the dark at 37 ℃. Then, the level of intracellular ROS was analyzed by flow cytometry.
MDA-MB-231 cells (70%~80% confluence) were seeded in a 6-well plate and exposed to different concentrations (2, 4 and 6 µmol•L-1) of compound 3s. After 48 h, the collected cells were suspended in PBS, and added with JC-1 working solution (5, 5', 6, 6'-tetrachloro-1, 1', 3, 3'-tetre- thylbenzimidalylcarbocyanineiodide). Following incubation at 37 ℃ in darkness, the treated cells were washed (cold PBS, twice) and analyzed immediately by flow cytometry.
MDA-MB-231 cells (1×106 cell/mL) were treated with 2, 4 and 6 µmol•L-1 of compound 3s for 48 h. Then the cells were harvested, lysed in ice-cold lysis buffer and centrifuged at 4 ℃. After that, the supernatant was obtained and the protein contents were determined using a BCA Protein Assay Kit. An equal amount of the proteins (50 µg) was loaded and separated by SDS-PAGE gel, followed by being transferred to a nitrocellulose membrane. Subsequently, the membranes were blocked in tris buffered saline tween (TBST) buffer with 0.5 g/mL skim milk at room temperature for 1 h. Appropriate primary antibodies were treated with the membranes overnight at 4 ℃, to make the target proteins in membranes be probed with primary antibodies. After being washed with TBST three times, the membranes were incubated with appropriate secondary antibodies at room temperature over 1 h. Eventually, the signal was observed via an enhanced chemiluminescence detection reagent and analyzed the relative optical density of each protein via laser scanning densitometry.
Xu Z., Zhao S. J., Liu Y.Eur. J. Med. Chem., 2019, 183:111700. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Zhang L., Xu Z.Eur. J. Med. Chem., 2019, 181:111587. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Magalhaes Moreira D. R., de Oliveira A. D., Teixeira de Moraes Gomes P. A., de Simone C. A., Villela F. S., Ferreira R. S., Leite A. C.Eur. J. Med. Chem., 2014, 75:467. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
De Melos J. L., Torres-Santos E. C., Faioes Vdos S., Del Cistia Cde N., Sant'Anna C. M., Rodrigues-Santos C. E., Echevarria A.Eur. J. Med. Chem., 2015, 103:409. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Linciano P., Moraes C. B., Alcantara L. M., Franco C. H., Pascoalino B., Freitas-Junior L. H., Costi M. P.Eur. J. Med. Chem., 2018, 146:423. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
El-Sharief M. A., Abbas S. Y., El-Bayouki K. A., El-Gammal E. W.Eur. J. Med. Chem., 2013, 67:263. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Zhang X., Guo H., Li Z., Song F., Wang W., Dai H., Wang J.Eur. J. Med. Chem., 2015, 101:419. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Banerjee D., Yogeeswari P., Bhat P., Thomas A., Srividya M., Sriram D.Eur. J. Med. Chem., 2011, 46:106. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Kesel A. J.Eur. J. Med. Chem., 2011, 46:1656. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Chavarria G. E., Horsman M. R., Arispe W. M., Kumar G. D., Chen S. E., Strecker T. E., Trawick M. L.Eur. J. Med. Chem., 2012, 58:568. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Pervez H., Khan N., Zaib S., Yaqub M., Naseer M. M., Tahir M. N., Iqbal J.Bioorg. Med. Chem., 2017, 25:1022. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Song S., You A., Chen Z., Zhu G., Wen H., Song H., Yi W.Eur. J. Med. Chem., 2017, 139:815. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
De Oliveira J. F., Lima T. S., Vendramini-Costa D. B., de Lacerda Pedrosa S. C. B., Lafayette E. A., da Silva R. M. F., de Lima M.Eur. J. Med. Chem., 2017, 136:305. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Vandresen F., Falzirolli H., Almeida Batista S. A., da Silva-Giardini A. P., de Oliveira D. N., Catharino R. R., da Silva C. C.Eur. J. Med. Chem., 2014, 79:110. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Lukmantara A. Y., Kalinowski D. S., Kumar N., Richardson D. R.Bioorg. Med. Chem. Lett., 2013, 23:967. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Rao V. A., Klein S. R., Agama K. K., Toyoda E., Adachi N., Pommier Y., Shacter E. B.Cancer Res., 2009, 69:948. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Malarz K., Mrozek-Wilczkiewicz A., Serda M., Rejmund M., Musiol R.Oncotarget, 2018, 9:17689. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Lovejoy D. B., Richardson D. R.Blood, 2002, 100:666. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Da Silva A. P., Martini M. V., de Oliveira C. M., Cunha S., de Carvalho J. E., Ruiz A. L., da Silva C. C.Eur. J. Med. Chem., 2010, 45:2987. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
鲍名凯, 杨益琴, 谷文, 徐徐, 曹明珍, 王石发, 有机化学, , 2014, 34:2146. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Bao M., Yang Y., Gu W., Xu X., Cao M., Wang S.Chin. J. Org. Chem., 2014, 34:2146(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
孙楠, 王欣, 丁志彬, 张齐, 徐徐, 徐海军, 王石发, 有机化学, , 2016, 36:2489. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Sun N., Wang X., Ding Z., Zhang Q., Xu X., Xu H., Wang S.Chin. J. Org. Chem., 2016, 36:2489(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
芮坚, 蔡涛, 杨金来, 杨益琴, 徐徐, 王石发, 南京林业大学学报(自然科学版), , 2017, 41:149. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Rui J., Cai T., Yang J., Yang Y., Xu X., Wang S.J. Nanjing. For. Univ., 2017, 41:149(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Ma C., Wang Y., Dong F., Wang Z., Zhao Y., Shan Y., Wang S.Chem. Biol. Drug Des., 2019, 94:1457. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Wang Y., Wang Z., Kuang H., Zhang Y., Gu W., Zhu Y., Wang S.Chem. Biol. Drug Des., 2019, 94:1281. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Sokolova A. S., Yarovaya O. C., Korchagina D. V., Zarubaev V. V., Tretiak T. S., Anfimov P. M., Salakhutdinov N. F.Bioorg. Med. Chem., 2017, 127:661. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Stavrakov G., Valcheva V., Philipova I., Doytchinova I.Eur. J. Med. Chem., 2013, 70:372. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Sokolova A. S., Yarovaya O. C., Korchagina D. V., Zarubaev V. V., Tretiak T. S., Anfimov P. M., Salakhutdinov N. F.Bioorg. Med. Chem., 2014, 22:2141. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Kuranov S. O., Tsypysheva I. P., Khvostov M. V., Zainullina L. F., Borisevich S. S., Vakhitova Y. V., Salakhutdinov N. F.Bioorg. Med. Chem., 2018, 26:4402. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Hossain M., Das U., Dimmock J. R.Eur. J. Med. Chem., 2019, 183:111687. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Dan W., Dai J.Eur. J. Med. Chem., 2020, 187:111980. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Zhu M., Wang J., Xie J., Chen L., Wei X., Jiang X., Bao M., Qiu Y., Chen Q., Li W., Jiang C., Zhou X., Jiang L., Qiu P., Wu J.Eur. J. Med. Chem., 2018, 157:1395. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
刘长辉, 文瑞明, 贺淼, 易先文, 叶晓琴, 方磊, 应用化学, , 2017, 38:900. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498Liu C., Wen R., He M., Yi X., Ye X., Fang L.Chin. J. Appl. Chem., 2017, 38:900(in Chinese). http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Zhang H., Qian Y., Zhu D., Yang X., Zhu H.Eur. J. Med. Chem., 2011, 46:4702. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Simon H. U., Haj-Yehia A., Levi-Schaffer F.Apoptosis, 2000, 5:415. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Circu M. L., Aw T. Y.Free Radical Biol. Med., 2010, 48:749. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Dharmaraja A. T.J. Med. Chem., 2017, 60:3221. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Yamada Y., Harashima H.Adv. Drug Delivery Rev., 2008, 60:1439. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Galluzzi L., Zamzami N., de La Motte Rouge T., Lemaire C., Brenner C., Kroemer G.Apoptosis, 2007, 12:803. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Jin Z., El-Deiry W. S.Cancer Biol. Ther., 2005, 4:139. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Martinou J. C., Desagher S., Antonsson B.Nat. Cell Biol., 2005, 2:41. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498
Xiong S., Mu T., Wang G., Jiang X.Protein Cell, 2014, 5:737. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=DE20101005498

Figure 2 Influence of compound 3s on cell cycle distribution in MDA-MB-231 cells
(a) Cells were treated with different concentrations (2, 4 and 6 µmol•L-1) of compound 3s for 48 h, which were then stained with propidium iodide (PI), and the DNA content was analyzed by flow cytometry. (b) Percentages of cells in various phases of the cell cycle

Figure 3 Compound 3s induced apoptosis in MDA-MB-231 cells
(a) Flow cytometry analysis of MDA-MB-231 cells treated with compound 3s (2, 4 and 6 µmol•L-1) for 48 h, respectively, and stained with Annexin V-APC/7-AAD. (b) Quantitative analysis of apoptotic cells in histogram

Figure 4 Effect of compound 3s on ROS production
(a) Analysis of the ROS levels by flow cytometry after the MDA-MB-231 cells were treated with compound 3s at indicated concentrations or in presence of NAC for 48 h and stained with DCFH-DA. (b) Quantification of the flow cytometric results

Figure 5 Determination of mitochondrial membrane potential in MDA-MB-231 cells
(a) Cells were treated with compound 3s (2, 4 and 6 µmol•L-1) for 48 h and observed the changes in mitochondrial membrane potential by flow cytometry. (b) Bar graph represents the percentage distribution of cells with red fluorescence or green fluorescence

Figure 6 Expression changes of apoptosis-related protein
(a) Western blot analysis of Bcl-2, Bax, Cytochrome C, Caspase-3 in MDA-MB-231 cells treated with compound 3s (2, 4 and 6 µmol•L-1) for 48 h, β-actin was used as the internal reference. (b) Statistical analysis of protein expression levels
Table 1. Antiproliferative activities of compounds 3a~3v
| Compound | IC50/(µmol•L-1) | |||
| MBA-MD-231 | RPMI-8226 | A549 | GES-1 | |
| 3a | 16.55±0.05 | 19.03±0.40 | 24.88±0.60 | > 50 |
| 3b | 48.91±0.22 | > 50 | > 50 | > 50 |
| 3c | 10.22±0.18 | 12.04±0.31 | 20.35±0.24 | > 50 |
| 3d | 37.40±0.14 | 46.88±0.28 | > 50 | > 50 |
| 3e | 15.96±0.50 | 14.33±0.22 | 29.63±0.02 | > 50 |
| 3f | 38.33±0.30 | > 50 | > 50 | > 50 |
| 3g | 11.22±0.21 | 11.90±0.18 | 16.46±0.35 | > 50 |
| 3h | > 50 | > 50 | > 50 | > 50 |
| 3i | 36.33±0.28 | > 50 | > 50 | > 50 |
| 3j | 39.46±0.24 | 48.46±0.33 | > 50 | > 50 |
| 3k | 24.62±0.22 | 10.64±0.29 | 17.65±0.25 | > 50 |
| 3l | 42.39±0.18 | > 50 | 48.39±0.40 | > 50 |
| 3m | 20.11±0.24 | 25.20±0.09 | 28.32±0.33 | > 50 |
| 3n | > 50 | > 50 | > 50 | > 50 |
| 3o | 46.57±0.14 | > 50 | > 50 | > 50 |
| 3p | > 50 | > 50 | > 50 | > 50 |
| 3q | > 50 | > 50 | > 50 | > 50 |
| 3r | 8.66±0.17 | 10.94±0.52 | 16.41±0.34 | > 50 |
| 3s | 3.90±0.04 | 6.56±0.27 | 10.64±0.19 | > 50 |
| 3t | 37.72±0.25 | 32.72±0.20 | 38.72±0.05 | > 50 |
| 3u | 12.00±0.09 | 11.00±0.23 | 18.97±0.18 | > 50 |
| 3v | 24.43±0.60 | 30.58±0.35 | 29.45±0.16 | > 50 |
| 1 | > 50 | > 50 | > 50 | > 50 |
| 2s | > 50 | > 50 | > 50 | > 50 |
| Etoposide | 3.48±0.08 | 1.49±0.36 | 15.22±0.56 | 6.86±0.06 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们