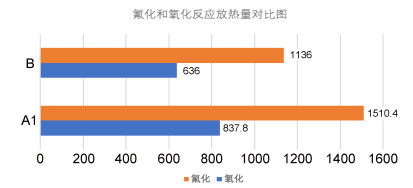
图 1.
每摩尔硼和铝氟化和氧化反应放热量
Figure 1.
Fluorination and oxidation heat release of per mole of B and Al

追求密度更大、能量更高的含能化合物始终是含能材料领域的永恒主题.自1771年英国人沃尔夫发明了世界首个合成炸药——三硝基苯酚(苦味酸)以来, 各国合成化学家在含能材料前进的道路上从未止步[1~4].从第一代含能材料的三硝基甲苯(TNT), 到以六硝基六氮杂异伍兹烷(CL-20)为代表的第三代含能材料, 其核心思想与策略都是在C、H、N等元素构成的链状、环状及笼型骨架中引入硝基基团, 包括硝胺基(NNO2)、硝酸酯基(ONO2)和多硝甲基(二硝甲基、三硝甲基和氟二硝甲基)等, 进而设计、合成新型高能量密度材料[5~15].在某种程度上, 含能材料的发展史可以认为是硝基化合物的开发史.硝基不仅能大幅提高CHON化合物密度, 更重要的是硝基是分子内“氧”的来源, 在爆轰反应中, 硝基中的氧将C、H等氧化成稳定氧化态(水、一氧化碳、二氧化碳等), 进而释放能量, 这也是含能材料与能源材料如煤、石油和天然气的本质区别.然而, 最近十几年, 各国在开发能量密度达到或超过CL-20的新型高能量密度化合物道路上却踌躇不前.究其原因, 一方面, 向单个分子中引入更多数量的硝基等含能基团时, 尽管密度和爆轰性能在一定程度上得到提升, 但同时也会导致目标化合物合成步骤长、合成难度大、制造成本高、感度高、分解温度低等问题, 严重限制其应用前景[16, 17]; 另一方面, 以硝基为代表的CHON类含能化合物能量密度存在理论上限, 高度对称的致密笼型高能化合物八硝基立方烷(ONC)[18, 19]和CL-20[20, 21]的密度和爆轰性能已达到很高的水平, 想要在此基础上进一步提高, 变得愈发困难.因此, 打破现有以硝基为基本能量单元的发展策略, 突破CHON类含能材料能量瓶颈, 具有重要的理论和实践意义.
氟是目前发现电负性最强的元素, 也是氧化能力最强的非金属单质, 其原子半径也显著小于氧原子.将N—F键替代硝基中N—O键形成的氟氮含能化合物具有以下独特优势: (1)氟原子较小的原子半径赋予含氟含能化合物更高的堆积密度, 二氟氨基化合物密度(ρ)理论上限可以达到2.303 g/cm3, 显著高于对应硝基化合物密度的理论上限2.166 g/cm3 [22]. (2) N—F键具有较低的键能(280 kJ/mol), 氟氮含能物质在燃烧分解过程中形成的气体产物H—F键能高(565 kJ/mol), 相对分子质量低, 生成热高, 有利于提高推进剂或炸药的燃气比容, 可大幅提高发射药、推进剂和混合炸药的能量水平[23, 24]. (3)高热值金属燃烧剂(铝粉、硼粉等)氟化反应比相应氧化反应放出更多的热量, 每摩尔铝氟化放热量是氧化放热量的1.80倍, 每摩尔硼氟化放热量是其氧化放热量的1.79倍[25~27], 如图 1所示. (4)氟的强氧化能力及氟化物独特的低熔点特性能够显著改善高热值可燃金属元素的燃烧或爆炸反应效率, 氧化铝的熔点高达2977 ℃, 燃烧过程中铝颗粒表面形成的难挥发氧化铝层严重制约着铝粉的充分燃烧, 而氟化铝沸点仅为1275 ℃, 燃烧高温能够轻易实现“剥壳效应”, 大大提高金属粉的燃烧效率.研究表明, 基于3, 3, 7, 7-四(二氟氨基)八氢化-1, 5-二硝基-1, 5-二氮杂辛烷(HNFX)、可燃剂BeH2配伍组成的推进剂配方, 理论比冲(Isp)高达3038 N·s/kg, 显示出诱人的应用前景[28].
将N—F键引入含能材料的研究非常早, 并且多集中在二氟氨基含能化合物的合成与反应特性研究[29~32].早在1936年, Juff等[33]报道了首个有机二氟氨基化合物二氟氨基三氟甲烷, 引起了美国高级工程局和国防部的关注.鉴于N—F含能化合物在开发推进剂用高性能氧化剂上显示出的巨大优势, 旨在探究其应用可行性的“Principia”项目于1958~1965间实施[34].在这期间, 利用F2、N2F4和HNF2等合成手段, 大量二氟氨基和偕二氟氨基化合物被合成出来, 如1, 2-二(二氟氨基)乙烷[35]、2, 2-二(二氟氨基)乙醇[36]、2, 3-二(二氟氨基)硝酸丙酯[37]、1, 1-双(二氟氨基)-3-甲基-4, 4, 4-三硝基丁烷[38]、三(二氟氨基)甲氧基-5, 5, 5-三硝基-3-丁硝胺[39]等.尽管如此, 由于当时合成的多数二氟氨基化合物结构中的α-H与二氟氨基极易脱去HF形成腈, 导致该类材料普遍存在结构稳定性差、感度高等问题, 致使相关的研究几乎停滞.进入20世纪90年代以来, 随着认识的深入和新型氟化试剂的开发, 以HNFX为代表的新型氟氮含能化合物重新引起各国学者的广泛关注, 美国海军空战武器中心Chapman教授和俄罗斯科学院相关人员在这方面做了大量工作, 陆续设计并合成出分子结构稳定的新型二氟氨基含能化合物.我国在氟氮含能化合物研究方面也有诸多亮点, 西安近代化学研究所郑远洋等[40]于1988年在国内报道了首个氟氮高能化合物四(二氟氨基亚硝胺基)乙烷, 并完成了密度、感度、爆发点和爆速等性能的研究.近期, 南京理工大学、黎明化工研究院等单位在氟氮含能粘合剂、增塑剂方面取得可喜成果[41~43].但与国外研究工作相比, 在研究的深度和广度方面, 还存在一定差距.
鉴于氟氮化合物作为含能材料的巨大潜力, 本文全面概述了近期氟氮含能化合物的合成研究进展, 以氟氮化合物结构特点进行分类, 着重介绍了氮氟唑类、二氟氨基二硝甲基化合物、偕二氟氨基化合物、二氟氨基聚合物等的合成途径, 并对典型氟氮含能化合物结构特点和爆轰性能进行了梳理, 以期为进一步开展相关研究工作提供参考.
含有C—F键的有机氟化物在许多尖端技术和重大工业项目及医药、农药和催化工业中都有着广泛而深入的研究和应用, C—F键合成方法和氟化试剂发展较为成熟.与之相比, N—F键合成途径和策略相对较少, 主要集中在氟气氟化和二氟氨基化两个方面. N—F键试剂是氟氮含能化合物合成以及氟氮化合物性能研究的基础, 故有必要对常见的N—F键合成方法和试剂进行简单介绍.
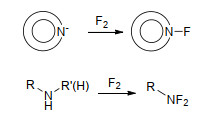
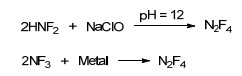
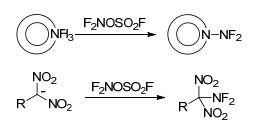
利用F2对相关底物进行氟化是合成氟氮化合物最直接、最简便的方法, 也是氟氮化合物中氟的终极来源. F2氟化主要通过以下2条途径生成相应的氟氮化合物: (1)通过对氮杂芳环氮负离子氧化, 生成相应N—F键氮杂芳环化合物[44, 45]; (2)对仲胺(尤其酰胺结构)、伯胺化合物氟化生成二氟氨基化合物[46~50], 如Scheme 1所示.其中, 对氨气、尿素等化合物氟化制备的小分子氟氮化合物HNF2、N2F4、NF3更是高效的二氟氨基化试剂, 利用这些二氟氨基试剂能够衍生出数量众多、性能各异的二氟氨基化合物.
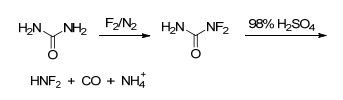
HNF2被广泛应用于合成二氟氨基化合物、偕二氟氨基化合物, 是一种重要的二氟氨基试剂和中间体. HNF2不稳定, 对撞击、摩擦均很敏感, 在结晶和融化过程中易发生爆炸[51, 52]. HNF2合成方法有多种, 包括N2F4法[53]、三苯基二氟氨基甲烷法[(Ph)3CNF2)][54, 55]和尿素法[56, 57], 其中通过尿素氟化、水解合成的方法最为常见, 路线如Scheme 2.
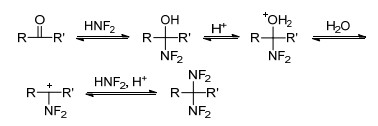
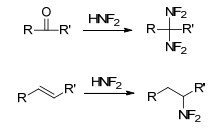
HNF2不仅能与具有羰基结构的醛、酮发生亲核加成反应[58, 59], 生成相应的偕二氟氨基化合物, 还能与不饱和烯、炔反应[60], 生成相应二氟氨基化合物, 如Scheme 3所示. HNF2与羰基反应如Scheme 4所示, 在强酸条件下, 羰基上氧质子化, 二氟化氨与羰基发生加成反应得到醇中间体, 随后经羟基质子化脱水、二氟化氨再次亲核进攻, 生成偕二氟氨基化合物.
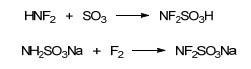
HNF2常温下为气体且危险性较高, 故开发安全性能好、常温下为液体或固体的二氟氨基化试剂显得非常必要. 1966年, Keith等[61]利用HNF2与SO3反应, 合成了室温下为液体的二氟氨基试剂F2NSO3H.尽管热稳定性和安全性相对HNF2有较大的提升, 但F2NSO3H在室温下仍会缓慢分解, 生成N2F4、SO2和H2SO4. 2006年, Haiges等[62]以NaSO3NH2为原料, 通过F2直接氟化, 成功获得了F2NSO3Na, 收率达到94% (Scheme 5). F2NSO3Na室温下为固体, 在干燥的环境中能够稳定存在, 其在合成方法简易程度、安全性等方面, 较HNF2、F2NSO3H均有较大提高.与HNF2相似, F2NSO3H和F2NSO3Na均能与酮、醛化合物生成相应的偕二氟氨基化合物[63~65].
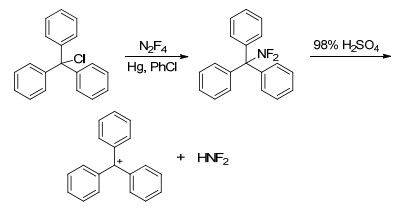
Ph3CNF2[54, 55, 66]是一种室温下稳定的固态二氟氨基化试剂, 其熔点为82 ℃~83 ℃, 且在空气中不潮解. Ph3CNF2合成路线如Scheme 6所示, 以三苯基氯甲烷为原料, 与N2F4反应生成Ph3CNF2, 收率可达88%. Ph3CNF2在浓硫酸中可分解成三苯甲基正离子和HNF2, 故Ph3CNF2在强酸下也能与酮反应, 生成相应的偕二氟氨基化合物.因此, (Ph)3CNF2可作为室温稳定、安全、可操作性强的二氟氨基化试剂, 具有广阔应用空间.唯一不足的是, 合成Ph3CNF2过程中仍需要使用危险性高的N2F4以及有毒金属汞.
F2NOSO2F是一种具有强氧化性的氟化试剂, 通过与碳、氮阴离子化合物发生氧化反应, 生成相应的二氟氨基化产物. 1963年, Lustig等[67]通过S2O6F2与N2F4之间自由基反应, 首次合成了F2NOSO2F, 并完成了结构和部分物化性能测试. F2NOSO2F熔点为-128.8 ℃, 沸点为-2.5 ℃, 液体密度为1.5 g/cm3.目前, 文献已利用F2NOSO2F与二硝甲基阴离子化合物、唑阴离子化合物反应, 成功合成了二氟氨基二硝甲基化合物和N—NF2唑类化合物[68~71], 该试剂已成为构建二氟氨基高能化合物的重要手段, 如Scheme 7所示.
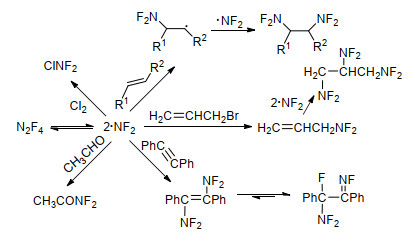
N2F4是一种无色气体状态的全氟氮化合物[72, 73], 其制备方法有多种, 但主要以HNF2氧化和NF3与金属氟化反应两种方法制备[51], 如Scheme 8所示. N2F4分子中N—N单键较长(0.14 nm), 键能较低, N—N单键解离能介于80~91 kJ/mol之间, 很容易生成二氟氨自由基[31].二氟氨自由基反应活性高, 能与烯烃、炔烃等不同类型结构的底物反应, 且产物复杂, 也是制备二氟氨基化合物的重要途径[35, 74~76], Scheme 9列举了部分示例.
除了上述介绍试剂外, CsOSO2OF[77]、Select-fluor™[78]及NF3[79]等也可作为N—F键或二氟氨基化试剂, 但限于篇幅, 不在赘述.
唑环(包括咪唑、吡唑、三唑、四唑等)化合物具有氮含量高、生成焓大、爆轰性能优异和相对钝感等特性, 在新型炸药、低特征信号推进剂、气体发生剂等领域具有广泛的应用前景.将唑环上氢用氟取代后会有以下好处: (1)消除唑类化合物酸性, 改善其相容性; (2)化合物的密度、氧平衡和生成焓进一步提高, 进而提升化合物爆速和爆压; (3)唑环芳香性能够与N—F键形成共轭体系, 进而增加结构稳定性.
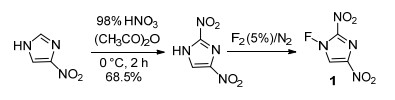
Forohar等[44]报道了硝基咪唑化合物氮氟化反应, 以4-硝基咪唑为原料, 在浓硝酸和乙酸酐体系下对其硝化合成2, 4-二硝基咪唑中间体, 再使用5% F2直接对其氟化, 获得了室温稳定的氟化产物2, 4-二硝基氟化咪唑(1), 合成路线如Scheme 10所示.但后续关于该化合物的密度、热稳定性、感度及爆轰等进一步性能数据并未见报道.
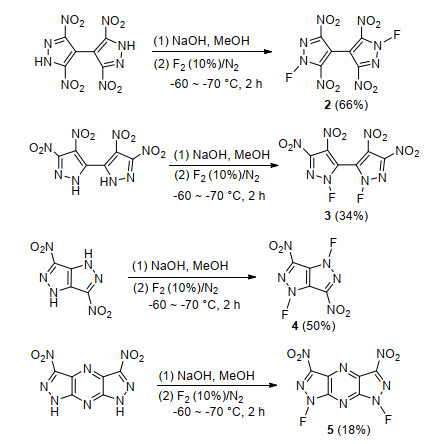
Dalinger等[45]尝试在甲醇或乙腈溶液中使用过量的CsOSO2F对多硝基联吡唑、多硝基并吡唑等的钠盐进行氟化, 期望得到吡唑氮氟化物, 但反应过程中并未检测到相关产物.实验发现, 反应结束后CsOSO2F已全部消耗, 且溶液呈酸性, 作者认为CsOSO2F首先与溶剂发生氟化反应, 生成HF并将吡唑钠酸化成很难被氟化的N—H结构, 导致氟化失败.随后该课题组使用10% F2对相应吡唑钠盐进行氟化, 为了减少副反应及HF带来不利因素, 反应加入NaF作为HF吸收剂, 在-60~-70 ℃低温条件下成功获得相应氟化物, 其中3, 3', 5, 5'-四硝基-1H, 1'H-4, 4'-联吡唑(2)收率最高, 达到66%, 其他介于18%~50%之间, 合成路线如Scheme 11所示.热分析结果显示, 合成的吡唑氮氟化产物分解点普遍较高, 其中1, 1'-二氟-3, 3', 4, 4'-四硝基-1H, 1'H-5, 5'-联吡唑(3)和1, 4-二氟-3, 6-二硝基吡唑[4, 3-c]并吡唑(4)热分解温度高于190 ℃, 表明N—F键的引入并没有大幅降低, 分子结构稳定性.目前, 多硝基唑类化合物氮氟化及氮氟化产物性能的文献较少, 该研究为后续相关研究提供了参考.
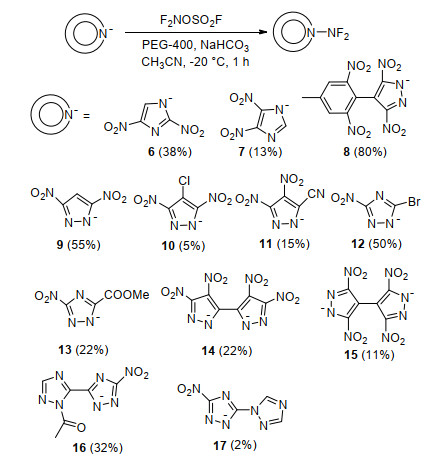
为了进一步提升N—F键在唑类含能化合物中的结构占比, Dalinger等[80]以多硝基取代的咪唑、吡唑和三唑盐为原料, 乙腈为溶剂, PEG-400作为相转移助剂, 使用二氟氨基试剂F2NOSO2F, 合成了咪唑、吡唑和三唑的N—NF2化合物, 其中化合物9, 10, 12, 17极度不稳定, 并未完成分离表征, 其收率根据19F NMR推测得到, 合成路线如Scheme 12所示.由于母体结构的不同, 这些硝基唑的N—NF2化收率差异较大, 介于5%~80%之间.
表 1列举了部分化合物的热分解温度(Tdec)、密度和爆轰性能, 并与黑索金(RDX)进行了对比.由于二氟氨基的引入, 该类化合物密度普遍较高, 化合物6、7和14的密度均超过1.90 g/cm3.但与Scheme 11中吡唑氮氟化产物相比, 该类化合物热稳定性普遍较差, 化合物11、14和15的分解温度不超过100 ℃.化合物6密度较高、氧平衡较好, 故其爆速(Vdet)达到8500 m/s, 爆压(P)超过30 GPa.另外, 化合物14的单晶结构表明N—F和Nring—Ndifuoroamino平均键长分别为0.1367和0.1410 nm, F—N—F和N—N—F平均键角为101.5°和104.2°, N—NF2键长较长, 解离能较低, 解释了该类化合物较高的感度和低的热分解温度的原因.
 下载:
导出CSV
下载:
导出CSV
| Substrate | Tdec/℃ | ΔΗf/(kJ·mol-1) | ρ/(g·cm-3) | Vdet/(m·s-1) | P/GPa |
| 6 | 70 | 117.2 | 1.92 | 8536 | 31.4 |
| 7 | 62 | 118.4 | 1.91 | 8494 | 31.0 |
| 10 | 127 | 228.0 | 1.80 | 7925 | 27.4 |
| 14 | 100 | 421.7 | 1.92 | 8257 | 30.9 |
| 15 | 84 | 458.8 | 1.88 | 8462 | 30.0 |
| RDX | 204 | 67 | 1.82 | 8850 | 34.9 |
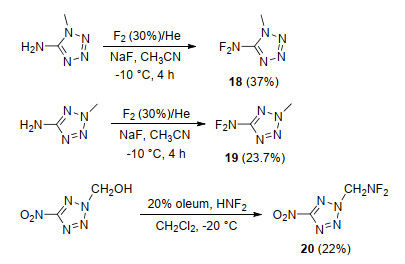
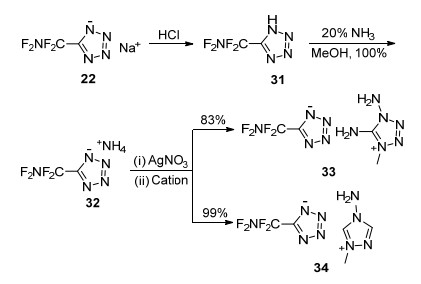
Fokin等[81]以1-甲基-5-氨基四唑、2-甲基-5-氨基四唑为原料, 在NaF的乙腈溶液中, 利用F2直接对其氟化, 成功获得了5-二氟氨基-1-甲基四唑(18)、5-二氟氨基-2-甲基四唑(19), 收率分别为37%和23.7%, 如Scheme 13所示.这是首次将二氟氨基引入到四唑结构中, 且得到的目标物能在常规条件下稳定存在.随后, 为了将二氟氨基引入到四唑烷烃基上, 该课题组又使用二氟化氨(HNF2)对2-羟甲基-5-硝基四唑进行氟化, 得到了2-二氟氨甲基-5-硝基四唑(20), 收率为22%.上述研究成功实现了向四唑环上碳以及四唑上氮甲基上引入二氟氨基, 丰富了氟氮唑含能化合物种类.
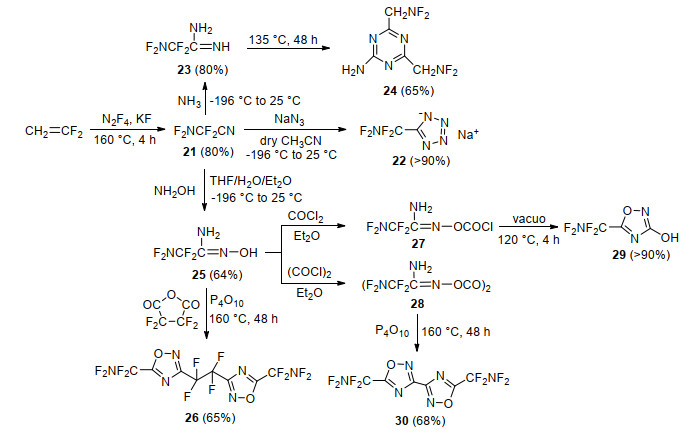
Shreeve等[82]以乙烯与N2F4为原料, 在高温和氟化钾作用下合成了二氟氨基二氟乙腈(21), 由于氰基较好的反应活性, 可以以此衍生出多种二氟氨甲基取代唑类化合物, 如Scheme 14所示.该课题组[83, 84]通过化合物21与氨发生加成反应, 合成了氨化产物23, 该化合物高温加热后聚合成二氟氨甲基取代的均三嗪化合物24.同样, 中间体21与羟胺反应, 获得了中间体25, 将其与酸酐或光气反应, 在高温下脱H2O或HCl后, 环化成二氟氨甲基取代的异呋咱化合物26、29和30.随后, 该课题组[85]通过中间体21与叠氮化钠发生环化反应, 高收率得到二氟氨基二氟甲基四唑钠盐化合物22.然而, 相关化合物的密度、热性能、感度及爆轰性能并未见报道.
鉴于四唑钠盐22具有较高生成焓及较好的能量水平, 2007年Shreeve课题组[86]通过钠盐22酸化获得二氟氨基二氟甲基四唑(31), 利用四唑酸性, 与氨水成盐得到其铵盐32.铵盐32与硝酸银作用生成银盐, 银盐与有机富氮盐1, 5-二氨基-4-甲基四唑和1-甲基-4-氨基-1, 2, 4-三唑的碘化物发生复分解反应, 生成了含二氟氨基的四唑富氮含能盐33~34 (Scheme 15).与化合物33和34相比, 铵盐22能量性能最好, 其实测密度达到1.88 g/cm3, 热分解温度为158 ℃, 理论爆速和爆压分别为8490 g/cm3和28.8 GPa, 显示出优异的爆轰性能.
在现有母体中引入新型高能基团是提升含能材料能量水平的有效途径.二氟氨基二硝甲基是一类结构稳定、能量较高的新型高能基团, 相比二硝甲基, 二氟氨基的引入不仅能够显著提高能量水平, 还能消除二硝甲基的酸性; 与三硝甲基相比, 二氟氨基更小的空间占位有望实现更好的结构稳定性和更大的堆积密度.
多硝基烷烃含能化合物具有种类丰富、研究起始时间早的特点, 将二氟氨基二硝甲基引入到烷烃中是早期开发新型高能材料的首选策略. Litvinov等[87]以F2NO-SO2F为二氟氨基试剂, 开展了二硝甲基烷烃化合物的二氟氨基化研究, 首次合成了系列具有二氟氨基二硝甲基结构的新型氧化剂二氟氨基三硝基甲烷[(NO2)3-CNF2] (35)、1-二氟氨基-1, 1, 2, 2, 2-五硝基乙烷[(NO2)3C-C(NO2)2NF2] (36)、氟代二氟氨基二硝基甲烷[(NO2)2C-F(NF2)] (37)、1, 3-双(二氟氨基)-1, 1, 3, 3-四硝基丙烷[CH2C(NO2)2NF2] (38)、1, 5-双(二氟氨基化)-1, 1, 3, 5, 5-五硝基-3-氮杂戊烷化合物{[NF2C(NO2)2CH2]2NNO2} (39).据Shreeve等[86]引述, 化合物39密度达到2.04 g/cm3, 这在非环含能化合物中是非常高的.二氟氨基二硝甲基的成功构建为新型高能基团开启了新的方向, 推动了后续二氟氨基二硝甲基化合物开发.
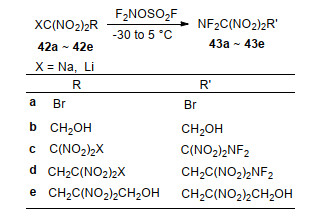
Fokin等[68]开展了反应底物结构对二硝甲基烷烃阴离子二氟氨基化收率的影响研究.以F2NOSO2F为二氟氨基化试剂, 分别完成了甲基/乙基羧酸酯二硝甲基锂盐、钠盐和钾盐在乙腈中的二氟氨基化反应(Scheme 16), 发现阳离子种类对反应收率影响较大, 其中锂盐收率最差, 未有目标产物生成, 钠盐收率最高, 收率均超过了50%.研究还发现钾盐化合物更易引发F2NO-SO2F发生歧化反应, 生成FNO和SO2F2, 不利于反应进行.基于上述结果, 作者尝试使用该方法对1, 1, 3, 3-四硝基丙烷二钠盐进行二氟氨基化, 但却导致了底物的剧烈分解.由此表明, 二硝甲基邻位基团(如羧酸酯等)对该反应也有一定的影响.
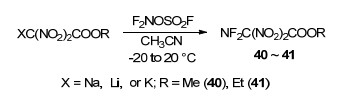
为了能够实现1, 1, 3, 3-四硝基丙烷二钠盐等化合物的二氟氨基化, Fokin等系统地开展了溶剂极性对二硝甲基钠盐、锂盐化合物的二氟氨基化的影响规律研究.由于该反应在大极性溶剂MeCN (ε=37.5)和小极性溶剂CH2Cl2 (ε=8.9)中都未成功, 因此该课题组尝试极性适中的Me2CO (ε=20.7)、MeCO2Et (ε=6.0)、二甘醇二甲醚(ε=7.5)以及混合溶剂环丁砜-CH2Cl2 (V/V=I:2, ε=24.7)、MeCN-CH2Cl2 (V/V=1:1、1:2、1:3, ε=26.1、21.2和19.9)作为反应溶剂.实验发现, 反应温度为-20~-40 ℃时, 在二甘醇二甲醚中会导致反应底物剧烈分解; 其他溶剂中随反应的进行, F2NOSO2F都会迅速被消耗, 底物二硝甲盐也很快溶解, 但只有在体积比为1:2和1:3的MeCN-CH2Cl2混合溶剂中分离获得相应的二氟氨基二硝甲基化合物(Scheme 17).产物的43a, 43d, 43e收率相对较好, 分别为65%, 41%和25%. 1, 2-双(二氟氨基)-1, 1, 2, 2-四硝基乙烷(43c)分子结构空间上高度拥挤, 导致其收率最低, 仅为5%~7%.
随后, 该课题组以2, 2, 4, 4-四硝基戊烷-1, 5-二醇(44)为原料, 在氧化镁的水溶液中使用F2NOSO2F, 完成了4-二氟氨基-2, 2, 4, 4-四硝基正丁醇(45)的合成, 反应过程中伴有副产物1, 3, -双(二氟氨基)-1, 1, 3, 3-四硝基丙烷(43d)的生成(Eq. 1).
|
|
(1) |
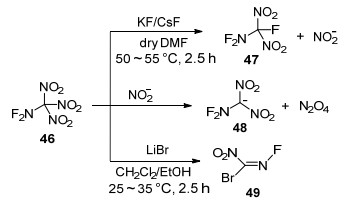
基于三硝甲基化合物与亲核试剂普遍具有较好的反应性, Khisamutdinov等[88]以无水N, N-二甲基甲酰胺(DMF)为溶剂, 开展了二氟氨基三硝基甲烷(46)与亲核试剂KF、CsF、LiBr反应研究(Scheme 18).结果发现, 与多硝基甲烷类似, 化合物46中一个硝基被氟取代, 生成了二氟氨基二硝基氟代甲烷(47), 反应收率与亲核试剂阳离子种类有关, KF作亲核试剂时收率仅为9%, 而采用CsF时收率可达40%.反应过程中生成的二氧化氮阴离子(NO2−)会与原料46发生反应, 生成二氟氨基二硝甲基阴离子48, 该阴离子在DMF中会进一步分解成氮氧化物, 因此导致总体收率较低.
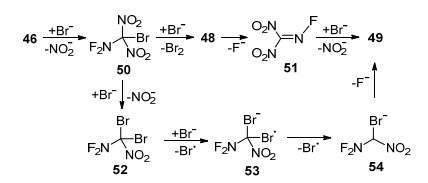
当该研究组尝试将二氟氨基三硝基甲烷与LiBr反应, 希望溴阴离子像氟离子一样取代一个硝基生成二氟氨基二硝基溴代甲烷(50)时, 结果却生成了N-氟代溴代硝基甲烷(49), 推测的两条可能的反应过程如Scheme 19所示.首先, 原料46与溴阴离子发生亲核取代反应, 生成中间体50, 与溴代三硝基甲烷一样, 50在溴的诱导下发生氧化还原反应生成不稳定的阴离子化合物48, 随后发生氟消除反应得到51, 最后再与溴阴离子发生取代生成49.另外一条可能的途径是, 46与溴阴离子连续发生两次取代反应生成52, 与四硝基甲烷生成二溴二硝基甲烷一样, 通过单电子转移得到的自由基阴离子化合物53进一步分解成硝基甲烷阴离子化合物54, 最后氟负离子离去转变成目标物49.
氮杂芳环含能化合物是近年研究的热点, 利用氮杂芳环反应位点向其引入硝基、硝胺基、硝酸酯、多硝甲基等含能基团, 结合氮杂芳环N—H键的酸性与各种富氮碱匹配, 能够实现含能材料性能的有效调节, 获得性能各异的新型含能材料.高能基团二氟氨基二硝甲基与氮杂芳环结合是探索高能量密度化合物的必然选择.
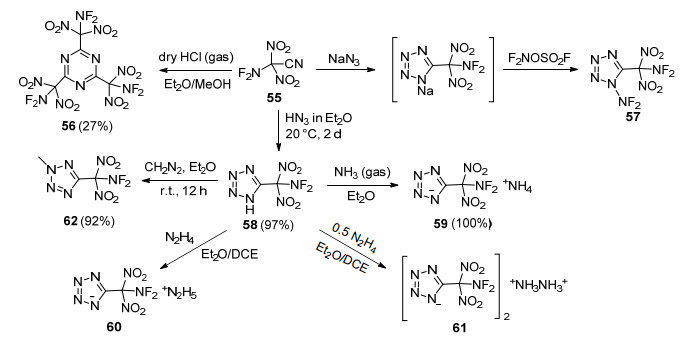
二氟氨基二硝基乙腈(55)[89]是合成二氟氨基化合物的重要中间体, 其结构稳定且氰反应活性高, 为构筑二氟氨基二硝甲基含能化合物提供基础. Fokin等[90]利用55在干燥氯化氢乙醇溶液中发生氰基聚合反应, 生成1, 3, 5-三(二氟氨基二硝甲基)均三嗪(56) (Scheme 20), 但该反应可重复性较差, 收率介于1%~30%之间.利用氰基与叠氮化钠环化反应得到其四唑钠盐中间体, 使用F2NOSO2F进一步对钠盐二氟氨基化, 获得了N—NF2四唑化合物(57), 但该化合物过于敏感, 并未完成其结构鉴定.随后, 该研究团队又将55与HN3在低温乙醚溶剂中合成了58, 收率高达97%.化合物54在室温下存放二周后会缓慢分解, 但能够稳定存在于醚溶液中.利用四唑酸性得到了其铵盐59和肼盐60~61, 与重氮甲烷反应生成了2-甲基-5-二氟氨基二硝甲基四唑(62)和1-甲基-5-二氟氨基二硝甲基四唑, 二者量约为16:1.铵盐59和甲基化产物62非常稳定, 铵盐熔点为131~132 ℃, 铵根离子与二氟氨基二硝甲基之间产生的分子间氢键有助于结构的稳定.
2015年, Dalinger等[69]开展了向吡唑N-二硝甲基的二氟氨化研究, 以3, 5-硝基-1-二硝甲基吡唑钾盐(63)为原料, 在-15 ℃的乙腈溶液中, 利用F2NOSO2F对其二氟氨基化, 首次将二氟氨基二硝甲基引入到吡唑环中, 收率达75% (Eq. 2).化合物64实测密度为1.92 g/cm3, 生成焓为193 kJ/mol, 分解温度为113 ℃, 理论爆速和爆压为8721 m/s和36.3 Gpa, 可作为潜在的高能氧化剂使用.
|
|
(2) |
2017年, Semenov等[70]采用同样的方法, 开展了两个异构体2, 2'-双(二硝甲基)-5, 5'-二硝基联(1, 2, 4-三唑)和2, 2'-双(二硝甲基)-5, 5'-二硝基联(1, 2, 3-三唑)的二氟氨基化研究, 使用其钠盐在更低的反应温度下(-30~-40 ℃)进行, 收率分别为70%和50%, 如Scheme 21所示.单晶结构显示, 2, 2'-双(二氟氨基二硝甲基)-5, 5'-二硝基联(1, 2, 4-三唑)(68)结构中两个三唑环共平面, 晶体密度为1.896 g/cm3. 2, 2'-双(二氟氨基二硝甲基)-5, 5'-二硝基联(1, 2, 3-三唑)(72)晶体结构显示两个硝基在分子同一侧, 有一定的空间位阻效应, 导致两个三唑环存在一定的夹角, 其晶体密度为1.936 g/cm3, 高于其异构体54, 表明取代基立体效应对含能分子堆积密度影响较大.
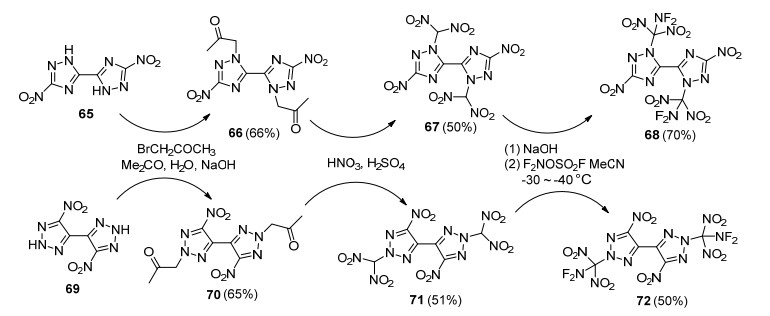
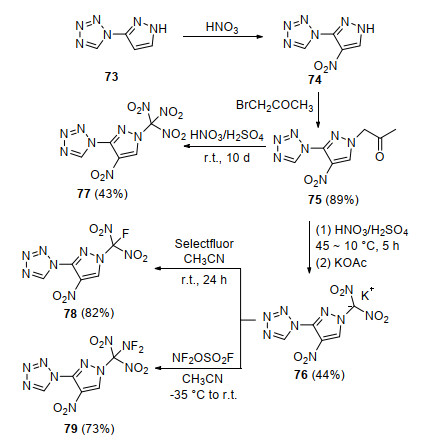
为了进一步提高二氟氨基二硝甲基吡唑化合物的能量, 更好探究其与氟二硝甲基、三硝甲基类似物结构和性能差异, Dalinger等[71]于2018年开展了二氟氨基二硝甲基、氟二硝甲基和三硝甲基取代的四唑联吡唑化合物的合成、结构与性能对比研究, 如Scheme 22所示.以四唑联吡唑(73)为母体, 通过硝化和取代两步反应得到关键中间体75, 使用100% HNO3/100% H2SO4对75再次硝化获得二硝甲基钾盐化合物76和三硝甲基产物77, 利用Selectfluor和F2NOSO2F分别对钾盐76氟化和二氟氨基化得到其氟二硝甲基产物78和二氟氨基二硝甲基产物79.通过化合物77、78和79单晶结构分析发现, 四唑和硝基处于吡唑环的邻位, 存在一定位阻效应, 导致四唑环和吡唑环具有较大的二面角(40°~60°).在-173 K下, 77、78和79晶体密度分别为1.857, 1.873和1.972 g/cm3, 证实了二氟氨基对含能分子密度的显著贡献.通过对晶体中分子间弱作用力分析发现, 氟原子参与分子间范德华作用的程度对化合物密度有直接影响, 79分子中两个氟原子与其他分子间硝基氧及四唑氮形成了数量可观的F…O和F…N作用, 有效地拉近了分子间距离, 从分子层面解释了79密度高的原因.
表 2列举了77、78和79的热分解温度、密度、爆速等性能.由表 2可知, 四唑的引入使得三个化合物的生成焓普遍较高, 但氟二硝甲基、三硝甲基和二氟氨基二硝甲基对生成焓贡献差别较大, 其中氟二硝甲基化合物78生成焓最小, 化合物77和79生成焓基本相当.让人意外的是, 二氟氨基化合物79分解温度高于77和78, 表明二氟氨基二硝甲基具有良好的结构稳定性.高的密度和生成焓赋予了化合物79优异爆轰性能, 其爆速接近9000 m/s, 爆压达36 GPa, 优于相应的氟二硝甲基化合物78和三硝甲基化合物77.化合物79更大的优势在于比冲, 其理论比冲(Isp=282s)显著高于季戊四醇四硝酸酯(PETN, 263s)、TNT (210 s)和RDX (266 s)等化合物.由此可以看出, 基于N—F键的高能化合物在密度、爆速、爆压和比冲方面优势明显, 佐证了该类化合物潜在的开发前景.
下载:
导出CSV
为了避免具有α-H结构的二氟氨基化合物自身发生HF消除反应, 以无α-H结构的偕二氟氨基化合物逐渐成为二氟氨基化合物研究热点.羰基等不饱和官能团与二氟氨基化试剂HNF2、NF2SO2F等发生亲核加成反应, 是获得偕二氟氨基含能化合物主要途径.
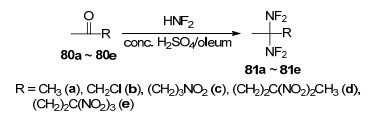
链状偕二氟氨基含能化合物是最早开发的一类偕二氟氨基含能化合物, 并证实了无α-H结构的偕二氟氨基化合物的结构稳定性, 促进了后续环状偕二氟氨基含能化合物的发展. Baum[58]以不同取代基的甲基酮链状化合物为底物, 在强酸条件下与HNF2发生亲核加成反应, 获得了多种结构稳定的偕二氟氨基链状化合物, 如Scheme 23所示.二氟氨基化反应收率受反应底物结构、反应体系酸性强度等影响较大.实验发现, 具有吸电子取代基的化合物需要强化反应条件, 如加大HNF2浓度、更高的反应温度、更强的酸性介质(如发烟硫酸)等.以5, 5, 5-三硝基-2-戊酮(80e)为例, 使用浓硫酸在回流温度反应4 h时收率为零; 当80e浓度为4 mL/mmol, 使用100%硫酸、HNF2过量8倍时, 室温下反应40 h的收率为53%.当底物浓度进一步降低至0.7 mL/mmol, 使用20%发烟硫酸、3倍HNF2, 室温下2 h的收率即可高达99.5%.由此可见, 该类型反应收率受反应条件强弱和底物结构影响较大.
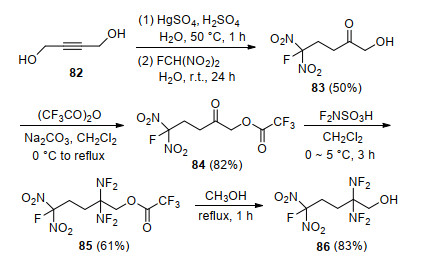
Coon等[63]以2-丁烯-1, 4-二醇为原料, 通过氧化、加成、酯化及二氟氨基化等反应, 报道了一种含偕二氟氨基和氟二硝甲基的高能增塑剂2, 2-双(二氟氨基)-5-氟-5, 5-二硝基-1-戊醇(86) (Scheme 24).化合物86外观呈浅黄色, 熔点为14 ℃, 沸点为79~80 ℃, 该化合物分子结构稳定, 且氟含量较高, 将其作增塑剂或氧化剂使用, 能够显著提高双基推进剂的比冲和燃速. Baum等[64]利用同样的方法, 对1, 3-双(2, 2, 2-氟二硝基乙氧基)丙酮进行二氟氨基化, 获得1, 3-双(2, 2, 2-氟二硝基乙氧基)-2, 2-偕二氟氨基丙烷, 分子能量和氧含量得到进一步提升.
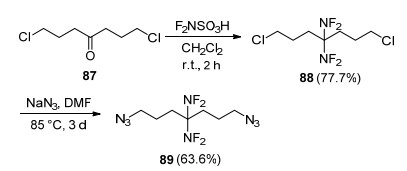
利用叠氮基高生成焓(356 kJ/mol)的特点, Flanagan等[65]设计合成了一种具有两个叠氮基和偕二氟氨基结构的化合物(89).以1, 7-二氯-4-庚酮为原料(87), 通过二氟氨基化和叠氮负离子取代反应合成了1, 7-二叠氮基-4, 4-偕二氟氨基庚烷(89), 如Scheme 25所示.为了进一步考察其应用可行性, 国内黎明化工研究院张明权等[91]开展了合成工艺优化, 确定了二氟氨基化反应原料配比, 将总收率提高到70%, 并报道了化合物89起始分解温度为195.2 ℃, 表明热稳定性良好.为了降低其感度, 张明权等采用复合降感剂, 将化合物89的撞击感度由5.9 cm改善至30 cm以上, 摩擦感度由100%降低至10%以下, 为化合物89的安全使用提供了保证.
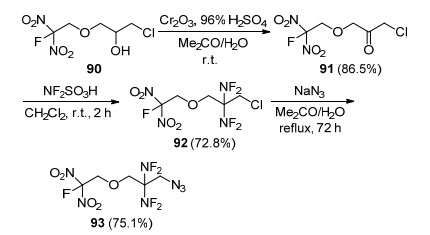
Frankel等[92]以3-氟二硝乙氧基-1-氯-2-丙醇(90)为原料, 通过氧化、二氟氨基化、取代等多步反应合成了1-叠氮-2, 2-双(氟二氨基)-3-氟二硝乙氧基丙烷(93) (Scheme 26), 该化合物分子中同时含有偕二氟氨基、叠氮基和氟二硝甲基, 因此具有高生成焓、富氧等特点, 在推进剂组分中可以作为含能增塑剂和高能氧化剂使用.
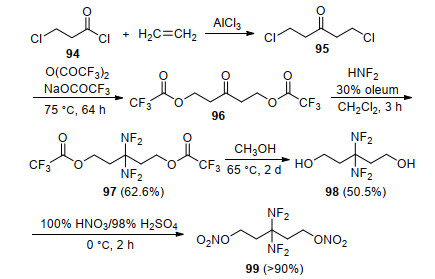
为了开发合成步骤简单且能量水平进一步提高的偕二氟氨基含能增速剂, Adolph等[93]以1, 5-二氯戊烷-3-酮(94)为原料, 通过三步反应合成了3, 3-双(二氟氨基)-1, 5-二硝酸酯基戊烷(99), 如Scheme 27所示.该分子含有二个硝酸酯基和一个偕二氟氨基, 密度为1.56 g/cm3, 玻璃化转变温度为-87 ℃, 初始分解温度为186 ℃, 峰温为196 ℃, 生成热为-320.0 kJ/mol, 其理论比冲可达278.9 s, 显著高于1, 2, 4-丁三醇三硝酸酯(BTTN, 262.3 s)和1, 3-双(2, 2, 2-氟二硝基乙氧基)-2, 2-偕二氟氨基丙烷(266.5 s).与其他偕氟二氨基增塑剂相比, 其在合成难易程度、热稳定性及能量水平等方面均具有明显优势.
以六元环和八元环为代表环状含能化合物(如RDX和HMX)在含能材料中占有重要地位, 致密对称的环结构赋予了此类化合物密度高、热稳定性好的特点.近年, 以HNFX为代表的环状偕二氟氨基含能化合物展现出了巨大的发展潜力.
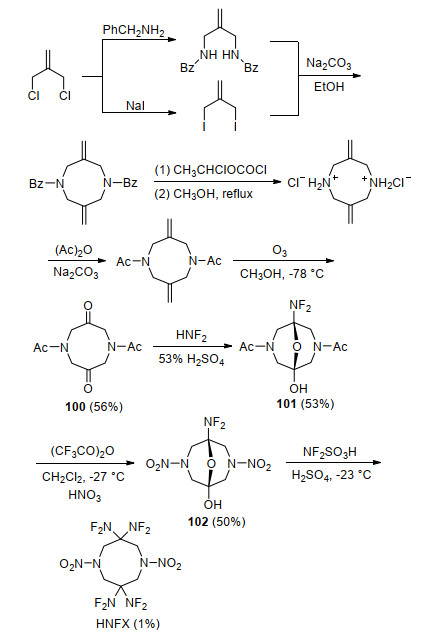
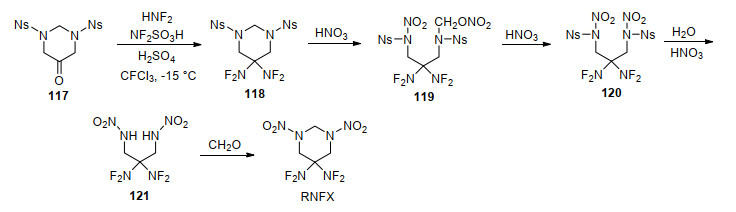
HNFX是类似HMX的八元环状偕二氟氨基硝胺化合物, 自其首次被设计出来便成为含能材料研究人员关注的焦点. HNFX的晶体密度为1.807 g/cm3, 初始分解温度为221 ℃, 峰温为231 ℃, 撞击感度与CL-20相当, 摩擦感度和静电感度略高于CL-20和PETN[29].目前发现的HNFX晶体密度仅为1.807 g/cm3, 这与Ammon等[94]预估的2.027 g/cm3差距较大.但相关研究人员普遍认为HNFX与HMX、CL-20一样存在多种晶型, 具有更高密度的晶型还有待进一步发现. Baum等[95]在一篇提交给美国海军研究局的报告中首次报道了HNFX合成路线, 如Scheme 28所示.在最后一步反应中, 由于中间体102分子中硝胺结构在强非硝酸条件下易分解, 致使最后一步反应收率仅为1%, 限制了进一步性能及应用可行性研究.
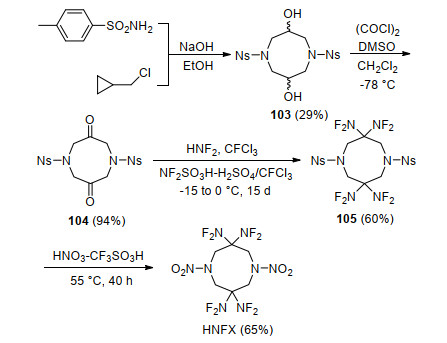
为了简化反应步骤并提高收率, Chapman等[96, 97]研究了N-保护基种类对氮杂辛烷二酮二氟氨基化反应的影响, 发现保护基的酸碱性对二氟氨基化和硝化反应具有重要影响.基于此, 该研究组以对硝基苯磺酰胺和环氧氯丙烷为原料, 首先合成了对硝基苯磺酰基为保护基中间体八氢-1, 5-二(4-硝基苯磺酰基)-1, 5-二氮杂辛烷-3, 7-二醇(103), 再利用斯文(Swern)氧化反应将其氧化成二酮化合物104, 该步收率为94%.随后通过优化反应温度以及向二氟氨基化反应体系中加入惰性试剂CFCl3, 将二氟氨基化收率提高至60%.最后, 利用筛选的高效硝化体系HNO3-CF3SO3H, 将HNFX最后一步收率由原来1%大幅提高到65%, 如Scheme 29所示.
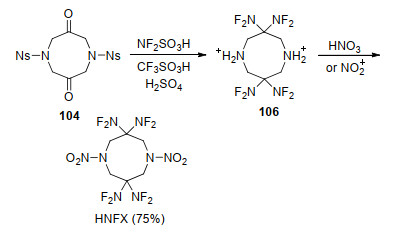
尽管上述方法在小试合成HNFX时较为成功, 然而放大后收率会显著下降.为此, Chapman等[98, 99]开发了从二酮化合物104到HNFX“一锅法”合成工艺, 如Scheme 30所示.通过向二氟氨基化体系中加入三氟磺酸, 将氮上的对硝基苯磺酰基在三氟磺酸作用下脱去, 成双氢取代的季铵正离子化合物106, 再向反应体系中加入98%~100%硝酸进行硝化反应, 一锅法合成HNFX, 将总收率提高到75%, 中间体均不需要分离, 大大简化了实验步骤, 为应用性能和基础应用研究提供了基础.
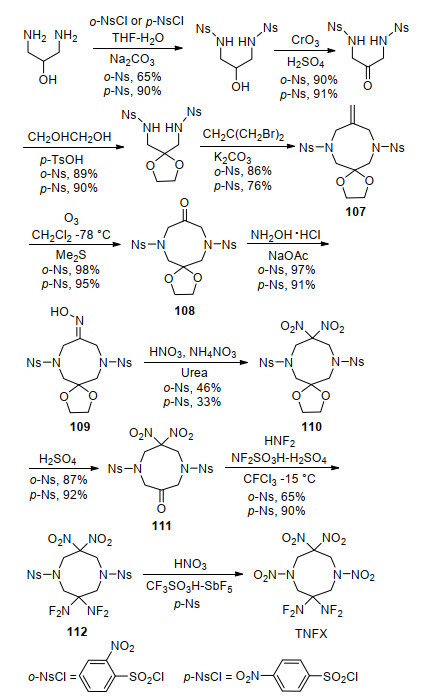
3, 3-双(二氟氨基)二八氢化-1, 5, 7, 7-四硝基-1, 5-二氮杂辛烷(TNFX)结构与HNFX类似, 将HNFX分子中一个偕二氟氨基换成二硝甲基即得到TNFX, 该化合物相比HNFX氧平衡得到改善. 2001年, Axenrod等[100]以2-羟基-1, 3-丙二胺为原料, 经氨基保护、羟基氧化、环化等构建了1, 5-二氮杂辛烷母体化合物107, 随后利用与HNFX相似的硝化、二氟氨基化等多步反应合成了TNFX, 其中二氟氨基化步骤收率可达90%, 如Scheme 31所示.研究发现, TNFX存在多晶现象, 三方晶系的晶体密度较低, 为1.712 g/cm3, 而正交晶系的晶体密度显著高于三方晶系, 达1.902 g/cm3, 这对寻找更高密度的HNFX具有指导意义.
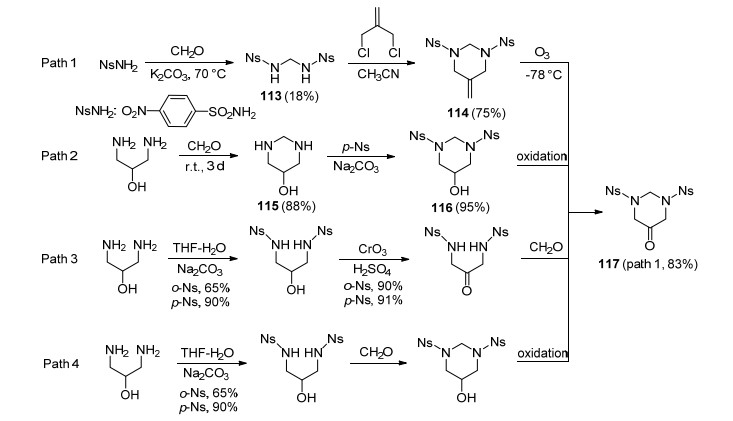
5, 5-双(二氟氨基)六氢-1, 3-二硝基嘧啶(RNFX)[101]是分子结构与RDX类似的六元环状偕二氟氨基硝胺化合物, 是Chapman课题组继HNFX之后合成的又一重要标志性化合物.合成六氢嘧啶酮是合成该化合物的关键步骤之一, 与HNFX合成思路相似, 首先构建对硝基苯磺酰基保护的六氢嘧啶酮117.公开的专利描述了两条路线:第一条路线是将对硝基苯磺酰胺先与甲醛作用, 生成硝基苯磺酰二胺化合物113, 收率仅为18%, 随后通过亲核取代环化和氧化反应得到117, 收率为83%.第二条路线是使用2-羟基丙二胺与甲醛缩合环化生成羟基取代的六氢嘧啶115, 收率为88%, 其与对硝基苯磺酰胺的反应收率高达95%, 但该专利并未报道后续氧化生成117的具体步骤.但将仲醇氧化成酮条件颇多, 如在合成HNFX时使用的斯文(Swern)氧化反应等, 作者认为第二条合成路线无论从收率、成本等方面考虑均更具优势.同时, 作者也设计另外两条合成路线, 供读者参考, 见Scheme 32所示.
参照合成HNFX和TNFX的二氟氨基化条件, 中间体117能够顺利转化成相应的偕二氟氨基中间体118.当使用98%硝酸对其硝解合成RNFX时, 六氢嘧啶环中处在两个氮中间的亚甲基发生开环反应, 生成中间体119, 在含有少量水的硝酸作用下, 119进一步发生去苯磺酰基化反应生成121, 其可以立即与等量甲醛发生环化反应, 得到目标化合物RNFX.该硝解过程与合成HNFX时硝化反应过程差别较大, 如Scheme 33所示.尽管该化合物的合成报道已过多年, 但其性能一直未见后续报道.该化合物分子结构较为完美, 硝胺基和二氟氨基在六元环中分布合理紧凑, 并且合成步骤相对简单, 作者认为该化合物是目前最具潜力的氟氮含能化合物之一.
除上述关注度较高的偕二氟氨基硝胺化合物外, Baum[58]以环状酮为底物, 利用HNF2/H2SO4体系合成了多种偕二氟氨基五元环和六元环烷烃化合物, 如表 3所示.该方法需要在强酸条件下使用过量的HNF2才能获得较好的收率.为了改善HNF2使用带来的安全隐患, Prakash等[66]于2002报道了新型二氟氨基化试剂三苯基二氟氨基甲烷, 并用该试剂合成了多种环状二氟氨基化化合物, 收率均能达到80%以上, 为偕二氟氨基化合物提供了一种安全、高效的合成方法.
下载:
导出CSV
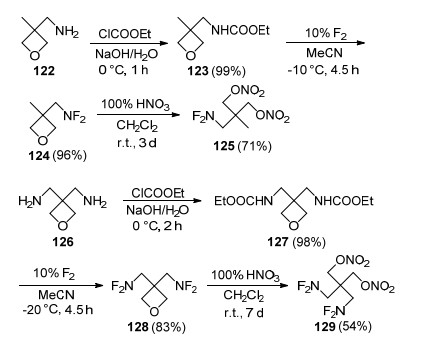
研究发现, 当二氟氨基与新戊基相连, 由于(CH3)3CCH2空间位阻效应, 抑制了二氟氨基与α-H发生消除HF反应, 是目前能够稳定存在具有α-H结构的二氟氨基化合物.由于分子结构稳定性的大幅提升, 因此可以使用F2直接氟化新戊基胺来合成二氟氨基化合物, 该方法工艺简单, 有利于放大实验. Manser等[50]以3-氨基甲基-3-甲基环氧丁烷(122)和3, 3-双(氨基甲基)环氧丁烷(126)为母体, 通过取代、氟化反应率先合成出了单二氟氨甲基取代的环氧丁烷化合物124和双二氟氨甲基取代的环氧丁烷化合物128, 在硝酸的二氯甲烷溶液对其硝化开环, 获得了同时具有硝酸酯基和二氟氨甲基结构的高能增塑剂2-双(二氟氨甲基)-1, 3-丙二醇二硝酸酯(125)和2-二氟氨甲基-2-甲基-1, 3-丙二醇二硝酸酯(129), 合成路线如Scheme 34所示.
2015年, 南京理工大学潘仁明等[102]对化合物124的合成进行了优化, 以三羟甲基乙烷为原料, 经溴代酯化、碱关环、高压胺化、酰胺保护和低温氟化五步反应, 得到了化合物124, 总收率为43%, 获得了经济可行的合成工艺. 2016年, 该课题组[103]完成了化合物124在浓硝酸、硝硫混酸、硫酸/硝酸钾及N2O5体系下开环硝化合成化合物125的工艺, 使开环硝化收率提高到65%;差示扫描量热(DSC)实验表明化合物125起始分解温度为162.6 ℃, 分解峰温为208.0 ℃, 同时还测定了该化合物与部分材料相容性, 结果显示化合物125与CL-20、AP及Al等均相容, 与RDX、HMX及B不相容.
2012年, 张明权等[43]完成了化合物129合成工艺优化和性能研究, 以3, 3-二氯甲基氧杂环丁烷为原料, 通过氨化、酯化、氟化反应完成了化合物129的合成, 总收率约43%.热分析表明, 化合物129熔点为43.5 ℃, 在15~200 ℃范围内不发生分解, 热稳定性良好; 撞击感度H50≈28.5 cm (5 kg落锤), 摩擦感度为100%(摆角90°, 压力9.92 MPa).同年, 潘仁明等[42]采用GJB 772A-97 502.1完成了化合物129与典型炸药、推进剂组分的化学相容性.结果表明, 化合物129与RDX、HMX、2, 6-二氨基-3, 5-二硝基-1-氧-吡嗪(LLM-105)、高氯酸铵(AP)、碱式碳酸铅的化学相容性达到1级, 与3, 4-二硝基呋咱基氧化呋咱(DNTF)、六硝基芪(HNS)、B粉、1号中定剂(二甲基二苯脲)的化学相容性达到2级, 与TNT、硝化棉(NC)-硝化甘油(NG)、Al化学不相容.
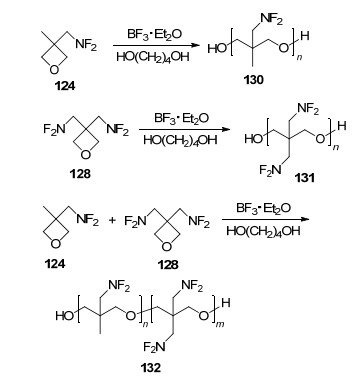
由于二氟氨基对推进剂比冲提升效果显著, 作为含能粘合剂使用的二氟氨基类聚合物早在20世纪60年代初便开始了研制工作, 由于结构不稳定以及合成难度大等问题, 并未得到实质发展.直到1995年, Manser等[104]在合成124和128单体基础上, 以二氯甲烷为溶剂, 1, 4-丁二醇/三氟化棚乙醚为催化引发剂, 成功制备出124、128的均聚物以及124~128共聚物, 如Scheme 35所示.均聚物131由于结构对称性好, 导致其结晶显著, 玻璃化转变温度(Tg)为138 ℃, 熔点为158 ℃, 初始分解温度为210 ℃.均聚物130以及共聚物132结构对称性明显降低, 在常温下均是液态, 均聚物130玻璃化转变温度为-21 ℃, 起始分解温度为191 ℃, 共聚物132起始分解温度为192 ℃, 二者作为含能粘合剂使用均有一定的优势.
2015年, 潘仁明课题组[102]开展了催化剂用量和反应温度对单体124聚合反应的影响, 获得的开环均聚优化工艺为:三氟化硼乙醚(TFBE)为阳离子催化剂, 1, 4-丁二醇(BDO)为引发剂, 聚合反应温度为-10 ℃, 反应时间为24 h, 获得的均聚物130的平均分子量(Mn)为3056, 多分散性指数(PDI)为1.73, 官能度(f)为1.87, Tg为-40.8 ℃.采用DSC法, 研究了共聚物130的热分解行为及动力学, 发现130的热分解由两个热失重阶段组成, 第一个阶段主要是HF的两步不完全解离, 第二阶段则是剩余的单氟亚胺基团与聚合物链段的分解.利用DSC法测得了130与火炸药中常见组分的化学相容性, 结果表明, 130与TNT、RDX、2, 4-二硝基苯甲醚(DNAN)、PETN、AP、Al相容性化为A级; 与HMX、CL-20、3-硝基-1, 2, 4-三唑-5-酮(NTO)、硝酸铵(AN)、Mg、B等相容性差, 为D级.
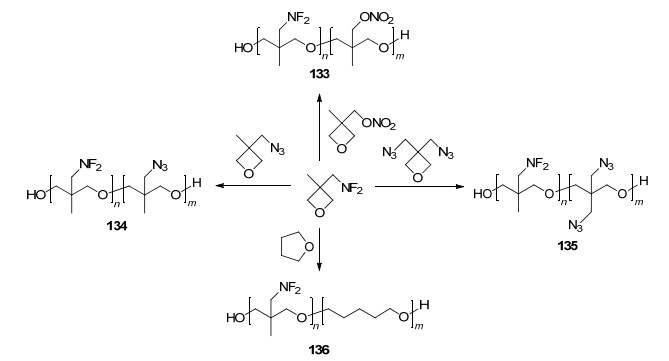
为了进一步提高生成热和氧平衡并有效降低玻璃化温度, 克服单一均聚物的性能缺陷, 潘仁明课题组[102]分别将3, 3-偕叠氮甲基氧杂环丁烷(BAMO)、3-偕叠氮甲基-3-甲基氧杂环丁烷(AMMO)、3-硝酸酯甲基-3-甲基氧杂环丁烷(NIMMO)、THF分别与124共聚, 成功将二氟氨基与叠氮基、硝酸酯结合, 合成出四种新型二氟氨基共聚物133、134、135和136, 合成路线如Scheme 36所示, 并完成了核磁、红外、Mn、PDI、f及Tg等研究(表 4).通过TG/DSC联用, 得到了共聚物的起始分解温度(Tonset)、热分解动力学及机理.该研究工作原创性强, 获得的新型二氟氨基粘合剂性能突出, 具有很高的学术和应用价值, 将我国二氟氨基粘合剂研究工作推上新的高度.
 下载:
导出CSV
下载:
导出CSV
| Substrate | Tonset /℃ | Mn/(g·mol-1) | PDI | f | Tg/℃ |
| 133 | 190 | 5772 | 1.30 | 1.98 | -24.3 |
| 134 | 175 | 4827 | 1.25 | 1.87 | -38.7 |
| 135 | 125 | 2675 | 1.79 | 2.69 | -28.4 |
| 136 | — | 2700 | 2.11 | 1.88 | -50.8 |
氟氮含能化合物在增加化合物密度、提升推进剂比冲以及提高金属粉燃烧效率等方面具有传统硝基含能化合物无法比拟的优势, 已成为开发下一代新型高能量密度材料的重要途径.经过研究人员几十年的努力, 氟氮含能化合物研究取得巨大进步, 已经开发出氟氮唑、二氟氨基二硝甲基、偕二氟氨基硝胺氮杂环等多种类型高能量密度化合物, 显示出诱人的性能和潜在的应用前景.然而, 目前仍然面临合成步骤长、反应条件苛刻、环境污染大以及氟氮化合物感度高、热稳定性不够好等问题, 导致很难批量生成和实际应用.因此, 为加快新型氟氮含能化合物研发速度和提升应用可行性, 建议可从以下几个方面开展工作: (1)开发结构稳定、安全性好且高效的新型二氟氨基化试剂, 避免使用稳定性差、易爆、环境污染大的二氟氨基化试剂, 如HNF2、N2F4等, 打破二氟氨基含能化合物合成条件限制, 加快二氟氨基含能化合物开发步伐. (2)采用共轭体系、分子内和分子间氢键、π-π堆积等策略, 改善氟氮含能化合物感度高、热稳定性差的问题, 从分子层面实现高能与降感的统一. (3)进一步探究氟氮含能化合物构效关系, 利用量子化学手段并结合晶体结构、感度和热性能实验数据, 阐明N—F键对氟氮含能化合物密度、感度、热稳定性的影响规律, 设计合成更高能量密度且结构稳定性好的新型氟氮化合物提供理论支撑. (4)加强HNFX、RNFX等具有潜在应用前景的氟氮含能化合物合成工艺优化和新路线探索研究, 寻找合成步骤短、安全性好、成本低、环境污染小的新工艺和新方法, 促进相关化合物性能和基础应用研究. (5)探索氟氮含能化合物在炸药爆轰和推进剂燃烧的反应过程和机理, 通过气体产物分析, 推测氟氮化合物氟原子在爆轰和燃烧过程中反应动力学和机理, 实现含氟氮化合物的炸药和推进剂配方最优化, 推进应用研究工作.
Gao, H.; Shreeve, J. M. Chem. Rev. 2011, 111, 7377. doi: 10.1021/cr200039c
Klapötke, T. M. Chemistry of High-energy Materials, 2nd ed., Walter de Gruyter, Berlin, 2012.
Badgujar, D. M.; Talawar, M. B.; Asthana, S. N.; Mahulikar, P. P. J. Hazard. Mater. 2008, 151, 289. doi: 10.1016/j.jhazmat.2007.10.039
薛琪, 毕福强, 张家荣, 张俊林, 王伯周, 张生勇, 有机化学, 2019, 39, 1244. doi: 10.6023/cjoc201811029Xue, Q.; Bi, F. Q.; Zhang, J. R.; Zhang, J. L.; Wang, B. Z.; Zhang, S. R. Chin. J. Org. Chem. 2019, 39, 1244 (in Chinese). doi: 10.6023/cjoc201811029
Wang, Y.; Liu, Y. J.; Song, S. W.; Yang, Z. J.; Qi, X. J.; Wang, K. C.; Liu, Y.; Zhang, Q. H.; Tian, Y. Nat. Commun. 2018, 9, 2444. doi: 10.1038/s41467-018-04897-z
Tang, Y. X.; Kumar, D.; Shreeve, J. M. J. Am. Chem. Soc. 2017, 139, 13684. doi: 10.1021/jacs.7b08789
Zhai, L. J.; Bi, F. Q.; Huo, H.; Luo, Y. F.; Li, X. Z.; Chen, S. P.; Wang, B. Z. Front. Chem. 2019, 7, 559. doi: 10.3389/fchem.2019.00559
张家荣, 毕福强, 廉鹏, 张俊林, 王伯周, 有机化学, 2017, 37, 2736. doi: 10.6023/cjoc201701014Zhang, J. R.; Bi, F. Q.; Lian, P.; Zhang, J. L.; Wang, B. Z. Chin. J. Org. Chem. 2017, 37, 2736 (in Chinese). doi: 10.6023/cjoc201701014
李云路, 薛梅, 王建龙, 曹端林, 马忠亮, 有机化学, 2016, 36, 1528. doi: 10.6023/cjoc201602011Li, Y. L.; Xue, M.; Wang J. L.; Cao, D. L.; Ma, Z. L. Chin. J. Org. Chem. 2016, 36, 1528 (in Chinese). doi: 10.6023/cjoc201602011
周静, 张俊林, 丁黎, 毕福强, 王伯周, 含能材料, 2019, 27, 708. doi: 10.11943/CJEM2018302Zhou, J.; Zhang, J. L.; Ding, L.; Bi, F. Q.; Wang, B. Z. Chin. J. Energ. Mater. 2019, 27, 708 (in Chinese). doi: 10.11943/CJEM2018302
冯晓军, 薛乐星, 曹芳洁, 刘谦, 李欣, 火炸药学报, 2019, 42, 608.Zhou, J.; Zhang, J. L.; Ding, L.; Bi, F. Q.; Wang, B. Z. Chin. J. Explos. Propellants 2019, 42, 608 (in Chinese).
Zhai, L. J.; Bi, F. Q.; Luo, Y. F.; Wang, N. X.; Zhang, J. L.; Wang, B. Z. Sci. Rep. 2019, 9, 4321. doi: 10.1038/s41598-019-39723-z
Xu, Y. G.; Shen, C.; Lin, Q. H.; Wang, P. C.; Jiang, C.; Lu, M. J. Mater. Chem. A 2016, 4, 17791. doi: 10.1039/C6TA08831G
Barton, L. M.; Edwards, J. T.; Johnson, E. C.; Bukowski, E. J.; Sausa, R. C.; Byrd, E. F. C.; Orlicki, J. A.; Sabatini, J. J.; Baran, P. S. J. Am. Chem. Soc. 2019, 141, 12531. doi: 10.1021/jacs.9b06961
Zhai, L. J.; Bi, F. Q.; Luo, Y. F.; Sun, L.; Huo, H.; Zhang, J. C.; Zhang, J. L.; Wang, B. Z.; Chen, S. P. Chem. Eng. J. 2020, 391, 123573. doi: 10.1016/j.cej.2019.123573
Vishnevskiy, Y. V.; Tikhonov, D.; Schwabedissen, J.; Stammler, H-G.; Moll, R.; Krumm, B.; Klapötke, T. M.; Mitzel, N. W. Angew. Chem., Int. Ed. 2017, 56, 9619.
Yu, Q.; Yin, P.; Zhang, J. H.; He, C. L.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M. J. Am. Chem. Soc. 2017, 139, 8816. doi: 10.1021/jacs.7b05158
Lukin, K. A.; Li, J.; Eaton, P. E.; Kanomata, N.; Hain. J.; Punzalan, E.; Gilardi, R. J. Am. Chem. Soc. 1997, 119, 9591. doi: 10.1021/ja970552q
Zhang, M. X.; Eaton, P. E.; Gilardi, R. Angew. Chem., Int. Ed. 2000, 39, 401. doi: 10.1002/(SICI)1521-3773(20000117)39:2<401::AID-ANIE401>3.0.CO;2-P
公绪滨, 孙成辉, 庞思平, 张静, 李玉川, 赵信岐, 有机化学, 2012, 32, 486. doi: 10.6023/cjoc1104225Gong, X. B.; Sun, C. H.; Pang, S. P.; Zhang, J.; Li, Y. C.; Zhao, X. Q. Chin. J. Org. Chem. 2012, 32, 486 (in Chinese). doi: 10.6023/cjoc1104225
Nair, U. R.; Sivabalan, R.; Gore, G. M.; Geetha, M.; Asthana S. N.; Singh, H. Combust., Explos. Shock Waves 2005, 41, 121. doi: 10.1007/s10573-005-0014-2
Ammon, H. L. Struct. Chem. 2001, 12, 205. doi: 10.1023/A:1016607906625
冯增国, 化学进展, 2000, 12, 171. doi: 10.3321/j.issn:1005-281X.2000.02.006Feng, Z. G. Prog. Chem. 2000, 12, 171 (in Chinese). doi: 10.3321/j.issn:1005-281X.2000.02.006
刘卉, 张英杰, 张路遥, 郑文芳, 潘仁明, 火炸药学报, 2019, 42, 363.Liu, H.; Zhang, Y. J.; Zhang, L. Y.; Zheng, W. F.; Pan, R. M. Chin. J. Explos. Propellants 2019, 42, 363 (in Chinese).
Yetter, R. A.; Dryer, F. L.; Rabitz, H.; Brown, R. C.; Kolb, C. E. Combust. Flame 1998, 112, 387. doi: 10.1016/S0010-2180(97)00123-5
Valluri, S. K.; Schoenitz, M.; Dreizin, E. Def. Technol. 2019, 15, 1.
李上文, 赵凤起, 袁潮, 罗阳, 高茵, 固体火箭技术, 2002, 25, 36. doi: 10.3969/j.issn.1006-2793.2002.02.009Li, S. W.; Zhao, F. Q.; Yuan, C.; Luo, Y.; Gao, Y. J. Solid Rocket Technol. 2002, 25, 36 (in Chinese). doi: 10.3969/j.issn.1006-2793.2002.02.009
Chapman, R. D. Struct. Bonding 2007, 125, 123.
李欢, 秦叶军, 李金华, 潘仁明, 王万军, 化学通报, 2012, 75, 1076.Li, H.; Qin, Y. J.; Li, J. H.; Pan, R. M.; Wang, W. J. Chem. Bull. 2012, 75, 1076 (in Chinese).
Klapötke, T. M. J. Fluorine Chem. 2006, 127, 679. doi: 10.1016/j.jfluchem.2006.03.001
Chen, J. F.; Yu, Y.; Li, Y. C.; Pang, S. P. J. Fluorine Chem. 2018, 205, 35. doi: 10.1016/j.jfluchem.2017.11.008
Ruff, O.; Giese, M. Eur. J. Inorg. Chem. 1936, 69, 598.
Davenas, A. J. Propul. Power 2003, 19, 1108.
Petry, R. C.; Freeman, J. P. J. Org. Chem. 1967, 32, 4034 doi: 10.1021/jo01287a068
Coon, C. L.; Ross, D. L. US 3732288, 1973.
Reed, S. F.; Shoults, R. D. J. Org. Chem. 1972, 37, 3326. doi: 10.1021/jo00986a027
Coon, C. L.; Ross, D. L. US 3714254, 1973.
Wiener, C.; Tyler, W. E. US 4128583, 1978.
郑远洋, 周继璋, 周大赉, 张明南, 兵工学报, 1988, 1, 59.Zheng, Y. Y.; Zhou, J. Z.; Zhou, D. L.; Zhang, M. N. Acta Armamentarii 1988, 1, 59 (in Chinese).
王万军, 李欢, 潘仁明, 朱卫华, 有机化学, 2019, 39, 170. doi: 10.6023/cjoc201808024Wang, W. J.; Li, H.; Pan, R. M.; Zhu, W. H. Chin. J. Org. Chem. 2019, 39, 170 (in Chinese). doi: 10.6023/cjoc201808024
李欢, 张路遥, 潘仁明, 王万军, 火炸药学报, 2012, 35, 37.Li, H.; Zhang, L. Y.; Pan, R. M.; Wang, W. J. Chin. J. Explos. Propellants. 2012, 35, 37 (in Chinese).
张明权, 刘红雨, 高宝柱, 张磊, 康玲, 张可人, 含能材料, 2012, 20, 314. doi: 10.3969/j.issn.1006-9941.2012.03.011Zhang, M. Q.; Liu, H. Y.; Gao, B. Z.; Zhang, L.; Kang, L.; Zhang, K. R. Chin. J. Energ. Mater. 2012, 20, 314 (in Chinese). doi: 10.3969/j.issn.1006-9941.2012.03.011
Laali, K. K.; Tanaka, M.; Forohar, F.; Cheng, M.; Fetzer, J. C. J. Fluorine Chem. 1998, 91, 185. doi: 10.1016/S0022-1139(98)00224-3
Dalinger, I. L.; Shkineva, T. K.; Vatsadze, I. A.; Popova, G. P.; Shevelev, S. A. Mendeleev Commun. 2011, 21, 48. doi: 10.1016/j.mencom.2011.01.020
Grakauskas, V.; Baum, K. J. Org. Chem. 1969, 34, 2840. doi: 10.1021/jo01262a010
Chapman, R. D.; Davis, M. C.; Gilardi, R. Synth. Commun. 2003, 35, 4173.
Sharts, C. M. J. Org. Chem. 1968, 33, 1008. doi: 10.1021/jo01267a014
Mcpake, C. B.; Murray, C. B.; Sandford, G. Aust. J. Chem. 2013, 66, 145. doi: 10.1071/CH12381
Archibald, T. G.; Manser, G. E. US 5789617, 1998.
Emelexus, H. J.; Shreeve, J. M.; Verma, R. D. Adv. Inorg. Chem. 1989, 33, 139. doi: 10.1016/S0898-8838(08)60195-6
Klapdor, M. F.; Willner, H.; Poll, W.; Mootz, D. Angew. Chem. 1996, 108, 320. doi: 10.1002/ange.19961080309
Freeman, J. P.; Kennedy, A.; Colburn, C. B. J. Am. Chem. Soc. 1960, 82, 5304. doi: 10.1021/ja01505a009
Petry, R. C.; Freeman, J. P. J. Am. Chem. Soc. 1961, 83, 3912.
Graham, W. H.; Parker, C. O. J. Org. Chem. 1963, 28, 850.
Grakauskas, V.; Baum, K. J. Am. Chem. Soc. 1970, 92, 2096. doi: 10.1021/ja00710a050
Banks, R. E.; Haszeldine, R. N.; Lalu, J. P. J. Chem. Soc., C 1966, 1514. doi: 10.1039/j39660001514
Baum, K. J. Am. Chem. Soc. 1968, 90, 7083. doi: 10.1021/ja01027a035
Fokin, A. V.; Kosyrev, Y. M.; Shevchenko, V. I. Russ. Chem. Bull. 1983, 31, 1626.
Graham, W. H.; Freeman, J. P. J. Am. Chem. Soc. 1967, 89, 716. doi: 10.1021/ja00979a058
Keith, J. N.; Douthart, R. J.; Sumida, W. K.; Solomon, I. J. Advanced Propellant Chemistry 1966, 141.
Haiges, R.; Wagner, R.; Boatz, J. A.; Yousufuddin, M.; Etzkorn, M.; Surya Prakash, G. K.; Christe, K. O.; Chapman, R. D.; Welker, M. F.; Kreutzberger. C. B. Angew. Chem., Int. Ed. 2006, 45, 5179. doi: 10.1002/anie.200601020
Coon, C. L.; Hill, M. E.; Ross, D. L. US 3759998, 1973.
Baum, K.; Grakauskas, V. US 4075246, 1978.
Flanagan, G. E.; Frankel, M. B.; Witucki, E. F. US 4141910, 1979.
Surya Prakash, G. K.; Etzkorn, M.; Olah, G. A.; Christe, K. O.; Schneidera, S.; Vij, A. Chem. Commun. 2002, 1712.
Lustig, M.; Cady, G. H. Inorg. Chem. 1963, 2, 388. doi: 10.1021/ic50006a036
Fokin, A. K.; Studnev, Y. N.; Rapkin, A. L.; Kuznetsova, L. D. Russ. Chem. Bull. 1996, 45, 2547. doi: 10.1007/BF01431113
Dalinger, I. L.; Shakhnes, A. K.; Monogarov, K. A.; Suponitsky, K. Y.; Sheremetev, A. B. Mendeleev Commun. 2015, 25, 429. doi: 10.1016/j.mencom.2015.11.010
Semenov, V. V.; Shevelev, S. A.; Bruskin, A. B.; Shakhnes, A. K.; Kuz'min, V. S. Chem. Heterocycl. Compd. 2017, 53, 728.
Dalinger, I. L.; Kormanov, A. V.; Suponitsky, K. Yu.; Muravyev, N. V.; Sheremetev, A. B. Chem. Asian J. 2018, 13, 1165.
Colburn, C. B.; Kennedy, A. J. Am. Chem. Soc. 1958, 80, 5004.
Baumgardner, C. L.; Lawton, E. L. Acc. Chem. Res. 1974, 7, 14. doi: 10.1021/ar50073a003
Reed Jr, S. F. J. Org. Chem. 1968, 33, 1861. doi: 10.1021/jo01269a034
Petry, R. C.; Parker, C. O.; Johnson, F. A.; Stevens, T. E.; Freeman, J. P. J. Org. Chem. 1967, 32, 1534. doi: 10.1021/jo01280a052
Petry, R. C.; Freeman, J. P. J. Am. Chem. Soc. 1961, 83, 3912.
Gakh, A. A.; Romaniko, S. V.; Ugrak, B. I.; Fainzilberg, A. A. Tetrahedron 1991, 47, 7447. doi: 10.1016/S0040-4020(01)89746-5
Majumder, U.; Armantrout, J. R.; Williams, R. V.; Shreeve, J. M. J. Org. Chem. 2002, 67, 8435. doi: 10.1021/jo026201y
Belter, R. K. J. Fluorine Chem. 2012, 132, 961.
Dalinger, I. L.; Vinogradov, V. M.; Shevelev, S. A.; Kuz'min, V. S.; Arnautova, E. A.; Pivina, T. S. Propellants, Explos., Pyrotech. 1998, 23, 212. doi: 10.1002/(SICI)1521-4087(199808)23:4<212::AID-PREP212>3.0.CO;2-Y
Fokin, A. K.; Studnev, Y. N.; Stolyarov, V. P.; Mel'nikov, A. A. Russ. Chem. Bull. 2000, 49, 949. doi: 10.1007/BF02494724
Marsden, H. M.; Shreeve, J. M. Inorg. Chem. 1987, 26, 169. doi: 10.1021/ic00248a033
John, E. O.; Shreeve, J. M. Inorg. Chem. 1988, 27, 3100. doi: 10.1021/ic00291a011
John, E. O.; Kirchmeier, R. L.; Shreeve, J. M. J. Fluorine Chem. 1990, 47, 333. doi: 10.1016/S0022-1139(00)82383-0
John, E. O.; Willett, R. D.; Scott, B.; Kirchmeier, R. L.; Shreeve, J. M. Inorg. Chem. 1989, 28, 893. doi: 10.1021/ic00304a018
Ye, C. f.; Gao, H. X.; Shreeve, J. M. J. Fluorine Chem. 2007, 128, 1410. doi: 10.1016/j.jfluchem.2007.07.006
Litvinov, B. V.; Fainzil'berg, A. A.; Pipekin, V. I.; Smirnov, S. P.; Loboiko, B. G.; Shevelev, S. A.; Nazin, G. M. Dokl. Akad. Nauk 1994, 336, 67.
Khisamutdinov, G. K.; Shevelev, S. A. Russ. Chem. Bull., Int. Ed. 2001, 50, 736. doi: 10.1023/A:1011345819626
Fokin, A. V.; Studnev, Y. N.; Kuznetsova, L. D. Dokl. Akad. Nauk 1996, 3, 358
Fokin, A. V.; Studnev, Y. N.; Kuznetsova, L. Russ. Chem. Bull. 1996, 45, 1952. doi: 10.1007/BF01457784
张明权, 刘红雨, 韦兴存, 张磊, 康玲, 化学推进剂与高分子材料, 2017, 15, 45.Zhang, M. Q.; Liu, H. Y, ; Wei, X. C.; Zhang, L.; Kang, L. Chem. Propellants Polym. Mater. 2017, 15, 45 (in Chinese).
Frankel, M. B.; Witucki, E. F. US 4341712, 1982.
Adolph, H. G.; Trivedi, N. J. US 6325876 B1, 2001.
Ammon, H. L.; Holden, J. R.; Du, Z. Structure and Density Predictions for Energetic Materials, 2002. http://www.chem.missouri.edu/thompson/MURI02/extended/Ammon_MURI_extended_abstract_4.pdf.
Baum, K.; Trivedi, N. J.; Lovato, J. M.; Iyer, V. K. Report NRO-1-1 (final), Fluorochem, Azusa, CA, 1993.
Chapman, R. D.; Welker, M. F; Kreutzberger, C. B. J. Org. Chem. 1998, 63, 1566. doi: 10.1021/jo9718399
Chapman, R. D.; Gilardi, R. D.; Welker, M. F.; Kreutzberger, C. B. J. Org. Chem. 1999, 64, 960. doi: 10.1021/jo9819640
Chapman, R. D.; Groshens, T. J. US 7563889 B1, 2009.
Chapman, R. D.; Groshens, T. J. US 8444783 B1, 2013.
Axenrod, T.; Guan, X. P.; Sun, J. G.; Qi, L.; Chapman, R. D.; Gilardi, R. D. Tetrahedron Lett. 2001, 42, 2621. doi: 10.1016/S0040-4039(01)00260-X
Chapman, R. D.; Nguyen, B. V. US 6310204 B1, 2001.
李欢, 博士论文, 南京理工大学, 南京, 2015.Li, H. Ph.D. Dissertation, Nanjing University of Science & Technology, Nanjing, 2015 (in Chinese).
梅莹, 硕士论文, 南京理工大学, 南京, 2016.Mei, Y. M.S. Thesis, Nanjing University of Science & Technology, Nanjing, 2016 (in Chinese).
Archibald, T. G.; Manser, G. E. Immoos, J. E. US 5240311, 1995.

图 1 每摩尔硼和铝氟化和氧化反应放热量
Figure 1 Fluorination and oxidation heat release of per mole of B and Al

图式 6 Ph3CNF2的合成及其与浓硫酸反应
Scheme 6 Synthesis of Ph3CNF2 and its reaction with conc. H2SO4

图式 15 二氟氨甲基四唑及其含能盐的合成
Scheme 15 Synthesis of difuoroaminomethyl-substituted tetrazole and its energetic salts

图式 16 偕二硝基乙酸酯阴离子二氟氨基化反应
Scheme 16 Difuoroamination of alkoxycarbonyl dinitromethanides

图式 20 二氟氨基二硝甲基取代的三嗪和四唑化合物的合成
Scheme 20 Synthesis of (difluoroamino)dinitromethyl-substituted triazine and tetrazole

图式 21 二氟氨基二硝甲基三唑化合物的合成
Scheme 21 Synthesis of (difluoroamino)dinitromethyl-substituted triazoles

图式 35 二氟氨基聚合物130, 131和132的合成
Scheme 35 Synthesis of NF2-based polymers 130, 131 and 132

图式 36 二氟氨基粘合剂133, 134, 135和136的合成
Scheme 36 Synthesis of NF2-based binders 133, 134, 135 and 136
表 1 N—NF2唑类化合物与RDX、HMX的性能对比
Table 1. Properties of N-difluoroaminoazoles compared with RDX and HMX
| Substrate | Tdec/℃ | ΔΗf/(kJ·mol-1) | ρ/(g·cm-3) | Vdet/(m·s-1) | P/GPa |
| 6 | 70 | 117.2 | 1.92 | 8536 | 31.4 |
| 7 | 62 | 118.4 | 1.91 | 8494 | 31.0 |
| 10 | 127 | 228.0 | 1.80 | 7925 | 27.4 |
| 14 | 100 | 421.7 | 1.92 | 8257 | 30.9 |
| 15 | 84 | 458.8 | 1.88 | 8462 | 30.0 |
| RDX | 204 | 67 | 1.82 | 8850 | 34.9 |
 下载: 导出CSV
下载: 导出CSV
表 3 环状偕二氟氨基烷烃衍生物
Table 3. Cyclic gem-bis(difluoramino) alkyl derivatives
下载: 导出CSV
表 4 二氟氨基粘合剂133, 134, 135和136的参数
Table 4. Parameters of NF2-based binders 133, 134, 135 and 136
| Substrate | Tonset /℃ | Mn/(g·mol-1) | PDI | f | Tg/℃ |
| 133 | 190 | 5772 | 1.30 | 1.98 | -24.3 |
| 134 | 175 | 4827 | 1.25 | 1.87 | -38.7 |
| 135 | 125 | 2675 | 1.79 | 2.69 | -28.4 |
| 136 | — | 2700 | 2.11 | 1.88 | -50.8 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们