Table 1.
Survey of the reaction conditionsa
Citation:
Song Mengmeng, Zhang Zhiguo, Zheng Dan, Li Xiang, Liang Rui, Zhao Xu'na, Shi Lei, Zhang Guisheng. Hypervalent Organoiodine Promoted Dearylation Reaction of N-Aryl Sulfonamides[J]. Chinese Journal of Organic Chemistry,
2020, 40(8): 2433-2441.
doi:
10.6023/cjoc202001007

高价碘试剂促进的N-芳基磺酰胺类化合物脱芳基反应
English
Hypervalent Organoiodine Promoted Dearylation Reaction of N-Aryl Sulfonamides
Abstract:
An efficient Dess-Martin periodinane (DMP)-promoted dearylation of N-arylsulfonamides was developed through a highly selective oxidative cleavage of the inert C(aryl)-N bonds in secondary sulfonamides while leaving the S-N bond unchanged. This metal-free reaction proceeds under mild conditions and provides access to various biologically important primary sulfonamides, some of which are otherwise unattainable using conventional aminolysis and hydrolysis methods. The concise and efficient dearylation reaction provides the use of an aryl group as a removable protecting sulfonamide group under metal catalyst-free conditions.
-
Key words:
- Dess-Martin periodinane (DMP)
- / C(aryl)-N bond cleavage
- / oxidation
- / metal-free reaction
- / sulfonamide
-
1. Introduction
Sulfonamides[1] are the bioactive moiety of sulfa drugs, [2] such as sulfadiazine (SD), sulfathiazole (ST), sulfamethoxazole (SMZ), sulfadimoxine (SDM), indapamide and prontosil.[3] They are also important intermediates in synthetic organic chemistry.[4] In general, the sulfonyl moiety of sulfonamides serves as the leaving group and delivers the sulfonyl or amino group to a new molecule accompanied by a S—N bond cleavage in reactions.[5] However, the regiospecific C—N bond cleavage of sulfonamides is relatively rare. In 1989, Hewson and co-workers[6] isolated six kinds of substituted p-benzo- quinones by treating N-arylsulfonamides with ceric ammonium nitrate (CAN) via an oxidation/dearomatization/ hydrolysis reaction (Eq. 1). An aryl transfer reaction from N-arylsulfonamides to three kinds of aryl acceptors via a regiospecific aryl C—N bond cleavage reaction was reported, in which the sulfonamide moiety also served as the leaving group (Eq. 2).[7] A challenging regiospecific C—N bond cleavage reaction of N-aliphatic substituted alkylsulfonamides was also achieved, and various N-alkyl- sulfonamides were easily dealkylated in moderate to good yields by ultrasonic irradiation in the presence of (diacetoxyiodo)benzene and iodine (Eq. 3).[8] Although remarkable progress has been achieved, it remains a challenge and highly desirable to develop a regiospecific C—N bond cleavage reaction[9] of sulfonamides under mild conditions.

(1) 
(2) 
(3) Hypervalent iodine reagents[10] are commercially available oxidants that have been widely used in many transformations in synthetic organic chemistry.[11] Water is also a useful reagent frequently serving as both solvent and reactant.[12] A regiospecific aryl C—N bond cleavage reaction of secondary N-arylamides in the presence of 2-iodoxy- benzoic acid (IBX)[13] in the mixed solvent system of hexa-fluoroisopropanol (HFIP) and H2O has been described recently.[14] A series of primary amides were obtained in good yields. Moreover, stereochemistry was notaffected during these transformations. The aryl group acts as a removable protecting group in this reaction.[15] In search of further applications, we deduced that it was also possible for N-arylsulfonamides to rapidly access a variety of useful primary sulfonamides via the oxidative dearomatization/hydrolysis process reported in our previous works, [14] which may be beneficial for pharmaceutical process chemistry.[16] Here, we report our recent synthesis of primary sulfonamides via a Dess-Martin periodinane (DMP) mediated[13] regiospecific oxidative cleavage of inert C(aryl)—N bonds of electronically neutral secondary N-aryl- sulfonamides under mild conditions (Eq. 4).

(4) 2. Results and discussion
Initially, N-arylsulfonamide (1r) was selected as a model substrate with IBX as the oxidant.[14a] It was found that the cleavage reaction can occur regiospecifically on the C(aryl)—N bond of 1r in the presence of 1.2 equiv. of IBX in a mixture of HFIP and H2O (V:V=2:1) at 25 ℃ for 4 h (Table 1, Entry 1). A higher yield (99%) of 2r was obtained when DMP, readily available by warming IBX in an acetic anhydride-acetic acid mixture, [13] was used, and the reaction time was shortened to 1 h (Table 1, Entry 2). The reaction time was shortened to 10 min when the reaction temperature was 40 ℃ (Table 1, Entry 3). To avoid the use of expensive HFIP solvents, the reaction was performed in water. As a result, most of 1r was recovered because DMP is insoluble in water (Table 1, Entry 4). Interestingly, a similar result was also observed when using only HFIP as the solvent (Table 1, Entry 5). These observations indicated the importance of using both solvents. Finally, it was found that a HFIP/H2O solvent volume ratio of 1:3 gave a satisfactory yield of 2r at 40 ℃ after 1.5 h (Table 1, Entry 8 vs. Entries 6 and 7). The conversion of 1r was significantly reduced at a lower DMP loading (Table 1, Entry 9). Different mixed solvents were also screened for the conversion, including trifluoroethanol (TFE)/H2O, EtOH/H2O, dimethyl sulfoxide (DMSO)/H2O, and MeCN/H2O. These solvents were less efficient than HFIP/H2O, and most of the starting material was recovered (Table 1, Entries 10~13). Other hypervalent iodine reagents, such as bis(trifluoroacetoxy)iodo]benzene (PIFA), PhIO, (diacetoxyiodo)benzene (PIDA), and hydroxy-(tosyloxy)iodo]benzene (HTIB), afforded slightly lower yields of 2r than that of DMP under the optimal conditions (Table 1, Entries 14~17 vs. Entry 8).
Table 1

 下载:
导出CSV
下载:
导出CSV

Entry Oxidant (equiv.) Solvent (V:V) T/℃ Time Yield/% of2r Recovered/% of1r 1 IBX (1.2) HFIP/H2O (2:1) 25 4 h 83 0 2 DMP (1.2) HIP/H2O (2:1) 25 1 h 99 0 3 DMP (1.2) HIP/H2O (2:1) 40 10 min 96 0 4 DMP (1.2) H2O 40 0.5 h 0 95 5 DMP (1.2) HIP 40 1 h 5 85 6 DMP (1.2) HIP/H2O (1:5) 40 2.5 h 80 18 7 DMP (1.2) HIP/H2O (1:1) 40 10 min 98 0 8 DMP (1.2) HIP/H2O (1:3) 40 1.5 h 97 0 9 DMP (0.6) HIP/H2O (1:3) 40 3 h 43 44 10 DMP (1.2) TFE/H2O (1:3) 40 1.5 h 5 84 11 DMP (1.2) EtOH/H2O (1:3) 40 1.5 h 0 96 12 DMP (1.2) DMSO/H2O (1:3) 40 0.5 h 0 97 13 DMP (1.2) MeCN/H2O (1:3) 40 0.5 h 0 98 14 PIFA (1.2) HIP/H2O (1:3) 40 1.5 h 81 12 15 PhIO (1.2) HIP/H2O (1:3) 40 1.5 h 72 19 16 PIDA (1.2) HIP/H2O (1:3) 40 1.5 h 77 11 17 HTIB (1.2) HIP/H2O (1:3) 40 1.5 h 67 26 a Unless otherwise indicated, all reactions were conducted with 1r (0.2 mmol) in 2 mL of solvent at 40 ℃. The substrate scope of this intramolecular C(aryl)—N bond cleavage reaction was subsequently investigated under the optimal conditions (Table 1, Entry 8). The key results are summarized in Table 2. The scope of the R2 group was initially investigated. This method was found to be applicable for Me groups in the para-, ortho- or meta-position (1a~1c), OMe (1d) and Cl (1e) group at the para-position, as well as Ph group (1f). However, the reaction did not work when R2 was a CO2Et group (1g), and most of the starting material was recovered. According to our previous studies, we deduced that the strong electron withdrawing group (EWG) CO2Et (1g) made the oxidation reaction difficult at the ortho-position of the amido group.[14] A similar result was also observed when the aryl group on the N atom was changed to a 4-methylbenzyl group (1h). We hypothesized that the benzylamine could not be oxidized to the corresponding imine under the current conditions, which inhibits the subsequent hydrolysis reaction.[14] 8-Aminoquinoline (AQ) group (1i) gave a 42% yield of the product.
Pleasingly, further investigation of the R1 group indicated that the reaction tolerated various substituted aryl groups with a 4-methylphenyl group serving as the leaving group. Substituted phenyl groups bearing a Me group in the para-, ortho- or meta-position afforded the desired products 2j~2l in 96%~97% yields. Other aryl groups with OMe (1m), Cl (1n), CN (1o), CF3 (1p), CO2Et (1q), and NO2 (1r) groups in the para-position afforded the desired products in 79%~97% high yields. Substrate 1s bearing two Cl groups in the meta-position behaved similarly and afforded the product 2s in 98% yield. Compound 1t also reacted smoothly and generated the corresponding amide 2t in 95% yield. It is noteworthy that heterocycles were tolerated for the transformations, but the thiophenyl-containing substrate (1u) gave a higher product yield than a pyridinyl-containing substrate (1v). Alkylsulfonamides 1w and 1x also reacted smoothly under the optimized conditions to afford the corresponding amides 2w and 2x in 96% and 92% yields, respectively. Further investigation of the 2-phenyl-N-(p-tolyl)ethene-1-sulfon- amide showed regiospecific cleavage of the aryl C—N bond under the optimized conditions, which resulted in the formation of 2-phenylethene-1-sulfonamide in 83% yield. A typical aliphatic sulfonic acid experiment with N-aryl camphor sulfonamide (1z) was also carried out. However, it gave a 27% low yield of amide 2z. Pleasingly, the sulfuric diamides 1a' and 1b' can be used in this protocol, delivering the desired products 2a' and 2b' in 76% and 59% yields, respectively. In these two transformations, we successfully utilized the properties of the electron- withdrawing and aliphatic groups to selectively cleave the 4-methylphenyl group directly attached to the nitrogen atom. It is noteworthy that the intermediates 3m and 2m were isolated in 10% and 58% yield, respectively, with 18% starting material recovered when the reaction of 1m was quenched after 5 min under the optimal reaction conditions (Eq. 5). This control experiment suggested that the mechanism of this transformation was an ortho-oxidation of the aryl group pathway, as discussed in our previous work.[14b]

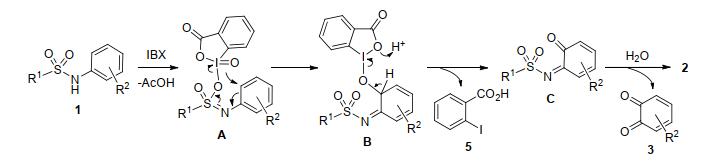
(5) Combined with some related previous literatures and our experiments results, [6, 8, 14a, 17] especially the cases of 1a, 1d, 1e, 1g, 1h and 1i, a possible mechanism was proposed (Eq. 6 and Scheme 1). The DMP was transformed to IBX by releasing two molecula AcOH in the presence of HFIP and H2O by warm (Eq. 6).[11c] Then, the IBX generated in situ oxidized the staring material 1 to generated the intermediate A. The ortho-oxidation process subsequently occurred via B and C, and the final product 2 formed after hydrolysis reaction (Scheme 1).[17]

(6) Table 2
Table 2. Extension of the reaction scopea下载:
导出CSV


a Unless otherwise indicated, all these reactions were conducted with 1 (0.2 mmol) and DMP (1.2 equiv.) in 2 mL of mixed solvent system (VHFIP:VH2O=1:3) at 40 ℃. b Compound 2c was conducted with 1 (0.2 mmol) and DMP (1.5 equiv.) in 2.0 mL of mixed solvent system (VHFIP:VH2O=1:3) at 40 ℃. c 96% of the starting material was recovered. d 97% of the starting material was recovered. e Compound 2m was reacted with 1 (0.2 mmol) and DMP (1.5 equiv.) in 2.0 mL of mixed solvent system (VHFIP:VH2O=1:3) at 40 ℃. f Compound 2v was reacted with 1 (0.2 mmol) and DMP (2.5 equiv.) in 2.0 mL of mixed solvent system (VHFIP:VH2O=1:1) at 60 ℃. Scheme 1
3. Conclusions
In summary, a DMP-promoted highly selective oxidation reaction was developed for the regiospecific cleavage of the inert C(aryl)—N bond in N-arylsulfonamides while leaving the S—N bond untouched. Under mild reaction conditions, a series of electronically neutral and structurally simple secondary N-aryl sulfonamides were rapidly transformed to useful primary sulfonamides in high yield. In contrast to the previously reported cleavage of the alkyl chain on the nitrogen atom of N-allylsulfonamides, an alternative new route to the cleavage of the aryl group of N-arylsulfonamides was presented. This work is also one in a series of projects on the use of an aryl group as a removable protecting group in the absence of a metal catalyst starting from secondary N-arylamides to primary amides, which are otherwise unattainable using conventional aminolysis and hydrolysis methods. We think these methodologies are highly valuable for biochemistry, organic synthetic chemistry, and industrial research.
4. Experimental section
4.1 General information
All reactions were carried out under air atmosphere, unless otherwise indicated. Starting materials 1 were synthesized according to the known methods reported in the literatures. Petroleum ether (PE) used refers to the 60~90 ℃ boiling point fraction of petroleum. Ethyl acetate is abbreviated as EA. 1H NMR and 13C NMR spectra[18] were recorded on Bruker Avance/600 (1H NMR: 600 MHz, 13C NMR: 150 MHz at 25 ℃) or Bruker Avance/400 (1H NMR: 400 MHz, 13C NMR: 100 MHz at 25 ℃) NMR spectrometers and TMS was used as internal standard. All high-resolution mass spectra (HRMS) were measured on a mass spectrometer by using electrospray ionization (ESI-oa-TOF), and the purity of all samples used for HRMS (> 95%) were confirmed by 1H NMR and 13C NMR spectroscopic analysis. Melting points were measured on a melting point apparatus equipped with a thermometer and were uncorrected. The literature value of melting point for all solid compounds were obtained from the Scifinder or references. All reactions were monitored by thin-layer chromatography (TLC) with GF254 silica gel coated plates. Flash chromatography was carried out on SiO2 (silica gel 200~300 mesh).
4.2 Synthesis
4.2.1 Synthesis of 1
Taking synthesis of 1a as an example, [17] to a round-bottom flask (25 mL) were added benzenesulfonyl chloride (0.593 mL, 4.66 mmol) and p-toluidine (499 mg, 4.66 mmol), and then the mixture was stirred well for 1 h in 0.5 mL of pyridine solution at 80 ℃ (the whole process was closely monitored by TLC). After the completion of the reaction, the reaction was quenched by 1 mol/L HCl solution (10 mL). The mixture was extracted with dichloromethane (5 mL×3). After filtration and concentration, the crude product was purified by a short flash silica gel column chromatography [eluent: V(ethylacetate):V(petroleum ether)=1:2] to give N-(p-tolyl)benzene- sulfonamide (1a) as light yellow solid (1128 mg, 98%). m.p. 119~121 ℃ (lit.[18] 118~120 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.76 (d, J=7.2 Hz, 2H), 7.53 (t, J=7.4 Hz, 1H), 7.43 (t, J=7.8 Hz, 2H), 7.03 (d, J=8.4 Hz, 2H), 6.95 (d, J=8.4 Hz, 2H), 2.27 (s, 3H).
N-(m-Tolyl)benzenesulfonamide (1b): White solid (1127 mg, 98%). m.p. 92~94 ℃ (lit.[18] 94~95 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.79 (d, J=7.2 Hz, 2H), 7.53 (t, J=7.4 Hz, 1H), 7.44 (t, J=7.4 Hz, 2H), 7.8 (t, J=7.7 Hz, 1H), 6.89 (dd, J=18.2, 9.4 Hz, 4H), 2.26 (s, 3H).
N-(o-Tolyl)benzenesulfonamide (1c): White solid (1093 mg, 95%). m.p. 123~124 ℃ (lit.[19] 122~123 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.74 (d, J=7.2 Hz, 2H), 7.55 (t, J=7.4 Hz, 1H), 7.43 (t, J=7.8 Hz, 2H), 7.31 (d, J=8.0 Hz, 1H), 7.17~7.11 (m, 1H), 7.08 (d, J=4.0 Hz, 2H), 6.53 (s, 1H), 1.99 (s, 3H).
N-(4-Methoxyphenyl)benzenesulfonamide (1d): White solid (1201 mg, 98%). m.p. 88~89 ℃ (lit.[20] 88~90 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.72 (d, J=7.2 Hz, 2H), 7.52 (t, J=7.4 Hz, 1H), 7.42 (t, J=7.7 Hz, 2H), 7.01~6.96 (m, 2H), 6.85 (s, 1H), 6.77~6.72 (m, 2H), 3.74 (s, 3H).
N-(4-Chlorophenyl)benzenesulfonamide (1e): Off-white solid (1244 mg, 100%). m.p. 121~122 ℃ (lit.[21] 120~121 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.77 (d, J=7.6 Hz, 2H), 7.56 (t, J=7.0 Hz, 1H), 7.45 (t, J=7.8 Hz, 2H), 7.20 (d, J=8.4 Hz, 2H), 7.05~6.97 (m, 3H).
N-Phenylbenzenesulfonamide (1f): White solid (1031 mg, 95%). m.p. 102~105 ℃ (lit.[20] 100~102 ℃); 1H NMR (600 MHz, CDCl3) δ: 7.78 (d, J=7.2 Hz, 2H), 7.53 (t, J=7.5 Hz, 1H), 7.44 (t, J=7.8 Hz, 2H), 7.25~7.22 (m, 2H), 7.12 (t, J=7.5 Hz, 1H), 7.07 (d, J=7.2 Hz, 2H), 6.82 (s, 1H).
Ethyl 4-(phenylsulfonamido)benzoate (1g): White solid (1351 mg, 95%). m.p. 138~141 ℃ (lit.[22] 141~143 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 10.86 (s, 1H), 7.82 (dd, J=7.8, 6.6 Hz, 4H), 7.62 (t, J=7.5 Hz, 1H), 7.56 (t, J=7.8 Hz, 2H), 7.22 (d, J=8.4 Hz, 2H), 4.23 (q, J=7.0 Hz, 2H), 1.26 (t, J=6.9 Hz, 3H).
N-(4-Methylbenzyl)benzenesulfonamide (1h): White solid (1119 mg, 92%). m.p. 82~83 ℃ (lit.[23] 84 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 8.10 (t, J=6.3 Hz, 1H), 7.81 (d, J=7.8 Hz, 2H), 7.63~7.56 (m, 3H), 7.09 (dd, J=20.4, 7.8 Hz, 4H), 3.93 (d, J=6.0 Hz, 2H), 2.25 (s, 3H).
N-(quinolin-8-yl)benzenesulfonamide (1i): Yellow green solid (1191 mg, 90%). m.p. 131~133 ℃ (lit.[24] 132.5~133 ℃); 1H NMR (600 MHz, CDCl3) δ: 9.25 (s, 1H), 8.71 (dd, J=4.2, 1.8 Hz, 1H), 8.03 (dd, J=8.5, 1.5 Hz, 1H), 7.90 (d, J=7.2 Hz, 2H), 7.82 (dd, J=6.9, 2.1 Hz, 1H), 7.43~7.35 (m, 4H), 7.32 (t, J=7.8 Hz, 2H).
2-Methyl-N-(p-tolyl)benzenesulfonamide (1j): Light yellow solid (1144 mg, 94%). m.p. 123~125 ℃ (lit.[25] 126 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.95 (d, J=8.1 Hz, 1H), 7.40 (t, J=7.5 Hz, 1H), 7.24 (s, 1H), 7.17 (s, 1H), 6.99 (d, J=8.3 Hz, 2H), 6.92 (d, J=8.5 Hz, 2H), 2.65 (s, 3H), 2.22 (s, 3H).
3-Methyl-N-(p-tolyl)benzenesulfonamide (1k): Yellow solid (1156 mg, 95%). m.p. 114~118 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.61 (s, 1H), 7.56 (d, J=7.2 Hz, 1H), 7.32~7.25 (m, 2H), 7.11 (s, 1H), 7.01 (d, J=8.4 Hz, 2H), 6.96 (d, J=8.8 Hz, 2H), 2.32 (s, 3H), 2.25 (s, 3H); HRMS (ESI) calcd for C14H16NO2S [M+H]+ 262.0896, found 262.0885.
4-Methyl-N-(p-tolyl)benzenesulfonamide (1l): White solid (1155 mg, 95%). m.p. 114~116 ℃ (lit.[26] 116~117.5 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.69 (d, J=8.4 Hz, 2H), 7.42 (s, 1H), 7.19 (d, J=8.4 Hz, 2H), 7.00 (s, 4H), 2.34 (s, 3H), 2.24 (s, 3H).
4-Methoxy-N-(p-tolyl)benzenesulfonamide (1m): White solid (1351 mg, 96%). m.p. 105~107 ℃ (lit.[26] 109.5~110 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.68 (d, J=8.8 Hz, 2H), 7.03 (d, J=8.0 Hz, 2H), 6.94 (d, J=8.4 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 6.63 (s, 1H), 3.82 (s, 3H), 2.27 (s, 3H).
4-Chloro-N-(p-tolyl)benzenesulfonamide (1n): Yellow solid (1296 mg, 99%). m.p. 84~85 ℃ (lit.[27] 88 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.68 (d, J=8.8 Hz, 2H), 7.39 (d, J=8.8 Hz, 2H), 7.04 (d, J=8.0 Hz, 2H), 6.96 (d, J=8.4 Hz, 3H), 2.28 (s, 3H).
4-Cyano-N-(p-tolyl)benzenesulfonamide (1o): Light yellow solid (887 mg, 70%). m.p. 149~152 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.85 (d, J=8.4 Hz, 2H), 7.72 (d, J=8.4 Hz, 2H), 7.05 (d, J=8.4 Hz, 3H), 6.95 (d, J=8.4 Hz, 2H), 2.28 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 143.2, 136.6, 132.9, 132.8, 130.3, 128.0, 123.0, 117.4, 116.7, 21.0; HRMS (ESI) calcd for C14H12N2NaO2S [M+Na]+ 295.0512, found 295.0511.
N-(p-Tolyl)-4-(trifluoromethyl)benzenesulfonamide (1p): Light yellow solid (1409 mg, 96%). m.p. 123~125 ℃ (lit.[25] 123.7~124.6 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.85 (d, J=8.4 Hz, 2H), 7.70 (d, J=8.4 Hz, 2H), 7.07 (d, J=8.0 Hz, 2H), 6.95 (d, J=8.4 Hz, 2H), 6.59 (s, 1H), 2.29 (s, 3H).
Ethyl 4-(N-(p-tolyl)sulfamoyl)benzoate (1q): Light yellow solid (929 mg, 97%). m.p. 108~120 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.11~8.05 (m, 2H), 7.83~7.77 (m, 2H), 7.03 (d, J=8.0 Hz, 2H), 6.98~6.92 (m, 2H), 6.86 (s, 1H), 4.39 (q, J=7.1 Hz, 2H), 2.27 (s, 3H), 1.39 (t, J=7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 165.3, 142.9, 136.1, 134.5, 133.3, 130.2, 130.1, 127.4, 122.9, 61.9, 21.0, 14.3; HRMS (ESI) calcd for C16H17NNaO4S [M+Na]+ 342.0770, found 342.0766.
4-Nitro-N-(p-tolyl)benzenesulfonamide (1r): Light yellow solid (1293 mg, 95%). m.p. 180~182 ℃ (lit.[28] 179~180 ℃); 1H NMR (600 MHz, CDCl3) δ: 8.27 (d, J=9.0 Hz, 2H), 7.89 (d, J=8.4 Hz, 2H), 7.08 (d, J=8.4 Hz, 2H), 6.95 (d, J=8.4 Hz, 2H), 6.57 (s, 1H), 2.30 (s, 3H).
3, 5-Dichloro-N-(p-tolyl)benzenesulfonamide (1s) White solid (1351 mg, 95%). m.p. 124~126 ℃ (lit.[29] 123~125 ℃); 1H NMR (400 MHz, CDCl3) δ: 10.30 (s, 1H), 7.94 (t, J=1.8 Hz, 1H), 7.66 (d, J=2.0 Hz, 2H), 7.08 (d, J=8.4 Hz, 2H), 6.97 (d, J=8.4 Hz, 2H), 2.21 (s, 3H).
N-(p-Tolyl)naphthalene-2-sulfonamide (1t): Light yellow solid (1329 mg, 96%). m.p. 107~109 ℃ (lit.[30] 127.5 ℃); 1H NMR (600 MHz, CDCl3) δ: 8.36 (d, J=1.2 Hz, 1H), 7.87 (t, J=8.4 Hz, 3H), 7.77 (dd, J=8.4, 1.8 Hz, 1H), 7.61 (t, J=7.2 Hz, 1H), 7.56 (t, J=7.2 Hz, 1H), 7.01 (d, J=17.4 Hz, 5H), 2.24 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 136.1, 135.6, 135.0, 133.7, 132.1, 130.0, 129.5, 129.4, 129.0, 130.0, 130.0, 127.6, 122.5, 20.9; HRMS (ESI) calcd for C17H15NNaO2S [M+Na]+ 320.0716, found 320.0714.
N-(p-Tolyl)thiophene-2-sulfonamide (1u): White solid (1351 mg, 95%). m.p. 113~115 ℃ (lit.[31] 115 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.52 (dd, J=5.0, 1.4 Hz, 1H), 7.47 (dd, J=4.2, 1.4 Hz, 1H), 7.07 (d, J=8.4 Hz, 2H), 7.04~6.98 (m, 3H), 6.79 (s, 1H), 2.29 (s, 3H).
N-(p-Tolyl)pyridine-3-sulfonamide (1v): White solid (1063 mg, 92%). m.p. 189~192 ℃ (lit.[32] 188~190 ℃); 1H NMR (400 MHz, CDCl3) δ: 10.32 (s, 1H), 8.83 (d, J=1.6 Hz, 1H), 8.76 (dd, J=5.0, 1.4 Hz, 1H), 8.08~8.06 (m, 1H), 7.60~7.58 (m, 1H), 7.05 (d, J=8.4 Hz, 2H), 6.97 (d, J=8.4 Hz, 2H), 2.19 (s, 3H); 13C NMR (150 MHz, DMSO) δ: 153.3, 147.0, 135.8, 134.7, 134.3, 134.1, 129.7, 124.3, 121.2, 20.3; HRMS (ESI) calcd for C12H13N2O2S [M+H]+ 249.0692, found 249.0691.
1-Phenyl-N-(p-tolyl)methanesulfonamide (1w): Brown solid (1143 mg, 94%). m.p. 107~108 ℃ (Lit.[33] 108 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.37~7.32 (m, 3H), 7.27 (d, J=1.6 Hz, 1H), 7.25 (s, 1H), 7.14 (d, J=8.0 Hz, 2H), 7.04 (d, J=8.4 Hz, 2H), 6.30 (s, 1H), 4.29 (s, 2H), 2.33 (s, 3H).
N-(p-Tolyl)butane-1-sulfonamide (1x): Brown solid (988 mg, 93%). m.p. 78~80 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.16~7.09 (m, 4H), 6.71 (s, 1H), 3.09~3.02 (m, 2H), 2.32 (s, 3H), 1.84~1.74 (m, 2H), 1.45~1.35 (m, 2H), 0.89 (t, J=7.4 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 135.3, 134.3, 130.3, 121.3, 51.1, 25.6, 21.6, 20.9, 13.6; HRMS (ESI) calcd for C11H17NNaO2S [M+Na]+ 250.0872, found 250.0872.
(E)-2-Phenyl-N-(p-tolyl)ethene-1-sulfonamide (1y):[34] Claybank liquid (1260 mg, 99%); 1H NMR (400 MHz, CDCl3) δ: 7.44 (d, J=15.2 Hz, 1H), 7.38~7.28 (m, 5H), 7.14 (d, J=8.4 Hz, 2H), 7.06 (d, J=8.0 Hz, 2H), 6.83 (d, J=15.6 Hz, 1H), 2.25 (s, 3H).
1-((1S, 4S)-7, 7-Dimethyl-2-oxobicyclo[2.2.1]heptan-1- yl)-N-(p-tolyl)methanesulfonamide (1z): White solid (1197 mg, 80%). m.p. 134~136 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.66 (s, 1H), 7.15 (dd, J=25.2, 8.4 Hz, 4H), 3.36 (d, J=15.6 Hz, 1H), 2.83 (d, J=15.6 Hz, 1H), 2.47~2.43 (m, 1H), 2.32 (s, 3H), 2.19~2.03 (m 4H), 1.98 (d, J=18.7 Hz, 1H), 1.52~1.43 (m, 1H), 0.95 (s, 3H), 0.86 (s, 3H); HRMS (ESI) calcd for C17H24NO3S [M+H]+ 322.1471, found 322.1481.
Ethyl 4-((N-(p-tolyl)sulfamoyl)amino)benzoate (1a'): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petroleum ether)=1:10] as a white solid (481 mg, 72%). m.p. 154~157 ℃ (lit.[35] 159~160 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 10.65 (s, 1H), 10.29 (s, 1H), 7.84 (d, J=8.4 Hz, 2H), 7.22 (d, J=9.0 Hz, 2H), 7.05 (d, J=8.4 Hz, 2H), 6.98 (d, J=8.4 Hz, 2H), 4.26 (q, J=7.0 Hz, 2H), 2.19 (s, 3H), 1.28 (t, J=7.2 Hz, 3H).
N-(4-Methylbenzyl)-N'-(4-methylphenyl)sulfuric diamide (1b'): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petroleum ether)=1:10] as a white solid (407 mg, 70%). m.p. 109~111 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.57 (s, 1H), 7.79 (t, J=6.4 Hz, 1H), 7.12~7.02 (m, 9H), 3.96 (d, J=6.0 Hz, 2H), 2.24 (d, J=1.6 Hz, 6H); 13C NMR (100 MHz, DMSO-d6) δ: 136.3, 136.1, 134.9, 131.6, 129.3, 128.7, 127.6, 118.8, 45.4, 20.7, 20.3; HRMS (ESI) calcd for C15H18N2NaO2S [M+Na]+ 313.0981, found 313.0981.
4.2.2 Synthesis of 2
Taking synthesis of 2a as an example, to a round-bottom flask (25 mL) was added N-(p-tolyl)benzenesulfonamide (1a) (49.4 mg, 0.2 mmol) and DMP (102 mg, 0.24 mmol), then the mixture was well stirred for 0.5 h in 2 mL of mixed-solvent (VHFIP:VH2O=1:3) at 40 ℃ (the whole process was closely monitored by TLC). After the reaction stopped, saturated sodium bicarbonate and carbon dichloride were added for extraction. Then the organic solvent was concentrated in vacuo. The residue was purified by flash column chromatography with EA and PE as eluent to give primary amide 2a as light yellow solid (29 mg, 93%).
Benzenesulfonamide (2a): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petro-leum ether)=1:5] as a light yellow solid (29 mg, 93%). m.p. 149~152 ℃ (lit.[36] 152~153 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 8.41 (d, J=7.8 Hz, 2H), 8.06 (d, J=7.8 Hz, 2H), 7.73 (s, 2H).
2-Methylbenzenesulfonamide (2j): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a light yellow solid (33 mg, 97%). m.p. 154~156 ℃ (lit.[37] 154~157 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.84 (d, J=8.0 Hz, 1H), 7.50~7.45 (m, 1H), 7.36 (t, J=7.6 Hz, 4H), 2.59 (s, 3H).
3-Methylbenzenesulfonamide (2k): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a light yellow solid (33 mg, 96%). m.p. 103~107 ℃ (lit.[37] 106~109 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.66~7.60 (m, 2H), 7.48~7.38 (m, 2H), 7.29 (s, 2H), 2.38 (s, 3H).
4-Methylbenzenesulfonamide (2l): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a light yellow solid (33 mg, 96%). m.p. 136~140 ℃ (lit.[37] 138.5 ℃); 1H NMR (400 MHz, CDCl3) δ: 7.73~7.68 (m, 2H), 7.36 (d, J=8.0 Hz, 2H), 7.26 (s, 2H), 2.37 (s, 3H).
4-Methoxybenzenesulfonamide (2m): The product was isolated by flash chromatography [eluent: V(ethyl-acetate):V(petroleum ether)=1:5] as a white solid (30 mg, 79%). m.p. 109~110 ℃ (lit.[38] 110~112 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 7.75 (d, J=9.0 Hz, 2H), 7.20 (s, 2H), 7.08 (d, J=8.4 Hz, 2H), 3.82 (s, 3H), 1.23 (s, 1H).
4-Chlorobenzenesulfonamide (2n): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a light yellow solid (62 mg, 92%). m.p. 138~140 ℃ (lit.[39] 142~144 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 7.82 (d, J=8.4 Hz, 2H), 7.65 (d, J=9.0 Hz, 2H), 7.45 (s, 2H).
4-Cyanobenzenesulfonamide (2o): The product was isolated by flash chromatography [eluent: V(ethyl-acetate):V(petroleum ether)=3:10] as a light red solid (33 mg, 92%). m.p. 161~164 ℃ (lit.[40] 167~168 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 8.07 (d, J=8.4 Hz, 2H), 7.97 (d, J=8.4 Hz, 2H), 7.65 (s, 2H).
4-(Trifluoromethyl)benzenesulfonamide (2p): The product was isolated by flash chromatography (eluent: V(ethylacetate):V(petroleum ether)=3:10) as a light yellow solid (41 mg, 92%). m.p. 175~176 ℃ (lit.[41] 176~177 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 8.03 (d, J=8.4 Hz, 2H), 7.98 (d, J=7.8 Hz, 2H), 7.61 (s, 2H).
Ethyl 4-sulfamoylbenzoate (2q): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a light brown solid (44 mg, 95%). m.p. 97~98 ℃ (lit.[42] 97~115 ℃); 1H NMR (400 MHz, DMSO-d6) δ: 8.13 (d, J=8.8 Hz, 2H), 7.95 (d, J=8.4 Hz, 2H), 7.56 (s, 2H), 4.35 (q, J=7.1 Hz, 2H), 1.33 (t, J=7.2 Hz, 3H).
4-Nitrobenzenesulfonamide (2r): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=1:5] as a light yellow solid (39 mg, 97%). m.p. 180~182 ℃ (lit.[43] 178~180 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 8.41 (d, J=7.8 Hz, 2H), 8.06 (d, J=7.8 Hz, 2H), 7.73 (s, 2H).
3, 5-Dichlorobenzenesulfonamide (2s): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a white solid (44 mg, 98%). m.p. 158~160 ℃ (lit.[29] 158~160 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 7.93 (t, J=2.1 Hz, 1H), 7.80 (d, J=1.8 Hz, 2H), 7.65 (s, 2H).
Naphthalene-2-sulfonamide (2t): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a light yellow solid (40 mg, 95%). m.p. 212~215 ℃ (lit.[44] 213~215 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 8.42 (d, J=1.2 Hz, 1H), 8.12 (dd, J=11.7, 8.7 Hz, 2H), 8.02 (d, J=7.8 Hz, 1H), 7.88 (dd, J=8.7, 2.1 Hz, 1H), 7.70~7.63 (m, 2H), 7.44 (s, 2H).
Thiophene-2-sulfonamide (2u): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a light yellow solid (31 mg, 96%). m.p. 140~142 ℃ (lit.[45] 142~144 ℃); 1H NMR (400 MHz, DMSO-d6) δ: 7.83 (dd, J=5.0, 1.4 Hz, 1H), 7.64 (s, 2H), 7.55 (dd, J=3.7, 1.4 Hz, 1H), 7.13 (dd, J=5.2, 3.6 Hz, 1H).
Pyridine-3-sulfonamide (2v): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petroleum ether)=3:10] a light yellow solid (7 mg, 21%). m.p. 108~109 ℃ (lit.[46] 109~110 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 8.98 (d, J=1.8 Hz, 1H), 8.78 (dd, J=4.8, 1.2 Hz, 1H), 8.21~8.17 (m, 1H), 7.67~7.58 (m, 3H).
Phenylmethanesulfonamide (2w): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petroleum ether)=3:10] as a light brown solid (33 mg, 96%). m.p. 96~99 ℃ (lit.[47] 99~100 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 7.39~7.33 (m, 5H), 6.84 (s, 2H), 4.26 (s, 2H).
Butane-1-sulfonamide (2x):[48] The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petroleum ether)=3:10] as a reddish brown semi-solid (25 mg, 92%). 1H NMR (400 MHz, CDCl3) δ: 4.72 (s, 2H), 3.24~3.04 (m, 2H), 1.91~1.79 (m, 2H), 1.54~1.42 (m, 2H), 0.96 (t, J=7.4 Hz, 3H).
(E)-2-Phenylethene-1-sulfonamide (2y): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petroleum ether)=2:10] as a light yellow solid (30 mg, 83%). m.p. 139~142 ℃ (lit.[49] 140~141 ℃); 1H NMR (600 MHz, DMSO-d6) δ: 7.69~7.66 (m, 3H), 7.45~7.41 (m, 5H), 7.31 (d, J=15.6 Hz, 2H), 7.23 (d, J=15.6 Hz, 2H), 7.11 (s, 3H).
((1S, 4S)-7, 7-Dimethyl-2-oxobicyclo[2.2.1]heptan-1-yl)- methanesulfonamide (2z): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petro-leum ether)=2:1] as a colorless semi-solid (12.4 mg, 27%). 1H NMR (400 MHz, DMSO-d6) δ: 6.86 (s, 2H), 3.39 (d, J=14.8 Hz, 1H), 2.96 (d, J=14.8 Hz, 1H), 2.45~2.28 (m, 2H), 2.05 (t, J=4.4 Hz, 1H), 1.98~1.84 (m, 2H), 1.56~1.34 (m, 2H), 1.04 (s, 3H), 0.80 (s, 3H).
Ethyl 4-(sulfamoylamino)benzoate (2a'): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a reddish brown solid (37 mg, 76%). m.p. 157~159 ℃; 1H NMR (600 MHz, DMSO-d6) δ: 10.11 (s, 1H), 7.86 (d, J=9.0 Hz, 2H), 7.34 (s, 2H), 7.21 (d, J=9.0 Hz, 2H), 4.27 (q, J=7.2 Hz, 2H), 1.30 (t, J=7.1 Hz, 3H); HRMS (ESI) calcd for C9H13N2O4S [M+H]+ 245.0591, found245.0583.
N-(4-Methylbenzyl)sulfuric diamide (2b'): The product was isolated by flash chromatography [eluent: V(ethylace-tate):V(petroleum ether)=3:10] as a colorless semi-solid (24 mg, 59%). 1H NMR (400 MHz, DMSO-d6) δ: 7.22 (d, J=8.0 Hz, 2H), 7.12 (d, J=8.0 Hz, 2H), 6.96 (t, J=6.4 Hz, 1H), 6.59 (s, 2H), 4.02 (d, J=6.8 Hz, 2H), 2.28 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 135.9, 135.6, 128.7, 127.7, 45.9, 20.7; HRMS (ESI) calcd for C8H12N2NaO2S [M+Na]+ 223.0512, found 223.0511.
4-Methoxy-N-(4-methyl-3, 6-dioxocyclohexa-1, 4-dien-1-yl)benzenesulfonamide (3m): The product was isolated by flash chromatography [eluent: V(ethylacetate):V(petro-leum ether)=1:10] as a yellow solid (6 mg, 10%). m.p. 112~115 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.86 (d, J=9.2 Hz, 2H), 7.63 (s, 1H), 6.99 (d, J=8.8 Hz, 2H), 6.54 (d, J=1.6 Hz, 1H), 6.48 (s, 1H), 3.87 (s, 3H), 2.02 (d, J=1.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 186.5, 181.5, 164.1, 148.4, 138.1, 129.9, 129.2, 114.7, 110.8, 55.8, 16.1; HRMS (ESI) calcd for C14H13NNaO5S [M+Na]+ 330.0407, found 330.0400.
Supporting Information 1H NMR and/or 13C NMR spectra of compounds 1 and 2. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
-
-
[1]
(a) Zhang, F.; Zheng, D.; Lai, L.; Cheng, J.; Sun, J.; Wu, J. Org. Lett. 2018, 20, 1167.
(b) Nematollahi, D.; Davarani, S. S. H.; Mirahmadpour, P.; Varmaghani, F. Chin. Chem. Lett. 2014, 25, 593.
(c) Sun, K.; Shi, Z.; Liu, Z.; Luan, B.; Zhu, J.; Xue, Y. Org. Lett. 2018, 20, 6687. -
[2]
For a List of Sulfa Drugs, See: http://www.thefullwiki.org/Sulfa_drugs.
-
[3]
(a) Wang, X.; Ahn, Y. M.; Lentscher, A. G.; Lister, J. S.; Brothers, R. C.; Kneen, M. M.; Gerratana, B.; Boshoff, H. I.; Dowd, C. S. Bioorg. Med. Chem. Lett. 2017, 27, 4426.
(b) Ascenzio, M. D.; Guglielmi, P.; Carradori, S.; Secci, D.; Florio, R.; Mollica, A.; Ceruso, M.; Akdemir, A.; Sobolev, A. P.; Supuran, C. T. J. Enzym. Inhib. Med. Chem. 2017, 32, 51.
(c) Luci, D. K.; Jameson, J. B.; Yasgar, A.; Diaz, G.; Joshi, N.; Kantz, A.; Markham, K.; Perry, S.; Kuhn, N.; Yeung, J.; Kerns, E. H.; Schultz, L.; Holinstat, M.; Nadler, J. L.; Taylor-Fishwick, D. A.; Jadhav, A.; Simeonov, A.; Holman, T. R.; Maloney, D. J. J. Med. Chem. 2014, 57, 495.
(d) Walsh, M. J.; Brimacombe, K. R.; Veith, H.; Bougie, J. M.; Daniel, T.; Leister, W.; Cantley, L. C.; Israelsen, W. J.; Vander Heiden, M. G.; Shen, M.; Auld, D. S.; Thomas, C. J.; Boxer, M. B. Bioorg. Med. Chem. Lett. 2011, 21, 6322.
(e) Al-Nadaf, A.; Sheikha, G. A.; Taha, M. O. Bioorg. Med. Chem. 2010, 18, 3088.
(f) Wydysh, E. A.; Medghalchi, S. M.; Vadlamudi, A.; Townsend, C. A. J. Med. Chem. 2009, 52, 3317.
(g) Johnson, S. L.; Chen, L. H.; Barile, E.; Emdadi, A.; Sabet, M.; Yuan, H.; Wei, J.; Guiney, D.; Pellecchia, M. S. Bioorg. Med. Chem. 2009, 17, 3352.
(h) Zheng, X.; Oda, H.; Takamatsu, K.; Sugimoto, Y.; Tai, A.; Akaho, E.; Ali, H. I.; Oshiki, T.; Kakuta, H.; Sasaki, K. Bioorg. Med. Chem. 2007, 15, 1014.
(i) Terrett, N. K.; Bell, A. S.; Brown, D.; Ellis, P. Bioorg. Med. Chem. Lett. 1996, 6, 1819. -
[4]
(a) Shi, M.; Yang, Y. H.; Xu, B. Synlett 2004, 1622.
(b) Liu, C. R.; Li, M. B.; Cheng, D. J.; Yang, C. F.; Tian, S. K. Org. Lett. 2009, 11, 2543.
(c) Liu, Z.; Larock, R. C. Org. Lett. 2003, 5, 4673.
(d) Fu, W.; Shen, R.; Bai, E.; Zhang, L.; Chen, Q.; Fang, Z.; Li, G.; Yi, X.; Zheng, A.; Tang, T. ACS Catal. 2018, 8, 9043.
(e) Gong, X.; Xia, H.; Wu, J. Org. Chem. Front. 2016, 3, 697.
(f) Yu, J.; Liu, S. S.; Cui, J.; Hou, X. S.; Zhang, C. Org. Lett. 2012, 14, 832.
(g) Xiang, Y.; Kuang, Y.; Wu, J. Org. Chem. Front. 2016, 3, 901.
(h) Li, Y. X.; Li, L. H.; Yang, Y. F.; Hua, H. L.; Yan, X. B.; Zhao, L. B.; Zhang, J. B.; Ji, F. J.; Liang, Y. M. Chem. Commun. 2014, 50, 9936.
(i) Chen, K.; Shi, B. F. Angew. Chem., Int. Ed. 2014, 53, 11950.
(j) Wang, H. Y.; Zhang, X.; Guo, Y. L.; Dong, X. C.; Tang, Q. H.; Lu, L. Rapid Commun. Mass Spectrom. 2005, 19, 1696.
(k) Feng, S. L.; Liu, C. Z.; Li, Q.; Yu, X. C.; Xu, Q. Chin. Chem. Lett. 2011, 22, 1021.
(l) Sun, K.; Li, Y.-L.; Feng, R.-R.; Mu, S.-Q.; Wang, X.; Zhang, B. J. Org. Chem. 2019, 84, 12792. -
[5]
Yao, B.; Zhang, Y. Tetrahedron Lett. 2008, 49, 5385. doi: 10.1016/j.tetlet.2008.06.114
-
[6]
Hewson, A. T.; Sharpe, D. A.; Wadsworth, A. H. Synth. Commun. 1989, 19, 2095. doi: 10.1080/00397918908052603
-
[7]
Wang, S. E.; Wang, L.; He, Q.; Fan, R. Angew. Chem., Int. Ed. 2015, 54, 13655. doi: 10.1002/anie.201508161
-
[8]
Katohgi, M.; Yokoyama, M.; Togo, H. Synlett 2000, 1055.
-
[9]
(a) Wang, Q.; Su, Y.; Li, L.; Huang, H. Chem. Soc. Rev. 2016, 45, 1257.
(b) Taniguchi, T.; Imoto, M.; Takeda, M.; Matsumoto, F.; Nakai, T.; Mihara, M.; Mizuno, T.; Nomoto, A.; Ogawa, A. Tetrahedron 2016, 72, 4132.
(c) Azzena, U.; Cattari, M.; Melloni, G.; Pisano, L. Synthesis 2003, 2811.
(d) Koreeda, T.; Kochi, T.; Kakiuchi, F. J. Organomet. Chem. 2013, 741~742, 148.
(e) Koreeda, T.; Kochi, T.; Kakiuchi, F. Organometallics 2013, 32, 682.
(f) Koreeda, T.; Kochi, T.; Kakiuchi, F. J. Am. Chem. Soc. 2009, 131, 7238.
(g) Ueno, S.; Chatani, N.; Kakiuchi, F. J. Am. Chem. Soc. 2007, 129, 6098. -
[10]
(a) Kamal, R.; Kumar, V.; Kumar, R. Chem.-Asian. J. 2016, 11, 1988.
(b) Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523.
(c) Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328.
(d) Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123.
(e) Zhdankin, V. V. ARKIVOC 2009, 1.
(f) Ladziata, U.; Zhdankin, V. V. ARKIVOC 2006, 26. -
[11]
(a) Wan, Y.; Zhang, Z.; Ma, N.; Bi, J.; Zhang, G. J. Org. Chem. 2018, 84, 780.
(b) Zhang, Z.; Gao, X.; Yu, H.; Zhang, G.; Liu, J. Adv. Synth. Catal. 2018, 360, 3406.
(c) Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155.
(d) Dess, D. B.; Martin, J. C. J. Am. Chem. Soc. 1991, 113, 7277.
(e) Zhang, Z.; Zhang, Y.; Huang, G.; Zhang, G. Org. Chem. Front. 2017, 4, 1372.
(f) Zhang, Z.; Gao, X.; Li, Z.; Zhang, G.; Ma, N.; Liu, Q.; Liu, T. Org. Chem. Front. 2017, 4, 404.
(g) Liu, Y.; Zhang, Z.; Wan, Y.; Zhang, G.; Li, Z.; Bi, J.; Ma, N.; Liu, T.; Liu, Q. J. Org. Chem. 2017, 82, 3901. -
[12]
(a) Onundi, Y.; Drake, B. A.; Malecky, R. T.; DeNardo, M. A.; Mills, M. R.; Kundu, S.; Ryabov, A. D.; Beach, E. S.; Horwitz, C. P.; Simonich, M. T.; Truong, L.; Tanguay, R. L.; Wright, L. J.; Singhal, N.; Collins, T. J. Green Chem. 2017, 19, 4234.
(b) Lu, L. H.; Wang, Z.; Xia, W.; Cheng, P.; Zhang, B.; Cao, Z.; He, W. M. Chin. Chem. Lett. 2019, 30, 1237.
(c) Xie, L. Y.; Duan, Y.; Lu, L. H.; Li, Y. J.; Peng, S.; Wu, C.; Liu, K. J.; Wang, Z.; He, W. M. ACS Sustainable Chem. Eng. 2017, 5, 10407. -
[13]
Frigerio, M.; Santagostino, M.; Sputore, S. J. Org. Chem. 1999, 64, 4537. doi: 10.1021/jo9824596
-
[14]
(a) Zhang, Z.; Zheng, D.; Wan, Y.; Zhang, G.; Bi, J.; Liu, Q.; Liu, T.; Shi, L. J. Org. Chem. 2018, 83, 1369.
(b) Zhang, Z.; Li, X.; Song, M.; Wan, Y.; Zheng, D.; Zhang, G.; Chen, G. J. Org. Chem. 2019. 84, 12792. -
[15]
Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis, John Wiley & Sons Inc., 1999.
-
[16]
Zhang, Y.; Li, Y.; Zhang, X.; Jiang, X. Chem. Commun. 2015, 51, 941. doi: 10.1039/C4CC08367A
-
[17]
Debnath, S.; Mondal, S. ChemistrySelect 2018, 3, 4129. doi: 10.1002/slct.201800435
-
[18]
Al-Shawabkeh, J. D.; Al-Nadaf, A. H.; Dahabiyeh, L. A.; Taha, M. O. Med. Chem. Res. 2014, 23, 127. doi: 10.1007/s00044-013-0616-2
-
[19]
Hosseinzadeh, R.; Tajbakhsh, M.; Mohadjerani, M.; Alikarami, M. J. Chem. Sci. 2010, 122, 143. doi: 10.1007/s12039-010-0015-x
-
[20]
Massah, A. R.; Sayadi, S.; Ebrahimi, S. RSC Adv. 2012, 2, 6606. doi: 10.1039/c2ra20418e
-
[21]
Zhang, W.; Xie, J.; Rao, B.; Luo, M. J. Org. Chem. 2015, 80, 3504. doi: 10.1021/acs.joc.5b00130
-
[22]
Chen, K.; Chen, W.; Han, B.; Chen, W.; Liu, M.; Wu, H. Org. Lett. 2020, 22, 1841. doi: 10.1021/acs.orglett.0c00183
-
[23]
Burmistrov, S. I. Ukr. Fiz. Zh. 1966, 32, 601.
-
[24]
Krasavin, I. A. Metody Polycheniya Khim. Reaktivov i Preparatov, Gos. Kom. Sov. Min. SSSR po Khim. 1962, 4~5, 69.
-
[25]
Ueda, Y.; Yano, H.; Momose, T. Chem. Pharm. Bull. 1964, 12, 5. doi: 10.1248/cpb.12.5
-
[26]
Shekhar, S. X.; Dunn, T. B.; Kotecki, B. J.; Montavon, D. K.; Cullen, S. C. J. Org. Chem. 2011, 76, 4552. doi: 10.1021/jo200443u
-
[27]
Gowda, B. T.; Jayalakshmi, K. L.; Shetty, M. Z. Naturforsch. 2004, 59a, 239.
-
[28]
You, C. R.; Yao, F.; Yan, T.; Cai, M. C. RSC Adv. 2016, 6, 43605. doi: 10.1039/C6RA04298H
-
[29]
Namba, K.; Zheng, X.; Motoshima, K.; Kobayashi, H.; Tai, A.; Takahashi, E.; Sasaki, K.; Okamoto, K.; Kakuta, H. Bioorg. Med. Chem. 2008, 16, 6131. doi: 10.1016/j.bmc.2008.04.040
-
[30]
Ueda, Y.; Yano, H.; Momose, T. Chem. Pharm. Bull. 1964, 12, 5. doi: 10.1248/cpb.12.5
-
[31]
Cremlyn, R. J.; Goulding, K. H.; Swinbourne, F. J.; Yung, K. M. Phosphorus Sulfur 1981, 10, 111.
-
[32]
Solov'ev, M. Y.; Filimonov, S. I.; Skorenko, A. V.; Ivanenkov, Ya. A.; Balakin, K. B.; Dorogov, M. V. Izv. Vyssh. Uchebn. Zaved., Khim. Khim. Tekhnol. 2004, 47, 28.
-
[33]
Kostsova, A. G. Zh. Obshch. Khim. 1953, 23, 949.
-
[34]
Maghsoodia, N. K.; Khazaelia, T.; Massah, A. R. J. Chem. Res. 2015, 39, 141. doi: 10.3184/174751915X14241022318075
-
[35]
Pantaine, L.; Richard, F.; Marrot, J.; Moreau, X.; Coeffard, V.; Grecka, C. Adv. Synth. Catal. 2016, 358, 2012. doi: 10.1002/adsc.201501139
-
[36]
Johnson, C. R.; Jonsson, E. U.; Bacon, C. C. J. Org. Chem. 1979, 44, 2055. doi: 10.1021/jo01327a001
-
[37]
Gregg, D. C.; Blood, C. A., Jr. J. Org. Chem. 1951, 16, 1255.
-
[38]
Bonk, J. D.; Amos, D. T.; Olson, S. J. Synth. Commun. 2007, 37, 2039. doi: 10.1080/00397910701356942
-
[39]
Illuminati, G. J. Am. Chem. Soc. 1956, 78, 2603. doi: 10.1021/ja01592a075
-
[40]
Amorosa, I. M. Farmaco 1949, 4, 290.
-
[41]
Yale, H.; Sowinski, F. J. Org. Chem. 1960, 25, 1824. doi: 10.1021/jo01080a612
-
[42]
Isozaki, M. Tokyo Kogyo Shikensho Hokoku 1950, 45, 295.
-
[43]
Baker, Robert H.; Dodson, R. M.; Riegel, B. J. Am. Chem. Soc. 1946, 68, 2636. doi: 10.1021/ja01216a063
-
[44]
Chhabra, S. R.; Mahajan, A.; Chan, W. C. J. Org. Chem. 2002, 67, 4017. doi: 10.1021/jo010456e
-
[45]
Tota, A.; John-Campbell, S. S.; Briggs, E. L.; Estevez, G. O.; Afonso, M.; Degennaro, L.; Luisi, R.; J. A. Org. Lett. 2018, 20, 2599. doi: 10.1021/acs.orglett.8b00788
-
[46]
Maslankiewicz, A.; Marciniec, K.; Pawlowski, M.; Zajdel, P. Heterocycles 2007, 71, 1975. doi: 10.3987/COM-07-11088
-
[47]
Moroda, A.; Togo, H. Tetrahedron 2006, 62, 12408. doi: 10.1016/j.tet.2006.09.112
-
[48]
Field, L.; Grunwald, F. A. J. Am. Chem. Soc. 1953, 75, 934. doi: 10.1021/ja01100a048
-
[49]
Truce, W. E.; Gunberg, P. F. J. Am. Chem. Soc. 1950, 72, 2401. doi: 10.1021/ja01162a014
-
[1]
-
Table 1. Survey of the reaction conditionsa
Entry Oxidant (equiv.) Solvent (V:V) T/℃ Time Yield/% of2r Recovered/% of1r 1 IBX (1.2) HFIP/H2O (2:1) 25 4 h 83 0 2 DMP (1.2) HIP/H2O (2:1) 25 1 h 99 0 3 DMP (1.2) HIP/H2O (2:1) 40 10 min 96 0 4 DMP (1.2) H2O 40 0.5 h 0 95 5 DMP (1.2) HIP 40 1 h 5 85 6 DMP (1.2) HIP/H2O (1:5) 40 2.5 h 80 18 7 DMP (1.2) HIP/H2O (1:1) 40 10 min 98 0 8 DMP (1.2) HIP/H2O (1:3) 40 1.5 h 97 0 9 DMP (0.6) HIP/H2O (1:3) 40 3 h 43 44 10 DMP (1.2) TFE/H2O (1:3) 40 1.5 h 5 84 11 DMP (1.2) EtOH/H2O (1:3) 40 1.5 h 0 96 12 DMP (1.2) DMSO/H2O (1:3) 40 0.5 h 0 97 13 DMP (1.2) MeCN/H2O (1:3) 40 0.5 h 0 98 14 PIFA (1.2) HIP/H2O (1:3) 40 1.5 h 81 12 15 PhIO (1.2) HIP/H2O (1:3) 40 1.5 h 72 19 16 PIDA (1.2) HIP/H2O (1:3) 40 1.5 h 77 11 17 HTIB (1.2) HIP/H2O (1:3) 40 1.5 h 67 26 a Unless otherwise indicated, all reactions were conducted with 1r (0.2 mmol) in 2 mL of solvent at 40 ℃.  下载: 导出CSV
下载: 导出CSV
Table 2. Extension of the reaction scopea
a Unless otherwise indicated, all these reactions were conducted with 1 (0.2 mmol) and DMP (1.2 equiv.) in 2 mL of mixed solvent system (VHFIP:VH2O=1:3) at 40 ℃. b Compound 2c was conducted with 1 (0.2 mmol) and DMP (1.5 equiv.) in 2.0 mL of mixed solvent system (VHFIP:VH2O=1:3) at 40 ℃. c 96% of the starting material was recovered. d 97% of the starting material was recovered. e Compound 2m was reacted with 1 (0.2 mmol) and DMP (1.5 equiv.) in 2.0 mL of mixed solvent system (VHFIP:VH2O=1:3) at 40 ℃. f Compound 2v was reacted with 1 (0.2 mmol) and DMP (2.5 equiv.) in 2.0 mL of mixed solvent system (VHFIP:VH2O=1:1) at 60 ℃.
下载: 导出CSV
-


 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 9
- 文章访问数: 2709
- HTML全文浏览量: 304
