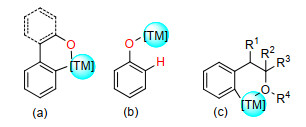
图 1.
含羟基、醚化合物的碳氢活化策略
Figure 1.
C—H activation strategy for hydroxyl and ether-containing compounds

通过碳氢活化的方法构建碳-碳键是化学家们研究的热点.在过去的二十年中, 由于基于导向基(DGs)策略的碳氢官能化反应能够实现高区域选择性碳氢官能团化, 因此碳氢活化领域的大部分研究都集中于基于导向基(DGs)的碳氢官能化反应.其中, 绝大部分的基于导向基(DGs)的碳氢活化反应是强螯合的“经典环金属化”反应[1].该反应的实质是催化剂与底物之间络合形成“热力学稳定”的五/六元环金属中间体.虽然该类反应能够有效实现碳氢官能团化反应, 但此类反应具有以下局限性:其一、底物通常限于含强σ供体或π受体的导向基的芳烃[2], 例如吡啶、噁唑啉、硫化物、磷化物等导向基的芳烃, 然而这类芳烃通常需要额外安装和拆卸的步骤, 因此具有较低的步骤经济性; 其二、基于强配位的反应会形成“热力学稳定”的五/六元环金属中间体, 因此含强配位导向基的芳烃往往难以实现远程导向; 其三、此类反应往往形成“热力学稳定”的五/六元环金属中间体, 因此随后的官能团化步骤反应性较低.
为了进一步拓宽基于导向基(DGs)策略的碳氢官能化反应的应用性, 以余金权为代表的科学家们发现了基于弱配位的碳氢官能团化反应[3].此类反应的导向基往往是一些有价值的官能团(例如羟基、醚、羧基、羰基、酰胺基等), 能够极大地提高碳氢活化反应的底物范围.但该类芳烃与过渡金属的配位能力较弱, 过渡金属络合物中间体的热力学稳定性也较差.因此, 将该类芳烃直接用于碳氢活化反应具有一定的挑战性.目前该类反应可以通过优化反应条件或引入配体提高反应活性和区域选择性.该反应类型的开发为新型化学反应和配体控制的碳氢活化反应提供了一个有力的工具.因此, 基于弱配位的碳氢活化反应成为近年来化学家们研究的热门方向.
众所周知, 在化合物中含氧原子的官能团非常多(例如羟基、羧基、酰胺、醛基、羰基等), 因此近年来在基于弱配位的碳氢活化反应领域中, 科研工作者们对过渡金属与氧原子之间的弱配位作用的研究最多.目前已证明Pd[4], Ru[5]和Ir[6]等催化剂与含氧导向基的芳烃具有良好的弱配位作用, 因此基于过渡金属-O弱配位的C(sp2)—H官能团化反应已取得重大进展.对近年来基于过渡金属-O弱配位策略的具有含氧官能团(羟基、醚、醛基、羰基、酰胺、羧基、磷酸酯、磺酸)的芳烃的C(sp2)—H烯基化反应进展进行了综述.
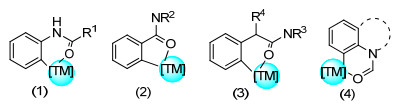
含羟基化合物的C(sp2)—H烯基化反应的研究主要集中于醇/酚羟基.含醇羟基导向基的芳烃会与过渡金属配位形成六元环络合物中间体, 从而实现C(sp2)—H烯基化(图 1a).此外, 苯酚的高区域选择性邻位烯基化反应也得到解决, 其C(sp2)—H活化策略主要通过苯酚与过渡金属形成配合物中间体(图 1b).醚导向的C(sp2)—H烯基化反应则是通过和过渡金属形成六元环络合物中间体, 从而活化邻位C(sp2)—H键(图 1c).对于含羟基/醚导向的直接烯基化反应进行了综述.
1997年Miura课题组[7a]以Pd(OAc)2作为催化剂, Cu(OAc)2作为氧化剂, N, N-二甲基甲酰胺(DMF)作为溶剂, 开创性地实现了邻芳基苯酚的C(sp2)—H苯乙烯基化反应(Eq. 1a).值得注意的是, 当丙烯酸酯类和丙烯酰胺类烯基作为底物参与反应时, 得到苯并吡喃类化合物.与Miura课题组不同的是, Sun课题组[7b]发现在乙酸环境中, 丙烯酸酯类烯烃源作为底物参与反应时, 没有生成苯并吡喃类化合物(Eq. 1b).相较于前者的研究, 该反应体系具有较好的基团耐受性(烷烃、甲氧基、卤素、芳基、酯键).但是, 含硝基的底物或1-苯己烯的底物参与反应仅以痕量转化为对应产物.
|
|
2010年, 余金权课题组[8a]首次发现叔醇导向的C(sp2)—H烯基化反应(Eq. 2a).该反应体系通过使用Pd催化剂, Li2CO3弱碱, 亮氨酸类配体实现.此外, 苯乙烯、乙烯基砜类、乙烯基磷酸二乙酯、1-己烯作为底物参与反应均能实现其邻位的烯基化.与Miura课题组的研究类似, 当使用丙烯酸酯类底物参与反应时, 生成了对应的苯并吡喃类产物.之后, 黄勇课题组[8b]开发了无碱添加剂的反应体系, 成功实现了该类化合物的烯基化反应(Eq. 2b).此外, 该反应体系还成功用于伯醇、仲醇或叔醇导向的烯基化反应.
|
|
2011年Gevorgyan课题组[9]开发了一种新型的硅醇导向基, 实现了苯酚邻位的C(sp2)—H烯基化(Eq. 3).与邻芳基苯酚和叔醇导向基的C(sp2)—H烯基化反应类似, 其活性位点为形成Pd—O六元环络合物对应的位点.此外, 该类导向基的优势在于能够在TBAF/THF中温和地实现导向基的去除, 形成苯酚类化合物.
|
|
(3) |
目前, 已有多种类型催化剂的反应体系用于苯酚邻位的端炔加成反应. 1995年, Hirama课题组[10a]创新性地使用SnCl4作为催化剂实现苯酚邻位炔烃加成反应(Eq. 4a). 2009年, Biswas课题组[10b]在GaCl3催化、甲苯回流下实现芳基乙炔与萘酚/苯酚反应, 得到相应的2-乙烯基萘酚/苯酚(Eq. 4b). 2013年Kumar课题组[10c]实现了In(OTf)3催化的萘酚邻位端炔的加成反应(Eq. 4c).此外, 该课题组还使用一锅法合成了2, 3-二芳基萘并呋喃, 扩展了该反应体系的应用范围.与前人工作不同的是, Takai课题组[11]将催化剂聚焦于金属Re和更加具有挑战性的非端炔类底物, 开发了中性条件下的Re2(CO)10催化苯酚邻位的非端炔的加成反应(Eq. 5).虽然上述反应成功实现了苯酚邻位的烯基化反应, 但是仅限于炔烃的加成反应. 2019年, 我们课题组[12]使用苯酚和端烯类底物, 高度区域选择性地实现了Pd(Ⅱ)催化苯酚邻位的烯基化反应(Eq. 6).该方法具有较好的基团耐受性(烷基、甲氧基、氰基、卤素、三氟甲基、羧基、硝基等)和底物耐受性(丙烯酸酯类、丙烯酰胺类、1-戊烯-3-酮、丙烯腈、苯乙烯类烯基源底物).为了验证该方法的实用性, 我们成功将该方法用于香豆素、氧茚的构建, 多肽的修饰和抗肿瘤药物的末期修饰.
|
|
2012年, 余金权课题组[13]提出了醚键导向的C(sp2)—H官能团化反应, 报道了醚导向的C(sp2)—H烯基化反应, 进一步证明了醚键与Pd的弱配位的作用可以用于C(sp2)—H活化(Eq. 7).
|
|
(7) |
羰基、醛基在小分子化合物和天然大分子中广泛存在, 其导向邻位的C(sp2)—H烯基化是化学家们研究的热门反应之一.芳基酮/醛中的羰基与过渡金属形成五元环金属中间体, 从而活化邻位C(sp2)—H键(图 2a), 而羰基导向的萘醌和吲哚衍生物的C(sp2)—H烯基化是和过渡金属配位形成六元环络合物中间体, 从而活化芳环上的C(sp2)—H键(图 2b, 2c).此外, 羰基导向的二茂铁C(sp2)—H烯基化反应是通过羰基与过渡金属配位形成五元环过渡金属络合物中间体实现(图 2d).对于芳香酮/醛、羰基导向的吲哚衍生物、苯醌和二茂铁的直接烯基化反应进行了综述.
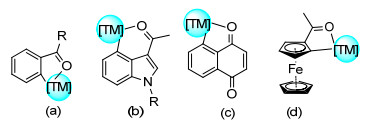
2007年Kakiuchi课题组[14]首次报道了Ru催化的芳香酮与烯基硼酸酯之间的偶联反应(Eq. 8).该反应以频哪酮作为溶剂回流实现.之后, 该课题组进一步扩大了该反应的底物范围, 2-丙烯基硼酸酯、1-丙烯基硼酸酯、芳基硼酸酯均具有良好的兼容性. 2011年, Jeganmohan课题组[15]实现了Ru催化的芳香酮与端烯的氧化偶联反应(Eq. 9).该反应体系具有较宽的底物范围, 对烷基、甲氧基取代的底物, 萘、环氧、色酮类底物和丙烯酸酯类、苯乙烯类烯基源均有良好的兼容性.之后, Glorius课题组[16]将催化剂转向过渡金属Rh, 证明苯乙酮和苯甲酰胺都是Rh催化烯基化反应的适宜底物(Eq. 10).此外, 缺电子和富电子苯乙烯类底物都具有良好的耐受性, 甚至能与1, 1-和1, 2-二取代烯烃偶联.之后, Maji课题组[17]进一步证明了高效、空气稳定的Co催化剂可用于羰基导向高区域选择性的烯基化反应(Eq. 11).除此之外, 该课题组还进一步研究苯乙酮、二芳基酮、色酮和与生物有关的查尔酮等底物的羰基导向的C(sp2)—H烯基化反应, 并通过该反应体系成功制备了肉桂酸乙酯、2-茚基酮、α-萘酚、木榄醇A.之后, 王从洋课题组[18]报道了Mn催化羰基导向的C(sp2)—H烯基化反应(Eq. 12).该反应对α、β、γ位取代的烯基类底物有较好的兼容度.
2007年, Shibata课题组[19a]将底物聚焦于非常富有挑战性的二炔, 利用[Rh(BIPHEP)]BF4]催化剂实现了二炔与芳族酮偶联反应, 得到环状1, 3-二烯产物(Eq. 13a). 2014年, Cheng课题组[19b]报道了CoBr2/1, 3-双(二苯基磷)丙烷(dppp)的催化体系, 实现了烯炔和含酮芳烃进行类似的加氢芳基化反应(Eq. 13b).其中, Zn、ZnI的作用是将Co(Ⅱ)盐还原为活性Co(I)催化剂.
|
|
2015年余金权课题组[20]使用1-苯基-2-丙酮、4-苯基-2-丁酮等更加远端的羰基作为导向基实现了芳烃邻位的烯基化反应(Eq. 14).
|
|
(14) |
2016年Szostak课题组[21]进一步实现了Ru催化的羰基导向的炔烃加成反应(Eq. 15).与Murai方法相比, 该系统进一步扩大了底物的范围.除此之外, 该课题组证明, 原位Ru(0)催化剂能够活化传统的Ru(0)无法活化的C(sp2)—H键, 对设计新型Ru(0)催化的反应体系有重要的意义.
|
|
(15) |
目前, 对于醛基导向的C(sp2)—H烯基化通常是通过引入氨基酸衍生物的瞬态导向基构建亚胺结构, 和过渡金属形成强配位的络合物中间体实现.若能直接通过弱配位策略实现醛基邻位烯基化反应是非常有意义的. 2018年, 秦华利课题组[22]报道了Rh催化的苯甲醛和苯甲酮的邻位乙烯基磺酰氟化反应(Eqs. 16a, 16b).值得注意的是, 该方法无需瞬态导向基, 是通过醛基/羰基与Rh之间的弱配位作用实现.为证明该反应体系的应用潜力,他们成功将其用于胆固醇衍生物的末期修饰.
|
|
2019年Ravikumar课题组[23a]报道了羰基导向金属Co催化的吲哚衍生物选择性C(4)—H官能团化反应(Eqs. 17a, 17b).除此之外, 当添加剂从Cu(OAc)2转化为Ag2CO3时, 实现了其与马来酰亚胺之间的偶联.今年, Banerjee课题组[23b]将催化剂转向金属Rh, 实现了羰基导向C(4)—H位吲哚的烯基化反应(Eq. 17c).
|
|
2014年张翱课题组[24a]将研究聚焦于底物1, 4-萘醌, 开发了Rh催化的1, 4-萘醌的烯基化反应(Eq. 18a).该方法具有较高的官能团兼容度、较宽的底物范围. 2019年Sharma课题组[24b]在酸性和碱性条件下, 分别实现了Rh催化的1, 4-萘醌与马来酰亚胺的C(5)—H区域选择性的烷基化和烯基化(Eq. 18b).同年, Ackermann课题组[24c]报道了Ru催化的萘醌的C(sp2)—H烯基化反应(Eq. 18c), 为制备多样性的具有生物学特性的化合物的策略提供了一种有效方法.
|
|
2012年Dixneuf课题组[25]报道了[RuCl2(p-cymene)]2催化体系下的二茂铁基烷基酮与丙烯酸酯类烯基源的烯基化反应(Eqs. 19a, 19b).与此同时, 在室温下二茂铁基苯基酮产生苯基单烯基化产物.
|
|
2011年Glorius课题组[26a]首次实现了Rh催化的苯甲酸甲酯的邻位烯基化反应(Eq. 20).之后, Chang课题组[26b]进一步实现了苯甲酸酯和苯甲醛的Rh催化的邻位的烯基化.该反应对多种苯甲酸酯和苯甲醛均具有很高的区域选择性.
Huestis课题组[27]报道了Rh催化含导向基芳烃的乙烯基璜酰氟化反应(Eq. 21).例如含羰基(含醛基、酮、酯、酰胺)导向的芳烃邻位乙烯基璜酰氟化反应.
|
|
(20) |
|
|
(21) |
酰胺键在化学中间体、药物和化学生物学中广泛存在, 对其精准位点的C(sp2)—H烯基化成为化学家们研究热点.目前, 对于酰胺键导向的C(sp2)—H烯基化主要用于乙酰苯胺、N-甲基苯甲酰胺及其衍生物等.其酰胺键上的氧原子与过渡金属弱配位, 实现了邻位的C(sp2)—H活化(图 3).对于乙酰苯胺、N-甲基苯甲酰胺、苯乙酰胺和酰胺导向的N杂环化合物及其衍生物的直接烯基化反应进行了综述.
乙酰苯胺的C(sp2)—H烯基化反应主要集中于Pd催化剂. 2002年Leeuwen课题组[28]实现了室温下乙酰苯胺邻位的C(sp2)—H烯基化反应(Eq. 22).该反应以Pd(OAc)2作为催化剂, 苯醌(BQ)作为氧化剂和催化剂稳定剂, 在常温下以进行, 产率中等(26%~91%).值得注意的是, TsOH作为添加剂能够加速该反应的进行.但是该反应具有较大的底物局限性, 仅限于甲基、甲氧基、三氟甲基取代的底物和丙烯酸酯类底物. 2005年Blacklock课题组[28b]进一步将底物扩展至丙烯酸丁酯.之后, Jutand课题组[28c]以电化学的方法实现了间甲基乙酰苯胺的邻位丙烯酸酯化和苯乙烯化反应. 2010年Lipshutz课题组[28d]实现了水作为唯一溶剂, 在无外加酸的情况下乙酰苯胺的邻位烯基化反应.该反应以[Pd(MeCN)4]-(BF4)2作为催化剂, AgNO3和BQ作为添加剂, PTS (polyoxyethanyl α-tocopheryl sebacate)/水作为溶剂.该方法最大的优点是绿色、环保和反应条件温和. 2011年, Hii课题组[28e]证明过氧化苯甲酸叔丁酯可以替代BQ加速反应, 并且Cu(OAc)2作为助催化剂能够增强反应性. 2012年Elizarov课题组[28f]通过脱乙酰基重氮化-Heck偶联成功实现了C(sp2)—H乙烯基化反应和导向基的去除.近年来, Youn课题组[28g]开发了一种乙酰苯胺/邻苯基乙酰苯胺的邻位C(sp2)—H烯基化策略.以氧化性较强的K2S2O8作为氧化剂, CF3COOH/CH2Cl2作为溶剂, 常温条件下实现了邻位的C(sp2)—H烯基化反应.该方法不仅能用于乙酰苯胺, 也能实现邻苯基乙酰苯胺的邻位C(sp2)—H烯基化, 而非六元环Pd络合物的活化位点.此外, 这种方法对于硝基、乙酰基取代的底物和丙烯酸酯类、丙烯酰胺类底物有较好的兼容性.
|
|
(22) |
2009年Brown课题组[29a]以1, 1-二甲基-3-苯基脲作为底物, 实现了其邻位的C(sp2)—H丙烯酸丁酯化反应(Eq. 23).之后, Yu课题组[29b]进一步扩大了底物范围, 实现了1-甲基-3苯基脲、1-乙基-3苯基脲、1-苯乙基-3苯基脲、1-丁基-苯基脲的C(sp2)—H烯基化反应.
|
|
(23) |
2019年, Jeganmohan课题组[30]报道了一种Rh催化的乙酰苯胺与马来酰亚胺的邻位氧化偶联反应(Eq. 24).值得注意的是, 该反应体系有少量的迈克尔型加成产物3-芳基琥珀酰亚胺生成.
Miura课题组[31a]在2012年报道了Ru催化的N, N-二苯基苯甲酰胺与二苯乙炔的加氢芳基化反应(Eq. 25).同年, 该课题组[31b]在乙酸条件下实现了Ru催化的苯甲酰胺和苯吡唑的炔烃加成反应, 得到了对应的邻烯基化产物.
|
|
(24) |
|
|
(25) |
2013年王细胜课题组[32a]报道了Rh催化、酰胺导向空气作为氧化剂的烯基化反应.这种反应体系具有良好的反应性、区域选择性和官能团耐受性(Eq. 26a). 2018年, 秦华利课题组[32b]通过N-甲氧基-N-甲基苯甲酰胺和乙烯基磺酰氟的高度选择性的氧化偶联来合成邻烯基化产物(Eq. 26b).
|
|
2016年, 钟国富课题组[33]报道了Ru催化苯甲酰胺与乙酸烯丙酯的直接烯基化策略.该方法在不使用任何氧化剂的情况下, 以优异的产率、良好的区域选择性和官能团相容性得到反式苯乙烯产物(Eq. 27).
|
|
(27) |
2020年, 李必杰课题组[34]开发了一种铱催化的、含磷配体控制的芳烃与端炔的反应, 获得了高选择性的C(sp2)—H烯基化产物(Eq. 28).值得注意的是, 炔烃上的硅基对催化活性有不可替代的作用.
|
|
(28) |
2018年Kumar课题组[35]报道了脂肪族伯乙酰胺是过渡金属催化C(sp2)—H活化中的有效远端导向基, 实现了其选择性的邻位C(sp2)—H烯基化反应(Eq. 29).该方法以Pd(OAc)2作为催化剂、BQ作为添加剂、O2作为唯一绿色氧化剂、三氟乙酸(TFA)作为溶剂下实现2-苯乙酰胺的邻位C(sp2)—H烯基化反应.该反应对于甲基、甲氧基、卤素具有良好的相容性, 并且对于丙烯酸酯类、乙烯基砜类、乙烯基磷酸酯类烯基具有良好的反应性.此外, 除了2-苯乙酰胺, 2-甲基-2-苯乙酰胺也能产生其对应的烯基化产物.
|
|
(29) |
2018年Ackermann课题组[36a]首次报道了Ru催化的芳基乙酰胺的C(sp2)—H烯基化反应(Eq. 30).值得注意的是, 该催化体系在三级、二级甚至一级酰胺上都是可行的. 2019年Prabhu课题组[36b]实现了Rh催化芳基乙酰胺的C(sp2)—H烯基化反应.该催化体系创新地以芳炔硅烷替代端炔, 在中性条件下, 通过脱硅反应途径, 实现了芳基乙酰胺的C(sp2)—H烯基化反应.
|
|
(30) |
2019年Jeganmohan课题组[37]开发出一种Rh催化乙酰胺导向且空气作为唯一的氧化剂的烯基化反应(Eqs. 31a, 31b).乙酰胺导向基团包括伯、仲、叔酰胺.该反应体系在1-金刚烷羧酸和二氯乙烷的存在下, 能与端烯进行氧化偶联.然而当添加剂和溶剂切换为乙酸铜和二甲醚时, 底物能与马来酰亚胺进行氧化偶联.
|
|
2012年, 李兴伟课题组[38]实现了Rh/Ru催化的C(sp2)—H烯基化反应(Eqs. 32a, 32b).该课题组发现Ru体系能够有效地与非端炔反应形成烯基化产物, 然而Rh催化体系能与端烯进行氧化偶联.该反应体系在合成有用的复合杂环化合物方面有重要的应用潜力.
|
|
2013年Oestreich课题组[39]报道了Pd催化吲哚类化合物选择性C-7位点的烯基化反应(Eq. 33).该反应在Pd催化剂、BQ和对甲苯磺酸(PTSA)添加剂、乙酸溶剂中实现.值得注意的是, α, β-不饱和底物和苯乙烯底物中均有良好的反应性.
|
|
(33) |
2013年宋飞杰课题组[40]实现了酰胺导向的咔唑类化合物的烯基化反应(Eq. 34).运用此反应体系能实现该类底物的丙烯酸酯化和苯乙烯化反应.
|
|
(34) |
在过去几年中, Frost[41a, 41b]和Ackermann等[41c]分别报道了Ru催化缺电子的烯烃(如丙烯酸酯类烯基)与噁唑烷酮、吡咯烷酮、噻唑烷酮、乙内酰脲、琥珀酰亚胺和吡唑酮的氧化偶联反应(Eq. 35).今年, 吴彦超课题组[42]开发了Ru催化吡咯C-2-位与端烯的区域选择性C(sp2)—H烯基化反应(Eq. 36).因吡咯是功能材料, 天然产物和生物分子中广泛存在的结构, 因此, 该策略为制备有价值的乙烯基吡咯提供了一种高效的工具.
|
|
(35) |
|
|
(36) |
羧酸类化合物是一类重要含氧化合物, 不仅在具有生物活性的天然产物(蛋白质组学、医药等)中广泛存在, 还能对其衍生成为酰胺、酯类等化合物[30].因此, 对于此类化合物进行碳氢活化研究具有十分重要的意义.其中, 过渡金属和羧基通常可以与过渡金属形成五/六元环络合物中间体, 从而实现C(sp2)—H烯基化反应(图 4).以下对于苯甲/乙酸、羧酸导向的二茂铁、远程羧酸导向的化合物及其衍生物的直接烯基化反应进行综述.
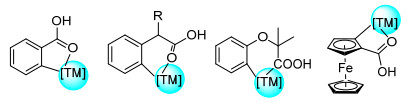
1998年, Muira课题组[43]使用Pd催化剂实现羧基导向的碳氢活化反应.该课题组利用苯甲酸作为底物, 在碱性条件下形成五/六元环酯(烯)结构. 2010年, 余金权课题组[44a]将研究转向Pd催化羧基导向的C(sp2)—H烯基化反应(Eq. 37a).首先, 该课题组以二苯乙酸盐作为底物, 利用氨基酸配体(Boc-Ile-OH)实现了其C(sp2)—H烯基化反应.同年, 余金权课题组[44b]首次以O2作为唯一绿色氧化剂, 实现了羧基导向的苯乙酸邻位单取代烯基化反应(Eq. 37b).该课题组发现, 当引入氨基酸衍生物作为配体, 含有不同位点取代基时, 可以调节其位置选择性.此外, 该反应体系具有广泛的底物谱, 如酮洛芬、布洛芬和萘普生等药物均能实现该反应.同年, 余金权课题组[44c]进一步实现了苯乙酸邻位的C(sp2)—H双取代的乙烯化反应(Eq. 37c).重要的是, 该课题组发现之前使用的苯醌(BQ)降低了反应速率.当使用Ac-Val-OH作为配体反应时间为6 h时, 双取代的乙烯基苯乙酸的转化率大于99%. 2012年Blackmond课题组[44d]通过动力学和NMR研究了苯乙酸盐邻位C(sp2)—H烯基化反应的机理, 确定了羧基与苯乙酸之间的弱配位作用.
|
|
2011年Miura课题组[45]将研究转向Rh催化剂.在银或铜盐的氧化剂下, 该课题组实现了苯甲酸和萘酸、α-、β-不饱和羧酸和杂环羧酸的高区域选择性的邻位烯基化反应(Eq. 38).
|
|
(38) |
近年来, 许多课题组将羧基导向C(sp2)—H烯基化反应转向Ru催化体系. 2016年Gooẞen课题组[46a]在Ru催化剂与碱式碳酸胍的反应体系下发现苯甲酸与炔烃能够反应, 生成相应的2-乙烯基苯甲酸(Eq. 39a).该反应体系适用于各种取代的富电子和缺电子的取代的底物.之后, 该课题组[46b]发现苯甲酸能够在50 ℃下与乙酸烯丙酯反应生成相应的烯丙基苯甲酸(Eq. 39b).但遗憾的是, 该方案仅适用于富含电子和不含电子的底物. 2018年, Jeganmohan课题组[46c]实现了Ru催化的苯甲酸与乙酸烯丙酯氧化偶联反应(Eq. 39c).但是, 在该反应中生成了绿色环保的五元杂环副产物. 2019年Baidya课题组[46d]在Ru催化剂下实现了高效一锅法(杂)芳香酸的不对称双官能化反应(Eq. 39d).该策略操作简单, 易于高收率和区域选择性地转化为官能化的芳烃.
|
|
2013年, 余金权课题组[47]开发了可用于远程羧酸导向的邻位C(sp2)—H烯基化反应(Eq. 40).该方法以Pd(OAc)2作为催化剂, Boc-Val-OH作为配体, KHCO3作为添加剂, O2作为氧化剂, tAmylOH作为溶剂实现.
|
|
(40) |
2020年, 吴养洁课题组[48]首次开发了Pd催化羧酸导向的二茂铁邻位烯基化反应.该反应体系以氨基酸衍生物为配体、O2作为唯一绿色氧化剂、TBAB为添加剂、Cs2CO3的碱性条件下实现(Eq. 41).
|
|
(41) |
有机磷、硫化合物由于在不同领域的应用而受到关注, 例如配位和材料化学、均相催化、药物和农用化学品, 聚合物添加剂和阻燃剂等.但是对此类化合物进行碳氢活化在近年来才开始被发掘.含磷/磺酸化合物的C(sp2)—H烯基化反应主要通过含磷酸、磺酸及其衍生物等导向基团的芳烃, 与过渡金属形成五/六元环配合物实现邻位C(sp2)—H烯基化(图 5).对于磷酸、磺酸导向的化合物及其衍生物的直接烯基化反应进行了综述.
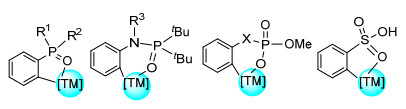
2013年Lee课题组[49a]首次使用Pd催化剂, 实现了磷酸导向邻位的C(sp2)—H烯基化反应(Eq. 42a).该反应产率较高(14%~98%), 烷基、烷氧基、卤素和酯基等取代的底物、萘环衍生物、联芳基衍生物都能很好地转化为对应的烯基化产物.此外, 该反应的实用性不仅限于丙烯酸乙酯类底物, 各种苯乙烯衍生物、乙烯基砜和磷酸乙烯基酯、苯基乙烯基酮等端烯都可以被成功用于C(sp2)—H烯基化反应.但是, 该方法不适用于苯丙烯和乙基乙烯基醚.同年, Kim课题组[49b]将研究转向苄基磷酸单酯, 成功实现了单磷酸导向的Pd催化邻位C(sp2)—H烯基化反应(Eq. 42b).值得注意的是, 该反应体系会产生单取代/双取代两种产物(单取代:双取代=48:47).此外, 甲氧基、氟、氯取代底物、萘环衍生物均能顺利产生对应烯基化产物.对于活性较高的烯基, 例如丙烯酸苄酯(91%)、磷酸乙烯基酯(76%), 苯乙烯衍生物(55%~96%)均能以较高产率获得.然而, 苯基乙烯基酮作为底物参与反应的产率较低(30%), 巴豆酸乙酯、烯丙基苯作为底物参与反应则反应无法进行.
|
|
同年, Yang课题组[50]通过使用P(O)Ph2作为新的导向基团, 成功实现了2-二苯基磷基2'-甲基联苯的C(sp2)—H烯基化反应(Eq. 43).该反应以较高的收率广泛用于缺电子烯基(32%~86%), 例如丙烯酸丁酯和丙烯酸苄酯, 亚磷酸亚烷基酯和乙烯基砜类烯基.但是, 该反应体系的基团相容性仅研究了甲基、甲氧基和硝基.
|
|
(43) |
2016年Oestreich课题组[51]将研究转向磷酸导向的苯胺衍生物的C(sp2)—H烯基化反应(Eq. 44).以对甲苯磺酸(PSTA)作为添加剂, 氧化性更强的K2S2O8作为氧化剂, 实现了C(sp2)—H烯基化反应.该反应在含有甲基和甲氧基的底物中顺利转化.但间位二取代甲基底物仅有痕量产物.此外, 对于丙烯酸酯类、苯乙烯及其衍生物、α-甲基苯乙烯类烯基均以中等产率转化为烯基化产物(39%~80%).此外, 作者将该导向基连接在二氢吲哚上, 并实现了C-7位选择性的C(sp2)—H烯基化.
|
|
(44) |
2013年Miura课题组[52a]发现了Rh催化的磷氧化物导向的邻位烯基化反应(Eq. 45a).与此同时, 该反应体系还能用于与炔基的氧化偶联, 从而选择性地生成磷异香豆素衍生物.同年, 该课题组[52b]发现Ru催化体系能实现与炔基加成生成烯基化产物(Eq. 45b).该反应体系能很好地兼容一、二和三芳基磷氧化物和二苯乙炔、二噻吩乙炔、非对称烷基(苯基)乙炔类底物.
|
|
2013年刘刚课题组[53a]报道了Rh催化以磺酸基团作为导向的邻位C(sp2)—H烯基化反应(Eq. 46a).该反应体系具有良好的反应性(34%~95%)和官能团(芳基、甲氧基、酯基、醛基、硝基)耐受性.除此之外, 该反应体系能够在较宽的烯烃范围(苯乙烯类烯烃、丙烯酸酯类烯烃、烷基烯烃、丙烯腈等), 成功且选择性地实现芳烃的烯基化反应.次年, Ackermann课题组[53b]研究转向了催化剂Ru, 成功实现了Ru催化的磺酸导向高区域选择性的邻位烯基化反应(Eq. 46b).该反应体系适用于多种烯基, 包括苯乙烯类、乙烯基砜类、丙烯腈类和乙烯基酮类等.此外, 各种的磺酸、磺酰氯和磺酰胺作为底物也能顺利生成对应的烯基化产物.
|
|
综上所述, 基于弱配位的含氧官能团的C(sp2)—H烯基化反应已经取得一定的成果.此外, 由于氨基酸类配体相较于其它配体价格低廉, 反应活性更高, 同时也能够增加区域选择性, 所以基于单保护氨基酸(MPAA)策略, 具有含氧官能团芳烃的弱配位碳氢官能团化反应, 受到众多化学家们的青睐.具有含氧官能团的芳烃的C(sp2)—H烯基化反应也趋向于天然底物, 以及无需预活化天然导向基的C(sp2)—H烯基化反应.与此同时, 由于芳环间位的反应活性较低, 目前实现其间位的碳氢活化仍然需要额外安装U型导向基.然而, 具有含氧官能团远程弱配位导向基的芳烃间位/对位区域选择性的C(sp2)—H烯基化的反应研究较少.因此, 此类反应的发展方向趋于发展多样性、无需预先活化的天然导向基、多位点(邻/间/对)、高区域选择性的C(sp2)—H烯基化反应.
(a) Wang, S.; Yan, F.; Wang, L.-S.; Zhu, L. Chin. J. Org. Chem. 2018, 38, 291 (in Chinese).
(汪珊, 严沣, 汪连生, 朱磊, 有机化学, 2018, 38, 291.)
(b) Wu, M.; Huang, X.-P.; Zhang, H.-B.; Li, P.-F. Chin. J. Org. Chem. 2019, 39, 3114 (in Chinese).
(吴梅, 黄新平, 张海兵, 李鹏飞, 有机化学, 2019, 39, 3114.)
(c) Dou, Y.-D.; Gu, X.-X.; Jiang, J.-J.; Zhu, Q. Prog. Chem. 2018, 30, 1317 (in Chinese).
(窦言东, 顾晓旭, 蒋建泽, 朱勍, 化学进展, 2018, 30, 1317.)
(a) Swamy, T.; Reddy, B. V. S.; Gree, R.; Ravinder, V. ChemistrySelect 2018, 3, 47.
(b) Giri, R.; Lan, Y.; Liu, P.; Houk, K. N.; Yu, J.-Q. J. Am. Chem. Soc. 2012, 134, 14118.
(c) Liu, L.; Wang, G.; Jiao, J.; Li, P. Org. Lett. 2017, 19, 6132.
Engle, K. M.; Mei, T.-S.; Wasa, M.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788. doi: 10.1021/ar200185g
Bras, J. L. B.; Muzart, J. Chem. Rev. 2011, 111, 1170. doi: 10.1021/cr100209d
Sarkar, S. D.; Liu, W.; Kozhushkov, S. I.; Ackermann, L. Adv. Synth. Catal. 2014, 356, 1461. doi: 10.1002/adsc.201400110
Kim, J.; Chang, S. Angew. Chem., Int. Ed. 2014, 53, 2203. doi: 10.1002/anie.201310544
(a) Miura, M.; Tsuda, T.; Satoh, T.; Nomura, M. Chem. Lett. 1997, 1103.
(b) Zhang, C.; Ji, J.; Sun, P. J. Org. Chem. 2014, 79, 3200.
(a) Lu, Y.; Wang, D. H.; Engle, K. M.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 5916.
(b) Li, L.; Liu, Q.; Chen, J.; Huang, Y. Synlett 2019, 30, 1366.
Huang, C. H.; Chattopadhyay, B.; Gevorgyan, V. J. Am. Chem. Soc. 2011, 133.
(a) Yamaguchi, M.; Hayashi, A.; Hirama, M. J. Am. Chem. Soc. 1995, 117, 1151.
(b) Yadav, J. S.; Reddy, B. V. S.; Sengupta, S.; Biswas, S. K. Synthesis 2009, 1301.
(c) Rao, V. K.; Shelke, G. M.; Tiwari, R.; Parang, K.; Kumar, A. Org. Lett. 2013, 15, 2190.
(a) Murai, M.; Yamamoto, M.; Takai, K. Org. Lett. 2019, 21, 3441.
(b) Murai, M.; Yamamoto, M.; Takai, K. Chem.-Eur. J. 2019, 25, 15189.
Dou, Y.-D.; Kenry.; Liu, J.; Jiang, J.-Z.; Zhu, Q. Chem.-Eur. J. 2019, 25, 6896. doi: 10.1002/chem.201900530
Li, G.; Leow, D.; Wan, L.; Yu, J.-Q. Angew. Chem., Int. Ed. 2013, 52, 1245. doi: 10.1002/anie.201207770
(a) Ueno, S.; Chatani, N.; Kakiuchi, F. J. Org. Chem. 2007, 72, 3600.
(b) Ueno, S.; Kochi, T.; Chatani, N.; Kakiuchi, F. Org. Lett. 2009, 11, 855.
Padala, K.; Jeganmohan, M. Org. Lett. 2011, 13, 6144. doi: 10.1021/ol202580e
Patureau, F. W.; Besset, T.; Glorius, F. Angew. Chem., Int. Ed. 2011, 50, 1064. doi: 10.1002/anie.201006222
(a) Sk, M. R.; Bera, S. S.; Maji, M. S. Adv. Synth. Catal. 2019, 361, 585.
(b) Sk, M. R.; Maji, M. S. Org. Chem. Front. 2020, 7, 19.
Ali, S.; Huo, J.; Wang, C. Org. Lett. 2019, 21, 6961. doi: 10.1021/acs.orglett.9b02554
(a) Tsuchikama, K.; Kuwata, Y.; Tahara, Y.; Yoshinami, Y.; Shibata, T. Org. Lett. 2007, 9, 3097.
(b) Santhoshkumar, R.; Mannathan, S.; Cheng, C.-H. Org. Lett. 2014, 16, 4208.
Li, G.; Wan, L.; Zhang, G.; Leow, D.; Spangler, J.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 4391. doi: 10.1021/ja5126897
Hu, F.; Szostak, M. Chem. Commun. 2016, 52, 9715. doi: 10.1039/C6CC04537E
Li, C.; Wang, S.-M.; Qin, H.-L. Org. Lett. 2018, 20, 4699. doi: 10.1021/acs.orglett.8b02037
(a) Banjare, S. K.; Nanda, T.; Ravikumar, P. C. Org. Lett. 2019, 21, 8138.
(b) Pradhan, S.; Mishra, M.; De, P. B.; Banerjee, S. Org. Lett. 2020, 22, 1720.
(a) Zhang, C.; Wang, M.; Fan, Z.; Sun, L.-P.; Zhang, A. J. Org. Chem. 2014, 79, 7626.
(b) Yakkala, P. A.; Giri, D.; Chaudhary, B.; Auti, P.; Sharma, S. Org. Chem. Front. 2019, 6, 2441.
(c) Dias, G. G.; Nascimento, T. A.; Almeida, A. A.; Bombaça, A. C. S.; Menna-Barreto, R. F. S.; Jacob, C.; Júnior, S. E. N. S.; Ackermann, L. Eur. J. Org. Chem. 2019, 13, 2344.
Singh, K. S.; Dixneuf, P. H. Organometallics 2012, 31, 7320.
(a) Patureau, F.; Besset, T.; Glorius, F. Angew. Chem., Int. Ed. 2011, 50, 1064.
(b) Park, S. H.; Kim, J. Y.; Chang, S. Org. Lett. 2011, 13, 2372.
Ncube, G.; Huestis, M. P. Organometallics 2019, 38, 76. doi: 10.1021/acs.organomet.8b00327
(a) Boele, M. D. K.; Strijdonck, G. P. F.; Vries, A. H. M.; Kamer, P. C. J.; Vries, J. G.; Leeuwen, P. W. N. M. J. Am. Chem. Soc. 2002, 124, 1587.
(b) Lee, G. T.; Jiang, X.; Prasad, K.; Repic, O.; Blacklock, T. J. Adv. Synth. Catal. 2005, 347, 1921.
(c) Amatore, C.; Cammoun, C.; Jutand, A. Adv. Synth. Catal. 2007, 349, 292.
(d) Lipshutz, B. H.; Nishikata, T. Org. Lett. 2010, 12, 1972.
(e) Liu, X. Z.; Hii, K. K. J. Org. Chem. 2011, 76, 8022.
(f) Schmidt, B.; Elizarov, N. Chem. Commun. 2012, 48, 4350.
(g) Kim, B. S.; Jiang, C.; Lee, D. J.; Youn, S. W. Chem. Asian J. 2010, 5, 2336.
(a) Rauf, W.; Thompson, A. L.; Brown, J. M. Chem. Commun. 2009, 26, 3874.
(b) Wang, L.; Liu, S.; Li, Z.; Yu, Y. Org. Lett. 2011, 13, 6137.
Tamizmani, M.; Gouranga, N.; Jeganmohan, M. ChemistrySelect 2019, 4, 2976. doi: 10.1002/slct.201900522
(a) Hashimoto, Y.; Hirano, T.; Satoh, T.; Kakiuchi, F.; Miura, M. Org. Lett. 2012, 14, 2058.
(b) Hashimoto, Y.; Hirano, K.; Satoh, T.; Kakiuchi, F.; Miura, M. J. Org. Chem. 2013, 78, 638.
(a) Wang, Y.; Li, C.; Li, Y.; Yin, F.; Wang, X.-S. Adv. Synth. Catal. 2013, 355, 1724.
(b) Wang, S.-M.; Li, C.; Leng, J.; Bukhari, S. N. A.; Qin, L.-H. Org. Chem. Front. 2018, 5, 1411.
Li, F.; Shen, C.; Zhang, J.; Wu, L.; Zhuo, X.; Ding, L.; Zhong, G. Adv. Synth. Catal. 2016, 358, 3932. doi: 10.1002/adsc.201600569
Sun, X.; Zhao, W.; Li, B.-J. Chem. Commun. 2020, 56, 1298. doi: 10.1039/C9CC08735D
Jaiswal, Y.; Kumar, Y.; Kumar, A. J. Org. Chem. 2018, 83, 1223. doi: 10.1021/acs.joc.7b02618
(a) Bu, Q; Rogge, T.; Kotek, V.; Ackermann, L. Angew. Chem., Int. Ed. 2018, 57, 765.
(b) Ramesh, V. B.; Muniraj, N.; Prabhu, K. R. Adv. Synth. Catal. 2019, 361, 3683.
Jambu, S.; Sivasakthikumaran, R.; Jeganmohan, M. Org. Lett. 2019, 21, 1320. doi: 10.1021/acs.orglett.8b04140
Zhao, P.; Niu, R.; Wang, F.; Han, K.; Li, X. Org. Lett. 2012, 14, 4166. doi: 10.1021/ol3018352
Jiao, L.-Y.; Oestreich, M. Org. Lett. 2013, 15, 5374. doi: 10.1021/ol402687t
Zhang, L.-Q.; Yang, S.; Huang, X.; You, J.; Song, F. Chem. Commun. 2013, 49, 8830. doi: 10.1039/c3cc44787a
(a) Leitch, J. A.; Wilson, P. B.; McMullin, C. L.; Mahon, M. F.; Bhonoah, Y. I.; Williams, H.; Frost, C. G. ACS Catal. 2016, 6, 5520.
(b) Leitch, J. A.; Cook, H. P.; Bhonoah, Y.; Frost, C. G. J. Org. Chem. 2016, 81, 10081.
(c) Ma, W.; Dong, H.; Wang, D.; Ackermann, L. Adv. Synth. Catal. 2017, 359, 966.
Chen, W.; Li, H.-J.; Li, Q.-Y.; Wu, Y.-C. Org. Biomol. Chem. 2020, 18, 500. doi: 10.1039/C9OB02421B
Miura, M.; Tsuda, T.; Pivasa-Art, S.; Nomura, M. J. Org. Chem. 1998, 63, 5211. doi: 10.1021/jo980584b
(a) Shi, B.-F.; Zhang, Y.-H.; Lam, J. K.; Wang, D.-H.; Yu, J.-Q. J. Am. Chem. Soc. 2010, 132, 460.
(b) Wang, D. H.; Engle, K. M.; Shi, B.-F.; Yu. J.-Q. Science 2010, 327, 315.
(c) Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2010, 49, 6169.
(d) Baxter, R. D.; Sale, D.; Engle, K. M.; Yu, J.-Q.; Blackmond, D. G. J. Am. Chem. Soc. 2012, 134, 4600.
Mochida, S.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2011, 76, 3024. doi: 10.1021/jo200509m
(a) Huang, L.; Biafora, A.; Zhang, G.; Bragoni, V.; Gooẞen, L. Angew. Chem., Int. Ed. 2016, 55, 6933.
(b) Trita, A. S.; Biafora, A.; Pichette-Drapeau, M.; Weber, P.; Gooẞen, L. J. Angew. Chem., Int. Ed. 2018, 57, 14580.
(c) Jambu, S.; Tamizmani, M.; Jeganmohan, M. Org. Lett. 2018, 20, 1982.
(d) Mandal, A.; Mehta, G.; Dana, S.; Baidya, M. Org. Lett. 2019, 21, 5879.
Dai, H.-X.; Li, G.; Zhang, X.-G.; Stepan, A. F.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 7567. doi: 10.1021/ja400659s
Huang, Y.; Pi, C.; Cui, X.; Wu, Y. Adv. Synth. Catal. 2020, 362, 1385. doi: 10.1002/adsc.201901195
(a) Chan, L. Y.; Kim, S.; Ryu, T.; Lee, P. H. Chem. Commun. 2013, 49, 4682.
(b) Meng, X.; Kim, S. Org. Lett. 2013, 15, 1910.
Wang, H.-L.; Hu, R.-B.; Zhang, H.; Zhou, A.-X.; Yang, S.-D. Org. Lett. 2013, 15, 5302. doi: 10.1021/ol402577p
Jiao, L. Y.; Ferreira, A. V.; Oestreich, M. Chem. Asian J. 2016, 11, 367. doi: 10.1002/asia.201500829
(a) Unoh, Y.; Hashimoto, Y.; Takeda, D.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2013, 15, 3258.
(b) Itoh, M.; Hashimoto, Y.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2013, 78, 8098.
(a) Dong, Y.; Liu, G. Chem. Commun. 2013, 49, 8066.
(b) Ma, W.; Mei, R.; Tenti, G.; Ackermann, L. Chem. Eur. J. 2014, 20, 15248.

图 1 含羟基、醚化合物的碳氢活化策略
Figure 1 C—H activation strategy for hydroxyl and ether-containing compounds

图 2 含羰基、醛基、酯基化合物的碳氢活化策略
Figure 2 C—H activation strategy for carbonyl, aldehyde, ester-containing compounds

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:







































 下载:
下载:


