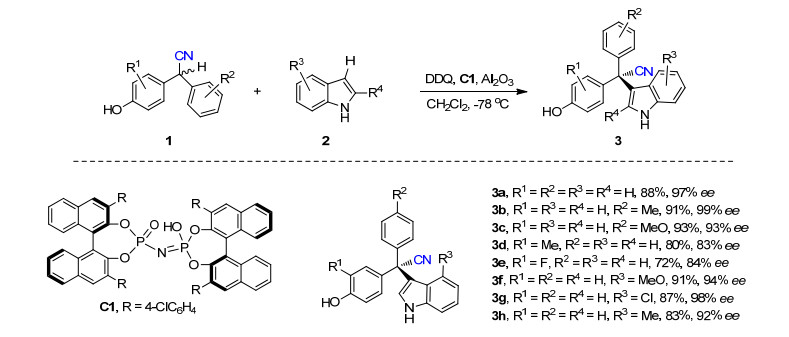
图式 1.
反应最优条件和部分底物实例
Scheme 1.
Optimal reaction conditions and some examples of substrates

三芳基甲烷在药物化学、材料科学和有机合成中都是重要的结构模块[1].由于芳环间缺乏足够的空间差异, 催化不对称合成这些分子是有机合成中一个长期的挑战[2].在这一领域, Sun课题组[3]发展了一种1, 1-二芳基三级醇或含1, 1-二芳基烯烃的杂芳烃对映选择性芳基化的策略, 但该策略需要对底物进行预官能团化.
氰基是常见的官能团, 广泛存在于生物活性天然产物和合成药物中.此外, 氰基可简单转化为其他官能团, 有较好的合成用途[4].对映选择性地构建氰基取代的全碳季碳中心, 现有的策略主要采用α-氰基碳负作为亲核试剂的方法[5].与之相比, 不对称氧化偶联两个不同组分的C—H, 实现全碳季碳中心的形成, 则研究较少.最近, 山东大学化学与化工学院刘磊课题组[6]报道了外消旋2, 2-二芳基乙腈和一系列富电子的(杂)芳烃的不对称氧化偶联, 可高效构建三芳基氰基取代的手性全碳季碳中心结构.同时, 该三芳基氰基甲烷可作为平台分子, 通过氰基转化合成相应的手性三芳基甲烷衍生物.
首先, 作者以不对称偶联外消旋的2, 2-二芳基乙腈1和吲哚2生成3a作为模型反应, 优化了反应条件.在2, 3-二氰基-5, 6-二氯苯醌(DDQ)作氧化剂, 碱性Al2O3作添加剂, 二氯甲烷为反应介质, C2-对称的亚胺二磷酸为催化剂, -78 ℃的条件下获得最高收率和对映选择性(Scheme 1).
随后, 作者考察了该方法的底物扩展性.其中, 芳基含不同取代基的2, 2-二芳基乙腈(3a~3e)获得72%~93%的收率以及99% ee.此外, 吲哚不同位置[C(4), C(5), C(6)及C(7)]带吸电子或供电子基团均能顺利地和1a氧化偶联(3f~3h), 也适合C(2)位取代的吲哚底物.此外, 该方法也适合其它(杂)芳烃底物, 如1-萘酚、苯酚以及2-取代的吡咯.除了2, 2-二芳基乙腈, 2, 2-二芳基乙酸酯也能用此方法与吲哚偶联, 生成三芳基取代的羧酸酯衍生物.
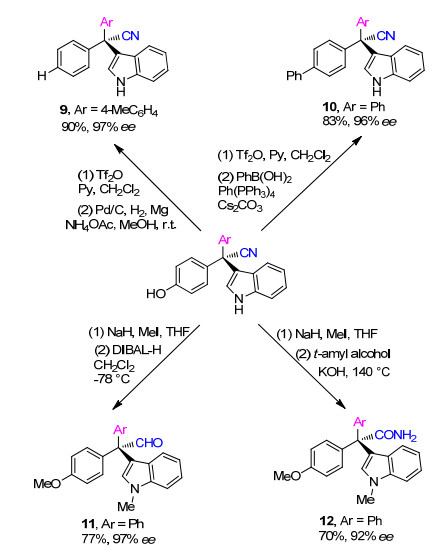
在此基础上, 作者对该方法的合成应用进行了考察(Scheme 2).酚羟基可通过三氟甲磺酸酐(Tf2O)活化, 再用Pd催化加氢除去(9); 此外, 3a在Tf2O除去酚羟基后可进一步在Pd催化下与PhB(OH)2发生偶联反应(10).氰基也可以转化成其他有价值的官能团.比如氰基可被DIBAL-H还原成醛(11).另外, 氰基还能用叔戊醇在碱性条件下水解生成相应的胺(12).在这些转化中, 没有观察到明显的对映异构纯度下降, 证明相应的方法在进一步转化为手性全碳季碳三芳基甲烷衍生物的可靠性.
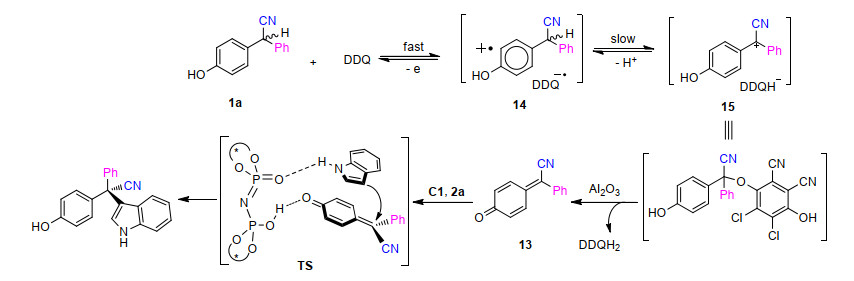
最后, 作者通过一系列控制实验对反应机理进行了考察(Scheme 3).外消旋的2, 2-二芳基乙腈1a用DDQ氧化, 得到90%产率的氰基-δ-苯基取代的p-醌甲基化物(p-QM, 13)和2, 3-二氯-5, 6-二氰基氢醌(DDQH2). 13在不加氧化剂DDQ的标准反应条件下也能得到3a, 并且ee值可与单次操作过程中的相当, 从而表明13是反应涉及的中间体(Scheme 3).然后作者分步进行不对称氧化偶联, 以了解这个反应的细节.首先, 在1a中只加氧化剂DDQ, 通过薄层色谱(TLC)监控发现底物氧化完全; 再将催化剂和2a加入反应混合物中, 3a的分离收率仅有6%, 而以86%的收率回收到了1a, 表明此氧化过程是可逆的. 13和DDQH2不反应, 因此15在碱性氧化铝中生成13是不可逆的.甲基保护的2, 2-二芳基乙腈底物不发生氧化偶联反应, 表明2, 2-二芳基乙腈底物中羟基部分在产生p-QM中间体中起重要作用.基于以上结果, 作者提出了一个合理的反应机理. 1a以可逆方式被DDQ氧化得到中间体15, 该中间体在反应体系中可能是稳定的. Al2O3可能会促进15的不可逆转化, 生成的DDQH2和p-QM 13一起参与随后的对映选择性1, 6-共轭加成, 该加成反应不受DDQH2的影响, 但这种新奇的加成作用方式的起因尚不清楚[7]. N-甲基保护的吲哚比2a的偶联活性差, 因此底物NH基团也很重要, 可能是作为氢键供体.基于以上分析, 作者提出了一个可能的过渡态, 这个过渡态中手性磷酸扮演双功能角色:活化两个耦合组分以及通过氢键控制远端立体中心的功能.
综上所述, 刘磊课题组发展了一种外消旋2, 2-二芳基乙腈与(杂)芳烃对映选择性氧化偶联的方法, 可以高效地构建具有全碳季碳立体中心的三芳基甲烷, 并具有出色的化学和对映选择性.该方法具有出色的官能团耐受性, 并且对于2, 2-二芳基乙腈和(杂)芳烃组分均具有广阔的应用范围.氰基的化学性质丰富, 可进一步方便地合成带有全碳季碳中心的手性三芳基甲烷衍生物.
Shchepinov, M. S.; Korshun, V. A. Chem. Rev. 2003, 32, 170.
Ellman, J.-A.; Owens, T.-D.; Tang, T.-P. Acc. Chem. Res. 2002, 35, 984. doi: 10.1021/ar020066u
Zhao, W.; Wang, Z.; Chu, B.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 1910. doi: 10.1002/anie.201405252
Kukushkin, V.-Y.; Pombeiro, A.-J.-L. Chem. Rev. 2002, 102, 1771. doi: 10.1021/cr0103266
Zheng, J.; Lin, L.; Dai, L.; Tang, Q.; Liu, X.; Feng, X. Angew. Chem., Int. Ed. 2017, 56, 13107. doi: 10.1002/anie.201705943
Wang, Z.; Zhu, Y.; Pan, X.; Wang, G.; Liu, L. Angew. Chem., Int. Ed. 2020, 59, 3053. doi: 10.1002/anie.201912739
Chu, W.-D.; Zhang, L.-F.; Bao, X.; Zhao, X.-H.; Zeng, C.; Du, J.-Y.; Zhang, G.-B.; Wang, F.-X.; Ma, X.-Y.; Fan, C.-A. Angew. Chem., Int. Ed. 2013, 52, 9229. doi: 10.1002/anie.201303928

图式 1 反应最优条件和部分底物实例
Scheme 1 Optimal reaction conditions and some examples of substrates

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

