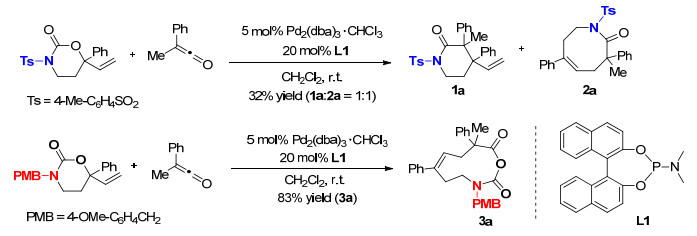
图式 1.
氮原子的保护基对[8+2]偶极环加成反应的影响
Scheme 1.
Effect of nitrogen atom protection on [8+2] dipolar cycloadditions

中环骨架广泛存在于许多天然产物与药物分子中.自从1960年Doering与Wiley[1]报道了利用[8+2]高阶环加成反应捕获庚富烯的工作后, 这一领域引起了化学家们的广泛关注.近年来, 许多[8+2]环加成反应被报道.但为了降低反应的复杂性和熵垒, 目前这些反应通常限制于构型固定的共轭8π组分, 并且, 大多只能得到包含十元环的并环化合物[2].良好的化学选择性以及对映选择性控制仍是该领域的一个巨大挑战.
2013年, 冯小明课题组[3]通过路易斯酸催化氮杂庚富烯与亚烷基丙二酸酯的不对称[8+2]环加成反应, 高效、高立体选择性地构建了具有环庚三烯结构单元的吡咯衍生物. 2017年, J rgensen小组[4]利用有机催化的方法实现了缺电子的庚富烯与环烯酮的不对称[8+2]环加成反应.尽管这一领域已经取得了重要的进展, 但是, 通过发展新的概念、新的催化体系, 高效、高立体选择性地构建十元单环骨架仍然是十分重要的.
近年来, 基于钯催化烯丙基烷基化的偶极环加成反应已经成为构建碳环与杂环化合物的有力工具[5].在这一领域, 乙烯基氨基甲酸酯通过与零价钯的氧化加成, 随后释放二氧化碳, 被广泛用作钯稳定偶极体的前体[6].基于对过渡金属催化的偶极环加成反应的认识, 肖文精、陆良秋课题组和蓝宇课题组考虑是否可以通过实现二氧化碳基团的保留, 进而为高阶偶极环加成反应提供可能.
近日, 该研究团队[7]结合钯催化与光活化, 发展了原位产生的1, 8-偶极子与烯酮之间的不对称偶极环加成反应, 实现了十元单环产物的高效、高对映选择性构建(Scheme 1).研究发现, 乙烯基氨基甲酸酯氮原子上的保护基对反应有重要的影响.当使用对甲苯磺酰基(Ts)作为取代基时, 只能产生六元环与八元环混合产物; 而当使用对甲氧基苄基(PMB)作为取代基时, 在相同条件下可以得到目标十元环化合物.随后, 研究人员进一步从不对称催化的角度对该反应进行了探究.通过一系列研究发现, 使用该课题组自主研发的手性P, S配体时, 该反应可以实现99%的产率以及96:4的对映体比例.最终, 他们得出结论, 手性联萘酚(BINOL)与手性二苯乙基两种手性骨架的匹配是实现高对映选择性的关键.随后, 在6W LED蓝光灯照射下, 使用α-重氮酮替代烯酮, 在相同反应条件下可以以49%的核磁收率以及96:4的对映体比例得到目标产物(Scheme 2).通过进一步调整反应操作, 可以以99%的核磁收率得到产物, 同时对映选择性得到保持.
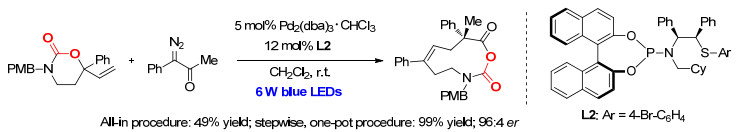
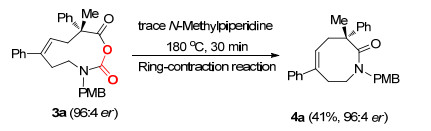
该方法的底物适用性广、官能团兼容性强, 对于不同的底物都可以实现较高的产率及对映体选择性.同时, 克级反应可以以81%的产率及96:4的对映体比例得到产物.此外, 在180 ℃与无溶剂的条件下, 使用微量的N-甲基哌啶处理产物, 可以将十元环产物转换为八元环产物, 产率适中, 并且实现了光学纯度的保持(Scheme 3)[8].
蓝宇等[7]的密度泛函理论计算表明, 钯对于底物乙烯基氨基甲酸酯的氧化加成过程是反应的决速步骤.同时, 通过分析反应的活化能差异发现, 当使用对甲氧基苄基作为取代基时, 底物通过释放二氧化碳产生两性离子中间体的过程相比于直接对烯酮的亲核加成过程更困难.当使用对甲基磺酰基作为取代基时, 却得到了相反的趋势, 对键级与键长的进一步比较也印证了这一结论.当使用对甲氧基苄基作为取代基时, 通过破坏碳-氮键来释放二氧化碳变得更加困难.在分子内环化一步, 通过线性选择性的烯丙基取代产生十元环产物相比于通过支链选择性过程产生八元环产物, 具有更低的活化能, 因此更容易形成.这些计算结果与实验观察一致.陆良秋团队首次报道了钯催化的乙烯基氨基甲酸酯与光催化生成的烯酮的不对称[8+2]高阶偶极环加成反应.该研究为构建含有手性季碳中心的十元化合物提供了一种条件温和、立体选择性高的新方法, 同时也为构建中型单环体系提供了新的思路.
von E. Doering, W.; Wiley, D. W. Tetrahedron 1960, 11, 183. doi: 10.1016/0040-4020(60)80069-5
Chen, Y. Y.; Ye, S. Y.; Jiao, L.; Liang, Y.; Sinha-Mahapatra, D. K.; Herndon, J. W.; Yu, Z. X. J. Am. Chem. Soc. 2007, 129, 10773. doi: 10.1021/ja072203u
Xie, M. S.; Liu, X. H.; Wu, X. X.; Cai, Y. F.; Lin, L. L.; Feng, X. M. Angew. Chem., Int. Ed. 2013, 52, 5604. doi: 10.1002/anie.201209601
Mose, R.; Preegel, G.; Larsen, J.; Jakobsen, S.; Iversen, E. H.; J rgensen, K. A. Nat. Chem. 2017, 9, 487. doi: 10.1038/nchem.2682
Butt, N. A.; Zhang, W. B. Chem. Soc. Rev. 2015, 44, 7929. doi: 10.1039/C5CS00144G
Leth, L. A.; Glaus, F.; Meazza, M.; Fu, L.; Th gersen, M. K.; Bitsch, E. A.; J rgensen, K. A. Angew. Chem., Int. Ed. 2016, 55, 15272. doi: 10.1002/anie.201607788
Zhang, Q. L.; Xiong, Q.; Li, M. M.; Xiong, W.; Shi. B.; Lan, Y.; Lu, L. Q.; Xiao, W. J. Angew. Chem., Int. Ed. 2020, 59, 14096. doi: 10.1002/anie.202005313
Dean, C. S.; Tarbell, D. S. J. Org. Chem. 1971, 36, 1180. doi: 10.1021/jo00808a003

图式 1 氮原子的保护基对[8+2]偶极环加成反应的影响
Scheme 1 Effect of nitrogen atom protection on [8+2] dipolar cycloadditions

图式 2 钯催化的不对称的[8+2]偶极环加成反应
Scheme 2 Palladium-catalyzed asymmetric [8+2] dipolar cycloadditions

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
