Table 1.
Optimization on reaction conditionsa
Citation:
Wang Guodong, Guo Yanhui, Wan Jieping. Base-Promoted, Metal- and Oxidant-Free C=C Bond Cleavage in Enaminones for Ambient Synthesis of NH2-Amidines[J]. Chinese Journal of Organic Chemistry,
2020, 40(3): 645-650.
doi:
10.6023/cjoc201912018

无金属和氧化剂温和条件下碱促进的烯胺酮碳-碳双键断裂合成NH2-结构脒类化合物
English
Base-Promoted, Metal- and Oxidant-Free C=C Bond Cleavage in Enaminones for Ambient Synthesis of NH2-Amidines
Abstract:
The C=C double bond cleavage of NH2-functionalized enaminones has been realized at room temperature to provide various N-sulfonyl amidines by reacting with sulfonyl azides. The reactions take place with good substrate tolerance in the presence of 1, 8-Diazabicyclo[5.4.0]undec-7-ene (DBU) without any metal or oxidant reagent. The 15N-labelling experiment on enaminone indicates that the sulfonyl azide component donates solely the sulfonamide fragment, and the reaction mechanism involving a key decomposition of the in situ generated 1, 2, 3-triazoline intermediate is convictively supported.
-
Key words:
- Enaminone
- / C=C bond cleavage
- / Metal-free
- / Oxidant-free
- / Amidines
-
1. Introduction
Effectively scissoring carbon-carbon bond represents one of the most important tools in current organic synthesis because of the ubiquitous presence of carbon-carbon bond in both saturated and unsaturated forms in nature.[1] The past decades have witnessed spectacular advances in synthetic organic reactions based on the key functionalization or activation of carbon-carbon bonds. Among the numerous known donors of reactive carbon-carbon bond, [2] the enaminones have been identified as a class of promising substrates in the designation of practical organic reactions by cleaving the C=C double bond in recent years.[3] As typical stable enamine species, the C=C double bonds in enaminones have been found to be capable of decomposing via the formation and reorganization of 1, 2-dioxetane[4]/1, 2-oxathietane intermediate, [5] free radical-based bond cleavage via DMF release, [6] ring opening and reconstruction of cyclopropane intermediate resulting from carbene cycloaddition, [7] and Pd-catalyzed tandem enamine hydrolysis and decarbonylation.[8]
On the other hand, as a typical transformation, the cycloaddition of the in situ formed unstable enamine intermediate and azide has been found as highly important route to cleave the carbon-carbon bond via the decomposition of the resulted triazoline ring, which provides effective accesses to amidines.[9] Accordingly, the employment of stable enamines such as enaminones has also received concerns in the development of synthetic method toward amidine scaffolds. Early in 1963, Fusco and co-workers[10] have observed the formation of N-sulfonyl amidines in the reactions of aryl stabilized N, N-disubstituted α-aminostyrene with tosyl azide. Later efforts in similar reactions, unexceptionally, can be used only for the synthesis of N-sulfonyl amidines featured with N, N-disubstituted substructure because only N, N-disubstituted α-aminostyrenes are the tolerable enamine substrates.[11] Clearly, such synthetic methods suffer from rigid restriction in the synthesis of amidine products with diversified amino functionalization, thus reflects the urgent requirement in developing new synthetic approaches allowing the synthesis of other amino group, such as free NH2-functionalized amidines.
Over past decade, an array of different tactics have been devised for the synthesis amidines. For instance, the condensation of amines with amide acetals or in situ activated amides, [12] the decomposition of in situ generated 1, 2, 3-triazoline ring given by copper-catalyzed alkyne-azide dipolar cycloaddition, [13] the activation of terminal alkynes, [14] catalytic C=N bond formation from the thioamide C=S bond[15] etc have been disclosed as useful methods. Amazingly, regardless the availability of these important synthetic routes, [16] most of them are limited in the synthesis of N, N-disubstituted or N-substituted amidines. Only rather a few transition-metal-catalyzed protocols, on the contrary, can be used for the synthesis of amidines bearing free NH2-group.[9c, 17] The synthesis of NH2-free amidines under transition-metal-free conditions, however, has not yet been realized. In combination of the urgent requirement to find transition-metal-free NH2-amidine synthesis and the highly versatile application of enaminone platform synthons in the synthesis of diversified organic products, [18] we thought that NH2-functionalized enaminones might be used as proper precursors to achieve such expected synthesis. Herein, we report the first metal-free method for the synthesis of NH2-featured N-sulfonyl amidines via the reactions of NH2-functionalized enaminones and sulfonyl azides, which significantly expands the frontiers of enaminones in the synthesis of structurally diverse amidines and sulfonylated scaffolds.[19]
2. Results and discussion
Originally, the readily available enaminone 1a and tosyl azide 2a were selected as substrates to probe practical reaction conditions. As presented in Table 1, by stirring in DMF at room temperature, the reaction of the two substrates in the presence of t-BuONa gave the target amidine product 3a with 21% yield, and 3a was not observed without using any additive (Entries 1, 2, Table 1). Following these results, a series of different base additives, including organic and inorganic ones, were then screened. Based on the in hand data, 1, 8-diazabicyclo[5.4.0]undec-7-ene (DBU) was identified as amongst the best additive by assisting the formation of 3a with 46% yield (Entries 3~7, Table 1). Heightening the reaction temperature led to inferior result (Entry 8, Table 1). Later on, this reaction was conducted independently in different media such as water, MeCN, toluene, EtOH, dimethyl sulfoxide (DMSO) and tetrahydrofuran (THF) (Entries 9~14, Table 1). However, DMF was yet the best medium. In further study, prolonging the reaction time to 12 h was found to improve the yield of the target product (Entries 15~16, Table 1). On the other hand, increasing the amount of tosyl azide 2a could also help in enhancing the product yield (Entries 17, 18, Table 1). Finally, when the loading of DBU was increased to 5 equiv., the target product was obtained with 91% yield (Entries 19, 20, Table 1).
Table 1

 下载:
导出CSV
下载:
导出CSV

Entry Additive Solvent Yieldb/% 1 t-BuONa DMF 21 2 — DMF 0 3 NaHCO3 DMF 20 4 NaOH DMF 19 5 DBU DMF 46 6 DMAP DMF 17 7 2, 6-Lutidine DMF 12 8c DBU DMF 26 9 DBU H2O 38 10 DBU MeCN 35 11 DBU Toluene 38 12 DBU EtOH 37 13 DBU DMSO 34 14 DBU THF 28 15d DBU DMF 61 16e DBU DMF 48 17d, f DBU DMF 52 18d, g DBU DMF 67 19d, g, h DBU DMF 81 20d, g, i DBU DMF 91 a General conditions: 1a (0.2 mmol), 2a (0.5 mmol), additive (0.4 mmol) in 2 mL of solvent, stirred at r.t. for 6 h. b Yield of isolated product based on 1a. c Reaction at 60 ℃. d Stirred for 12 h. e Stirred for 24 h. f With 0.3 mmol of 2a. g With 1.0 mmol of 2a. h With 0.8 mmol of DBU. I With 1.0 mmol of DBU. With the optimized reaction conditions, the application scope of this C=C bond cleavage reaction in the synthesis of NH2-functionalized N-sulfonyl amidines was systematically investigated (Table 2). Firstly, the enaminone 1a was employed to react with a plethora of aryl sulfonyl azides, respectively. As expected, the reaction displayed broad tolerance to the aryl substructure. Azides functionalized with phenyl containing electron donating (3a~3b) and withdrawing (3c~3d) group in the para-position, the phenyls with ortho- and meta-substituent (3e~3g) and polysubstituted phenyl (3h), as well as naphthyl/heteroaryl (3i~3j) were all transformed into the target products with good to excellent yields. On the other hand, enaminones 1 with alkyl (3k~3n), alkoxyl (3o~3p), halogen (3q) substituted β-phenyl also participated the reaction to provide the products with satisfactory results. Furthermore, equivalent enaminones featured with heteroaryl (3r) and fuse aryl (3s) were also practical substrates. Finally, employing methyl sulfonyl azide to react with enaminone 1a did not provide the expected amidine product under the standard conditions.
Table 2
Encouraged by the successful synthesis of the diverse amidine products, the gram scale reaction for the synthesis of 3a was then performed. We were delighted to find that authentically practical result was provided by the scale-up synthesis with up to 78% yield of 3a (Eq. 1).

(1) Later on, for the sake of probing the reaction mechanism, several control experiments were then designed and conducted. First, the model reaction for 3a synthesis was conducted in the presence of free radical scavenger 2, 2, 6, 6-Tetramethylpiperidinooxy (TEMPO) and 2, 6-di-t-butyl-p-cresol (BHT), respectively. Both scavengers, however, exhibited no inhibition effect to the reaction even at the loading of 4 equiv. (Eqs. 2, 3), suggesting against the formation of any free radical intermediate in the reaction. In addition, the reaction of 15N-labelled enaminone and tosyl azide 2a gave 15N-labelled product 15N-3a, confirming that the amino group in the enaminone substrate was retained during the reaction process (Eq. 4).

(2) 
(3) 
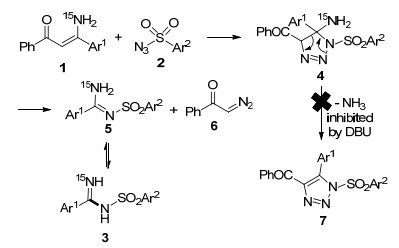
(4) On the basis of the results obtained in hand, a plausible mechanism for the reaction is proposed. As outlined in Scheme 1, initially, the enaminone and sulfonyl azide couples provide 1, 2, 3-triazoline intermediate 4 via the well documented dipolar cycloaddition. The possible transformation of this intermediate to 1, 2, 3-triazole 7 by eliminating ammonium can be inhibited by the presence of DBU. Instead, the other transformation route of ring decomposition providing amidine 5 and the diazoketone 6 is favored under the present reaction conditions. The tautomerization of 5 then yields the final amidine products 3.
Scheme 1
3. Conclusions
In conclusion, by employing NH2-functionalized enaminone esters as the C=C bond donors, we have successfully established the first metal-free protocol toward the synthesis of free NH2-featured N-sulfonyl amidines by cleaving C=C double bond. Besides the metal-free operation, the ambient reaction conditions as well as the practical scale-up synthesis constitute also notable advantages of this work.
4. Experimental section
4.1 General experimental information
All experiments were carried out under air atmosphere. Enaminones 1[20] and tosyl azides 2 (except the commercially available 2a)[21] were synthesized following literature processes. All other chemicals and solvents used in the experiments were obtained from commercial sources and used directly without further treatment. 1H NMR and 13C NMR spectra were recorded in 400 MHz apparatus and the frequencies for 1H NMR and 13C NMR test are 400 MHz and 100 MHz, respectively. The chemical shifts were reported with TMS as internal standard. Melting points were tested in an X-4A instrument without correcting temperature and the HRMS were obtained under ESI model in a mass spectrometer equipped with TOF analyzer.
4.2 General procedure for the synthesis of amidines 3
To a 25 mL round-bottom flask were added enaminone 1 (0.2 mmol), sulfonyl azide 2 (1.0 mmol), DBU (1.0 mmol) and DMF (2.0 mL). Then the mixture was stirred at the room temperature for 12 h (TLC). Upon completion, 5 mL of water was added, and the resulting mixture was extracted with ethyl acetate (8 mL×3). The organic phases were combined and washed with small amount of water for three times. After drying with anhydrous Na2SO4, the solid was filtered and the solvent in the acquired solution was removed under reduced pressure. The resulting residue was subjected to flash silica gel column chromatography to provide pure products with the elution of mixed petroleum ether/ethyl acetate (V:V=5:1 or 3:1).
N-Tosylbenzimidamide (3a):[22] 49.8 mg, 91% yield. White solid, m.p. 153~155 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.30 (s, 1H), 7.86 (d, J=8.4 Hz, 2H), 7.78 (d, J=7.6 Hz, 2H), 7.49 (t, J=7.4 Hz, 1H), 7.37 (t, J=7.8 Hz, 2H), 7.26 (d, J=8.0 Hz, 2H), 6.68 (s, 1H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.8, 143.0, 139.3, 133.3, 132.7, 129.4, 128.7, 127.4, 126.4, 21.5.
N-(4-Methoxyphenylsulfonyl)benzimidamide (3b): 49.0 mg, 84% yield. White solid, m.p. 109~111 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.26 (s, 1H), 7.90 (d, J=8.8 Hz, 2H), 7.78 (d, J=7.6 Hz, 2H), 7.48 (t, J=7.4 Hz, 1H), 7.36 (t, J=7.4 Hz, 2H), 6.92 (d, J=8.8 Hz, 2H), 6.88 (s, 1H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.7, 134.1, 133.3, 132.7, 128.7, 128.5, 128.5, 127.5, 114.0, 55.6; ESI-HRMS calcd for C14H15N2O3S [M+H]+ 291.0798, found 291.0799.
N-(4-Chlorophenylsulfonyl)benzimidamide (3c): 46.1 mg, 78% yield. White solid, m.p. 98~100 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.29 (s, 1H), 7.90 (d, J=8.8 Hz, 2H), 7.78 (d, J=7.6 Hz, 2H), 7.51 (t, J=7.4 Hz, 1H), 7.46~7.35 (m, 4 H), 6.80 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 163.2, 140.6, 138.7, 133.0, 132.9, 129.1, 128.8, 127.9, 127.5; ESI-HRMS calcd for C13H12ClN2O2S [M+H]+ 295.0303, found 295.0304.
N-(4-Bromophenylsulfonyl)benzimidamide (3d): 49.9 mg, 74% yield. White solid, m.p. 102~103 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.29 (s, 1H), 7.83 (d, J=8.4 Hz, 2H), 7.77 (d, J=7.2 Hz, 2H), 7.59 (d, J=8.4 Hz, 2H), 7.52 (t, J=7.4 Hz, 1H), 7.39 (t, J=7.6 Hz, 2H), 6.85 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 163.3, 141.2, 133.0, 132.9, 132.1, 128.8, 128.0, 127.5, 127.2; ESI-HRMS calcd for C13H12BrN2O2S [M+H]+ 338.9797, found 338.9798.
N-(o-Tolylsulfonyl)benzimidamide (3e): 40.0 mg, 73% yield. White solid, m.p. 68~70 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.25 (s, 1H), 8.06 (d, J=8.0 Hz, 1H), 7.79 (d, J=7.2 Hz, 2H), 7.50 (t, J=7.4 Hz, 1H), 7.45~7.35 (m, 3H), 7.29 (d, J=7.2 Hz, 2H), 6.63 (s, 1H), 2.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.8, 140.2, 137.6, 133.3, 132.7, 132.4, 132.3, 128.8, 127.7, 127.4, 125.8, 20.4; ESI-HRMS calcd for C14H15N2O2S [M+H]+ 275.0849, found 275.0852.
N-(2-Chlorophenylsulfonyl)benzimidamide (3f): 38.4 mg, 65% yield. White solid, m.p. 91~92 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.43 (s, 1H), 8.20 (dd, J=7.8, 1.4 Hz, 1H), 7.79 (d, J=7.2 Hz, 2H), 7.56~7.43 (m, 3H), 7.39 (d, J=7.6 Hz, 3H), 6.73 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 164.1, 139.4, 133.3, 133.2, 132.8, 132.6, 131.6, 129.7, 128.8, 127.5, 126.9; ESI-HRMS calcd for C13H12Cl-N2O2S [M+H]+ 295.0303, found 295.0305.
N-(3-Chlorophenylsulfonyl)benzimidamide (3g): 41.3 mg, 70% yield. White solid, m.p. 79~80 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.33 (s, 1H), 7.97 (s, 1H), 7.88 (d, J=7.2 Hz, 1H), 7.80 (d, J=7.2 Hz, 2H), 7.60~7.47 (m, 2H), 7.43 (t, J=7.4 Hz, 3H), 6.64 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 163.3, 143.8, 134.9, 133.1, 132.9, 132.5, 130.2, 128.9, 127.5, 126.6, 124.6; ESI-HRMS calcd for C13H12ClN2O2S [M+H]+ 295.0303, found 295.0302.
N-(Mesitylsulfonyl)benzimidamide (3h): 49.3 mg, 82% yield. White solid, m.p. 90~92 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.17 (s, 1H), 7.76 (d, J=7.6 Hz, 2H), 7.49 (t, J=7.4 Hz, 1H), 7.39 (t, J=7.6 Hz, 2H), 6.92 (s, 2H), 6.40 (s, 1H), 2.71 (s, 6 H), 2.28 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 161.8, 141.6, 138.7, 136.4, 133.6, 132.5, 131.6, 128.8, 127.3, 22.7, 20.9; ESI-HRMS calcd for C16H19N2-O2S [M+H]+ 303.1162, found 303.1166.
N-(Naphthalen-2-ylsulfonyl)benzimidamide (3i). 49.7 mg, 80% yield. White solid, m.p. 103~105 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.52 (s, 1H), 8.38 (s, 1H), 7.97 (d, J=8.4 Hz, 1H), 7.90 (t, J=8.4 Hz, 2H), 7.85 (d, J=8.0 Hz, 1H), 7.68 (d, J=8.0 Hz, 2H), 7.61~7.51 (m, 2H), 7.13 (d, J=8.0 Hz, 2H), 6.74 (s, 1H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.1, 143.7, 139.1, 134.7, 132.1, 130.2, 129.4, 129.3, 129.1, 128.5, 127.8, 127.5, 127.3, 127.1, 122.4, 21.5; ESI-HRMS calcd for C17H15-N2O2S [M+H]+ 311.0849, found 311.0850.
N-(Quinolin-8-ylsulfonyl)benzimidamide (3j): 45.2 mg, 73% yield. White solid, m.p. 107~109 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.18 (s, 1H), 8.98 (s, 1H), 8.77 (s, 1H), 8.55~8.42 (m, 2H), 8.26 (d, J=8.0 Hz, 1H), 7.91~7.71 (m, 3H), 7.70~7.60 (m, 1H), 7.58~7.49 (m, 1H), 7.48~7.38 (m, 2H); 13C NMR (100 MHz, DMSO-d6) δ: 164.0, 151.4, 144.0, 139.6, 137.3, 134.3, 133.7, 132.6, 130.4, 129.0, 128.9, 128.2, 126.1, 122.7; ESI-HRMS calcd for C16H14N3O2S [M+H]+ 312.0801, found 312.0803.
4-Methyl-N-tosylbenzimidamide (3k): 50.7 mg, 88% yield. White solid, m.p. 167~168 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.27 (s, 1H), 7.86 (d, J=8.0 Hz, 2H), 7.68 (d, J=8.0 Hz, 2H), 7.26 (d, J=8.0 Hz, 2H), 7.18 (d, J=8.0 Hz, 2H), 6.53 (s, 1H), 2.39 (s, 3H), 2.36 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.7, 143.5, 142.9, 139.5, 130.4, 129.4, 129.3, 127.4, 126.4, 21.5; ESI-HRMS calcd for C15H17N2O2S [M+H]+ 289.1005, found 289.1008.
N-(4-Chlorophenylsulfonyl)-4-methylbenzimidamide(3l): 46.0 mg, 75% yield. White solid, m.p. 151~152 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.27 (s, 1H), 7.90 (d, J=8.4 Hz, 2H), 7.68 (d, J=8.4 Hz, 2H), 7.43 (d, J=8.8 Hz, 2H), 7.19 (d, J=8.0 Hz, 2H), 6.69 (s, 1H), 2.37 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.2, 143.9, 140.8, 138.6, 130.0, 129.5, 129.0, 127.9, 127.5, 21.5; ESI-HRMS calcd for C14H14ClN2O2S [M+H]+ 309.0459, found 309.0460.
4-Methyl-N-(naphthalen-2-ylsulfonyl)benzimidamide(3m): 50.6 mg, 78% yield. White solid, m.p. 160~162 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.52 (s, 1H), 8.38 (s, 1H), 7.97 (d, J=8.4 Hz, 1H), 7.90 (t, J=8.4 Hz, 2H), 7.85 (d, J=8.0 Hz, 1H), 7.68 (d, J=8.0 Hz, 2H), 7.61~7.49 (m, 2H), 7.13 (d, J=8.0 Hz, 2H), 6.74 (s, 1H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.1, 143.7, 139.1, 134.7, 132.1, 130.2, 129.4, 129.3, 129.1, 128.5, 127.8, 127.5, 127.3, 127.1, 122.4, 21.5; ESI-HRMS calcd for C18H17N2O2S [M+H]+ 325.1005, found 325.1006.
4-Methyl-N-(quinolin-8-ylsulfonyl)benzimidamide (3n): 45.0 mg, 69% yield. White solid, m.p. 164~166 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 8.22 (s, 1H), 8.05 (d, J=2.8 Hz, 1H), 7.84 (s, 1H), 7.62~7.50 (m, 2H), 7.34 (d, J=8.0 Hz, 1H), 6.88~6.78 (m, 3H), 6.73 (q, J=4.0 Hz, 1H), 6.32 (d, J=8.0 Hz, 2H), 1.40 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 163.8, 151.4, 144.0, 143.0, 139.7, 137.2, 133.6, 131.4, 130.3, 129.4, 129.0, 128.3, 126.1, 122.7, 21.4; ESI-HRMS calcd for C17H16N3O2S [M+H]+ 326.0958, found 326.0961.
4-Methoxy-N-tosylbenzimidamide (3o): 48.8 mg, 80% yield. White solid, m.p. 164~166 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.24 (s, 1H), 7.86 (d, J=8.0 Hz, 2H), 7.77 (d, J=8.8 Hz, 2H), 7.26 (d, J=8.0 Hz, 2H), 6.87 (d, J=9.2 Hz, 2H), 6.54 (s, 1H), 3.82 (s, 3H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.4, 162.2, 142.8, 139.6, 129.4, 129.3, 126.4, 125.2, 114.0, 55.5, 21.5; ESI-HRMS calcd for C15H17N2O3S [M+H]+ 305.0954, found 305.0958.
N-(4-Chlorophenylsulfonyl)-4-methoxybenzimidamide(3p): 46.6 mg, 72% yield. White solid, m.p. 72~74 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.23 (s, 1H), 7.90 (d, J=8.4 Hz, 2H), 7.77 (d, J=8.8 Hz, 2H), 7.43 (d, J=8.4 Hz, 2H), 6.87 (d, J=8.8 Hz, 2H), 6.69 (s, 1H), 3.82 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 163.5, 162.7, 141.0, 138.5, 129.5, 129.0, 127.9, 124.8, 114.1, 55.5; ESI-HRMS calcd for C14H14ClN2O3S [M+H]+ 325.0408, found 325.0409.
4-Chloro-N-tosylbenzimidamide (3q): 47.5 mg, 77% yield. White solid, m.p. 108~109 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.30 (s, 1H), 7.85 (d, J=8.4 Hz, 2H), 7.72 (d, J=8.4 Hz, 2H), 7.34 (d, J=8.8 Hz, 2H), 7.28 (d, J=10.0 Hz, 2H), 6.67 (s, 1H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 161.7, 143.3, 139.1, 131.6, 129.7, 129.5, 129.0, 128.9, 126.4, 21.5; ESI-HRMS calcd for C14H14Cl-N2O2S [M+H]+ 309.0459, found 309.0456.
N-Tosylfuran-2-carboximidamide (3r): 38.0 mg, 72% yield. White solid, m.p. 145~146 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.19 (s, 1H), 7.85 (d, J=8.0 Hz, 2H), 7.51 (s, 1H), 7.26 (d, J=8.0 Hz, 2H), 7.22 (d, J=3.2 Hz, 1H), 6.73 (s, 1H), 6.53~6.45 (m, 1H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 153.2, 146.5, 146.0, 143.0, 139.3, 129.4, 126.4, 116.9, 113.0, 21.5; ESI-HRMS calcd for C12H13N2O3S [M+H]+ 265.0641, found 265.0642.
N-Tosyl-2-naphthimidamide (3s): 55.0 mg, 85% yield. White solid, m.p. 181~183 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 9.21 (s, 1H), 8.52 (s, 1H), 8.32 (s, 1H), 8.04 (d, J=7.6 Hz, 1H), 8.02~7.96 (m, 2H), 7.93~7.87 (m, 3H), 7.69~7.57 (m, 2H), 7.38 (d, J=8.0 Hz, 2H), 2.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 163.1, 142.9, 140.2, 135.1, 132.4, 131.1, 129.9, 129.5, 129.2, 128.7, 128.5, 128.1, 127.4, 126.6, 124.6, 21.4; ESI-HRMS calcd for C18H17N2O2S [M+H]+ 325.1005, found 325.1007.
Supporting Information The 1H NMR and 13C NMR spectra of all products, the HRMS spectrum of 15N-3a. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
-
-
[1]
For selected reviews, see:
(a) Kim, D.-S.; Park, W.-J.; Jun, C.-H. Chem. Rev. 2017, 117, 8977.
(b) Souillart, L.; Cramer, N. Chem. Rev. 2015, 115, 9410.
(c) Ruhland, K. Eur. J. Org. Chem. 2012, 2683。
(d) Liang, Y.-F.; Jiao, N. Acc. Chem. Res. 2017, 50, 1640.
(e) Wu, X.; Zhu, C. Chem. Rec. 2018, 18, 587.
(f) Wan, J.-P.; Gao, Y.; Wei, L. Chem. Asian J. 2016, 11, 2092. -
[2]
For selected examples, see:
(a) Roque, J. B.; Kuroda, Y.; Göttemann, L. T.; Sarpong, R. Science 2018, 361, 171.
(b) He, C.; Guo, S.; Huang, L.; Lei, A. J. Am. Chem. Soc. 2010, 132, 8273.
(c) Ren, R.; Wu, Z.; Zhu, C. Chem. Commun. 2016, 52, 8160.
(d) Dieskau, A. P.; Holzwarth, M. S.; Plietker, B. J. Am. Chem. Soc. 2012, 134, 5048.
(e) Souillart, L.; Parker, E.; Cramer, N. Angew. Chem. Int. Ed. 2014, 53, 3001. -
[3]
For selected reviews in enaminone-based synthesis, see:
(a) Fu, L.; Wan, J.-P. Asian J. Org. Chem. 2019, 8, 767.
(b) Wan, J.-P.; Gao, Y. Chem. Rec. 2016, 16, 1164.
(c) Elassar, A.-Z. A.; El-Khair, A. A. Tetrahedron 2003, 59, 8463.
(d) Greenhill, J. V. Chem. Soc. Rev. 1977, 277. -
[4]
(a) Wasserman, H. H.; Ives, J. L. J. Am. Chem. Soc. 1976, 98, 7868.
(b) Cao, S.; Zhong, S.; Xin, L.; Wan, J.-P.; Wen, C. ChemCatChem 2015, 7, 1478.
(c) Yu, Q.; Zhang, Y.; Wan, J.-P. Green Chem. 2019, 21, 3436.
(d) Wan, J.-P.; Lin, Y.; Cao, X.; Liu, Y.; Wei, L. Chem. Commun. 2016, 52, 1270.
(e) Yang, Y.; Zhong, G.; Fan, J.; Liu, Y. Eur. J. Org. Chem. 2019, 4422. -
[5]
Gan, L.; Gao, Y.; Wan, J.-P. J. Org. Chem. 2019, 84, 1064. doi: 10.1021/acs.joc.8b02670
-
[6]
(a) Zhou, P.; Hu, B.; Li, L.; Rao, K.; Yang, J.; Yu, F. J. Org. Chem. 2017, 82, 13268.
(b) Tian, L.; Guo, Y.; Wei, L.; Wan, J.-P.; Shou, S. Asian J. Org. Chem. 2019, 8, 1484.
(c) Liu, Y.; Xiong, J.; Wei, L.; Wan, J.-P. Adv. Synth. Catal. 2020, 362, 877. -
[7]
(a) Ni, M.; Zhang, J.; Liang, X.; Jiang, Y.; Loh, T.-P. Chem. Commun. 2017, 53, 12286.
(b) Chen, J.; Guo, P.; Zhang, J.; Rong, J.; Sun, W.; Jiang, Y.; Luo, T.-P. Angew. Chem. Int. Ed. 2019, 58, 12674. -
[8]
Hu, W.; Zheng, J.; Li, M.; Wu, W.; Liu, H.; Jiang, H. Chin. J. Chem. 2018, 36, 712. doi: 10.1002/cjoc.201800127
-
[9]
(a) Chang, S.; Lee, M.; Jung, D. Y.; Yoo, E. J.; Cho, S. H.; Han, S. K. J. Am. Chem. Soc. 2006, 128, 12366.
(b) Xu, X.; Li, X.; Ma, L.; Ye, N.; Weng, B. J. Am. Chem. Soc. 2008, 130, 14084.
(c) Ding, R.; Chen, H.; Xu, Y.-L.; Tang, H.-T.; Chen, Y.-M.; Pan, Y.-M. Adv. Synth. Catal. 2019, 361, 3656.
(d) Zheng, X.; Wan, J.-P. Adv. Synth. Catal. 2019, 361, 5690. -
[10]
Fusco, R.; Bianchetti, G.; Pocar, D.; Ugo, R. Chem. Ber. 1963, 96, 802. doi: 10.1002/cber.19630960321
-
[11]
(a) Kato, N.; Hamada, Y.; Shioiri, T. Chem. Pharm. Bull. 1984, 32, 2496.
(b) Gao, T.; Zhao, M.; Meng, X.; Li, C.; Chen, B. Synlett 2011, 1281.
(c) Efimov, I.; Beliaev, N.; Beryozkina, T.; Slepukhin, P.; Bakulev, V. Tetrahedron Lett. 2016, 57, 1949.
(d) Bakulev, V. A.; Beryozkina, T.; Thomas, J.; Dehaen, W. Eur. J. Org. Chem. 2018, 262. -
[12]
(a) Chandna, N.; Chandak, N.; Kumar, P.; Kapoor, J. K.; Sharma, P. K. Green Chem. 2013, 15, 2294.
(b) Chen, S.; Xu, Y.; Wan, X. Org. Lett. 2011, 13, 6152.
(c) Yang, W.; Huang, D.; Zeng, X.; Luo, D.; Wang, X.; Hu, Y. Chem. Commun. 2018, 54, 8222.
(d) Chen, J.; Long, W.; Fang, S.; Yang, Y.; Wan, X. Chem. Commun. 2017, 53, 13256. -
[13]
(a) Bae, I.; Han, H.; Chang, S. J. Am. Chem. Soc. 2005, 127, 2038.
(b) Yao, B.; Shen, C.; Liang, Z.; Zhang, Y. J. Org. Chem. 2014, 79, 936.
(c) Murugavel, G.; Punniyamurthy, T. J. Org. Chem. 2015, 80, 6291.
(d) Shang, Y.; He, X.; Hu, J.; Wu, J.; Zhang, M.; Yu, S.; Zhang, Q. Adv. Synth. Catal. 2009, 351, 2709. -
[14]
(a) van Vliet, K. M.; Polak, L. H.; Siegler, M. A.; van der Vlugt, J. I.; Guerra, C. F.; de Bruin, B. J. Am. Chem. Soc. 2019, 141, 15240.
(b) Liu, B.; Ning, Y.; Virelli, M.; Zanoni, G.; Anderson, E. A.; Bi, X. J. Am. Chem. Soc. 2019, 141, 1593. -
[15]
(a) Aswad, M.; Chiba, J.; Tomohiro, T.; Hatanaka, Y. Chem. Commun. 2013, 49, 10242.
(b) Hajibabaei, K.; Boeini, H. Synlett 2014, 2044. -
[16]
For other known methods, see:
(a) Mo, D.-L.; Pecak, W. H.; Zhao, M.; Wink, D. J.; Anderson, L. L. Org. Lett. 2014, 16, 3696.
(b) Zhou, M.; Li, J.; Tian, C.; Sun, X.; Zhu, X.; Cheng, Y.; An, G.; Li, G. J. Org. Chem. 2019, 84, 1015.
(c) Kim, J.; Stahl, S. S. J. Org. Chem. 2015, 80, 2448. -
[17]
Yi, F.; Sun, Q.; Sun, J.; Fu, C.; Yi, W. J. Org. Chem. 2019, 84, 6780. doi: 10.1021/acs.joc.9b00538
-
[18]
(a) Shang, Z.; Chen, Q.; Xing, L.; Zhang, Y.; Wait, L.; Du, Y. Adv. Synth. Catal. 2019, 361, 4926.
(b) Wu, M.; Jiang, Y.; An, Z.; Qi, Z.; Yan, R. Adv. Synth. Catal. 2018, 360, 4236.
(c) Guo, Y.; Xiang, Y.; Wei, L.; Wan, J.-P. Org. Lett. 2018, 20, 3971.
(d) Wan, J.-P.; Cao, S.; Liu, Y. Org. Lett. 2016, 18, 6034.
(e) Cheng, D.; Deng, Z.; Yan, X.; Wang, M.; Xu, X.; Yan, J. Adv. Synth. Catal. 2019, 361, 5025.
(f) Wan, J.-P.; Tu, Z.; Wang, Y. Chem.-Eur. J. 2019, 25, 6907.
(g) Luo, T.; Xu, H.; Liu, Y. ChemistrySelect 2019, 4, 10621.
(h) Yang, L.; Wei, L.; Wan, J.-P. Chem. Commun. 2018, 54, 7475.
(i) Yan, R.; Li, X.; Yang, X.; Kang, X.; Xiang, L.; Huang, G. Chem. Commun. 2015, 51, 2573.
(j) Zhao, Q.; Xiang, H.; Xiao, J.-A.; Xia, P.-J.; Wang, J.-J.; Chen, X.; Yang, H. J. Org. Chem. 2017, 82, 9837.
(k) Bai F.; Hu, D.; Liu, Y.; Wei, L. Chin. J. Org. Chem. 2018, 38, 2054 (in Chinese).
(白飞成, 胡德庆, 刘云云, 韦丽, 有机化学, 2018, 38, 2054.)
(l) Fu, L.; Cao, X.; Wan, J.-P.; Liu, Y. Chin. J. Chem. 2020, 38, 254. -
[19]
(a) Xie, L.-Y.; Fang, T.-G.; Tan, J.-X.; Bang, B.; Cao, Z.; Yang, L.-H.; He, W.-M. Green Chem. 2019, 21, 3858.
(b) Wan, J.-P.; Zhong, S.; Guo, Y.; Wei, L. Eur. J. Org. Chem. 2017, 4401.
(c) Yu, H.; Bao, P.; Wang, L.; Lv, X.; Yang, D.; Wang, H.; Wei, W. Chin. J. Org. Chem. 2019, 39, 463 (in Chinese).
(岳会兰, 鲍鹏丽, 王雷雷, 吕晓霞, 杨道山, 王桦, 魏伟, 有机化学, 2019, 39, 463.)
(d) Du, Y.; Wei, L.; Liu, Y. Heteroatom Chem. 2017, 28, e21401.
(e) Dong, D.; Chen, W.; Chen, D.; Li, L.; Li, G.; Wang, Z.; Deng, Q.; Long, S. Chin. J. Org. Chem. 2019, 39, 3190 (in Chinese).
(董道青, 陈文静, 陈德茂, 李丽霞, 李光辉, 王祖利, 邓企, 龙姝, 有机化学, 2019, 39, 3190.) -
[20]
Zhu, Z.; Tang, X.; Li, J.; Li, X.; Wu, W.; Deng, G.; Jiang, H. Chem. Commun. 2017, 53, 3228. doi: 10.1039/C7CC00260B
-
[21]
Chandna, N.; Kaur, F.; Kumar, S.; Jain, N. Green Chem. 2017, 19, 4268. doi: 10.1039/C7GC01593C
-
[22]
Baeten, M.; Maes, B. U. W. Adv. Synth. Catal. 2016, 358, 826. doi: 10.1002/adsc.201501146
-
[1]
-
Table 1. Optimization on reaction conditionsa
Entry Additive Solvent Yieldb/% 1 t-BuONa DMF 21 2 — DMF 0 3 NaHCO3 DMF 20 4 NaOH DMF 19 5 DBU DMF 46 6 DMAP DMF 17 7 2, 6-Lutidine DMF 12 8c DBU DMF 26 9 DBU H2O 38 10 DBU MeCN 35 11 DBU Toluene 38 12 DBU EtOH 37 13 DBU DMSO 34 14 DBU THF 28 15d DBU DMF 61 16e DBU DMF 48 17d, f DBU DMF 52 18d, g DBU DMF 67 19d, g, h DBU DMF 81 20d, g, i DBU DMF 91 a General conditions: 1a (0.2 mmol), 2a (0.5 mmol), additive (0.4 mmol) in 2 mL of solvent, stirred at r.t. for 6 h. b Yield of isolated product based on 1a. c Reaction at 60 ℃. d Stirred for 12 h. e Stirred for 24 h. f With 0.3 mmol of 2a. g With 1.0 mmol of 2a. h With 0.8 mmol of DBU. I With 1.0 mmol of DBU.  下载: 导出CSV
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 17
- 文章访问数: 1980
- HTML全文浏览量: 362
