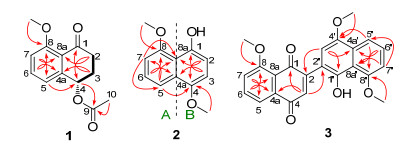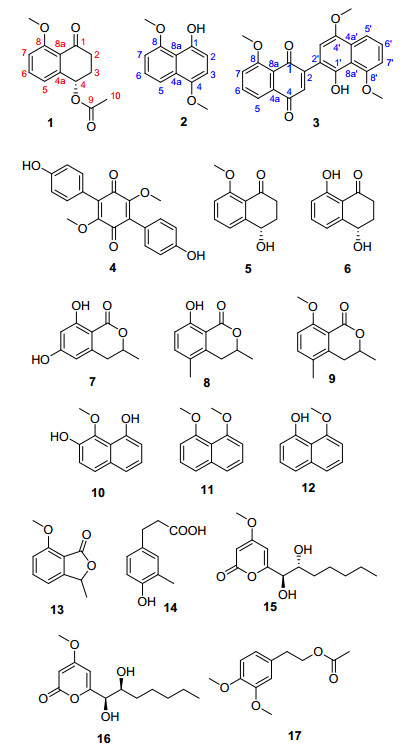
Figure 1.
Structures of compounds 1~17 isolated from Hypoxylon rubiginosum FS521
Cytotoxic Secondary Metabolites from a Sea-Derived Fungal Strain of Hypoxylon rubiginosum FS521
Jie Zhang , Yuchan Chen , Zhaoming Liu , Bohong Guo , Xiaoxia Gao , Hongxin Liu , Weimin Zhang

The genus Hypoxylon (Xylariaceae), which belongs to ascomycetes, contains more than 150 species. Previously, the Hypoxylon species were reported to produce secondary metabolites, such as bezo[j]fluoranthenes and their derivatives, [1, 2] azaphilones, [3] cytochalasins, [4] and various aromatic compounds.[5] Owing to our continuing interests in the discovery and development of new constituents with bioactive potentials from fungal strains, [6~9] a phytochemical study on the EtOAc extract of the fungus Hypoxylon rubiginosum FS521, resulted in the isolation of one new compound named hypoxone A (1) together with two new natural products 4, 8-dimethoxy-1-naphthol (2) and 1'-hydroxy-4', 8, 8'-trimethoxy[2, 2']binaphthalenyl-1, 4- dione (3), and fourteen known compounds, 3, 6-dimethyl- atromentin (4), [10] xylarenone (5), [11] regiolone (6), [12] methylsclerone (7), [13] 5-methylmellein (8), [14] 3, 5-dimethyl-8- methoxy-3, 4-dihydrosiocomarin (9), [15] 9-methoxynaphtha- lene (10), [13] 1, 8-dimethoxy naphthalene (11), [16] 8-methoxy-1-naphthol (12), [17] 7-methoxy-3-methylisoben- zofuran (13), [18] 3-methy-4-hydroxyphey ipropanoid (14), [19] nodulisporipyrone A (15), [20] nodulisporipyrone B (16), [20] 4-[2-[(2, 2-dichloro-l-methylethenyl)oxy]ethyl]-l, 2- dimethoxybenzene (17)[21] (Figure 1). Herein, the details of the isolation, structural elucidation, and cytotoxic activity of these compounds were described.
Compound 1 was isolated as yellow powder. Its molecular formula was determined as C13H14O4 based on the HRESIMS with pseudo-molecular peak at m/z 257.0785 [M+Na]+, implying seven degrees of unsaturation. The 1H NMR spectrum of 1 revealed the presence of an aromatic ring system at [δH 7.03 (d, J=8.0 Hz, 1H, H-5), 7.50 (d, J=8.0 Hz, 1H, H-6), 7.00 (d, J=8.0 Hz, 1H, H-7)], one oxygenated methylene at δH 6.05 (dd, J=3.6, 5.7 Hz, 1H, H-4), one methyl signal resonance at δH 2.10 (s, 3H, H-10), and a methoxy functional group at δH 3.93 (s, 3H, 8-OMe). The 13C NMR combined with HSQC data (Table 1) of 1 exhibited 13 carbon characteristic signals including two methyl, two methylenes, four methines, and five quaternary carbons.
 下载:
导出CSV
下载:
导出CSV
| No. | 1 | 2 | Synthetic 2[22] | |||||
| δH | δC | δH | δC | δH | δC | |||
| 1 | 196.1, C | 147.9, C | 147.9, C | |||||
| 2 | 2.90 (m), 2.64 (m) | 35.6, CH2 | 6.78 (d, 8.3) | 106.3, CH | 6.76 (d, 8.4) | 106.1, CH | ||
| 3 | 2.28 (m) | 27.8, CH2 | 6.78 (d, 8.3) | 106.3, CH | 6.76 (d, 8.4) | 106.1, CH | ||
| 4 | 6.05 (dd, 3.6, 5.7) | 69.8, CH | 148.1, C | 148.1, C | ||||
| 4a | 143.0, C | 127.9, C | 127.8, C | |||||
| 5 | 7.03 (d, 8.0) | 120.3, CH | 7.85 (d, 8.5) | 115.9, CH | 7.85 dd, 8.6, 0.8) | 115.5, CH | ||
| 6 | 7.50 (t, 8.0) | 134.6, CH | 7.34 (t, 8.5) | 125.2, CH | 7.32 (dd, 8.6, 7.7) | 125.2, CH | ||
| 7 | 7.00 (d, 8.0) | 112.6, CH | 6.85 (d, 8.5) | 104.9, CH | 6.84 (dd, 7.7, 0.8) | 104.9, CH | ||
| 8 | 160.0, C | 155.9, C | 155.9, C | |||||
| 8a | 124.4, C | |||||||
| 9 | 170.4, C | |||||||
| 10 | 2.10 (s) | 21.3, CH3 | ||||||
| 4-OMe | 3.93 (s) | 56.0, CH3 | 3.92 (s) | 56.0, CH3 | ||||
| 8-OMe | 3.93 (s) | 56.2, CH3 | 4.06 (s) | 56.1, CH3 | 4.03 (s) | 56.1, CH3 | ||
The 1H-1H COSY spectrum indicated the presence a continued fragment C-2/C-3/C-4. Based on the 1H-1H COSY fragment, the HMBC correlations from H-2 to C-4 (δC 69.8) and C-8a (δC 124.4) coupling with H-3 to C-1 (δC 196.1) and C-4a (δC 143.0) established the presence of a cyclohexanone in the molecule. Moreover, the HMBC correlations from H-2 to C-4 (δC 69.8) and C-8 (δC 160.0), H-3 to C-1 and C-4a (δC 143.0), H-5 to C-4 and C-8, H-7 to C-5 (δC 120.3) and C-8a (δC 124.4), and H-6 to C-4a and C-8 further confirmed the existence of a 1, 2, 3-trisustituted phenyl functionality in 1. Moreover, the methoxy moiety was concluded to be located at the C-8 position by the
HMBC correlation from 8-OMe to C-8. A closer comparison of its 1H NMR and 13C NMR data with those of the co-isolated known compound xylarenone (5) strongly suggested that they should share the same tetralone scaffold attributed to the similarity of their NMR profiles in most cases, [11] whereas the obvious difference between compounds 1 and 5 was the presence of an additional acetyl group in 1. This deduction was further confirmed by the critical HMBC correlation (Figure 2) from H-4 to C-9 (δC 170.4). The configuration of C-4 for compound 1 was tentatively assigned to be S conformation, which was identical to that of 5 on the basis of their similar optical rotation ([α]D20 +22.5 (c 0.1, MeOH) for 1 and [α]D20 +21.3 (c 0.03, CHCl3) for 5).[11] Therefore, the complete structure of compound 1 was successfully established and given a trivial name as hypoxone A.
Compound 2 was obtained as a yellow needle. Its molecular formula was established as C12H12O3 with seven indices of hydrogen deficiency, based on the protonated molecular ion peak [M+H]+ determined at m/z 205.0854 with the aid of the high-resolution electrospray ionization mass spectroscopy (HRESIMS). The 1H NMR spectrum of 2 revealed that a 1, 2, 3-trisubstituent aromatic ring system [δH 7.85 (1H, d, J=8.5 Hz, H-5), 7.34 (1H, t, J=8.5 Hz, H-6), 6.85 (1H, d, J=8.5 Hz, H-7)], a 1, 2, 3, 4-tetrasubsti- tuent benzene ring δH 6.78 (2H, d, J=8.3 Hz, H-2, 3), as well as two methoxy signals at δH 3.93 (3H, s, 4-OMe) and 4.06 (3H, s, 8-OMe). The 13C NMR combined with HSQC data (Table 1) of 2 exhibited 12 carbon signals, including two methyls, five methines, and five quaternary carbons. The complete structure of 2 was determined by the analysis its HMBC spectrum. The HMBC correlations from H-7 to C-5 and C-8a, H-5 to C-7 and C-8a, in conjunction with H-6 to C-4a and C-8 further confirmed the presence of the 1, 2, 3-trisubstituent benzene ring A. Meanwhile, the HMBC correlations from H-2 to C-4 and C-8a, H-3 to C-1 and C-4a indicated the ring B in the molecule. Finally, the HMBC correlations from 4-OMe to C-4 and 8-OMe to C-8 further confirmed that the two methoxys should be located at C-4 and C-8 positions, respectively. Therefore, compound 2 was assigned to be a naphthol derivative as 4, 8- dimethoxy-1-naphthol. Compound 2 was previously reported as a synthetic compound, [22] and it was isolated as a natural product and fully characterized by spectral methodologies including NMR and HRESIMS for the first time in our study.
Compound 3 was obtained as a black powder. Its molecular formula was established as C23H18O6 by the HRESIMS, with fifteen indices of hydrogen deficiency based on the protonated-molecular ion [M+H]+ deter- mined at m/z 391.1179 referring to HRESIMS spectrum. The 1H NMR and HSQC spectra of 1 showed characteristic signals ascribed to two sets of aromatic protons at [δH 7.78 (1H, d, J=7.7 Hz, H-4a), 7.68 (1H, t, J=7.7 Hz, H-5), 7.32 (1H, d, J=7.7 Hz, H-6)] and [δH 7.87 (1H, d, J=8.5 Hz, H-5'), 7.39 (1H, t, J=8.5 Hz, H-6'), 6.87 (1H, d, J=8.5 Hz, H-7')], suggesting the existence of two trisubstituted aryl rings, along with three methoxy functionalities [δH 3.96 (3H, s, 4'-OMe), 4.01 (3H, s, 8-OMe), 4.03 (3H, s, 8'-OMe)]. Analysis of its 13C NMR spectroscopic data further revealed 23 carbon resonances, including twelve quaternary carbons, eight methines, and three methyls. Moreover, the detailed interpretation of their 1H-1H COSY and HMBC spectra disclosed that 3 was also a synthetic compound, which was previously reported by Takeya' group.[23] Compound 3 was also isolated as a natural product and extensively established by spectroscopic strategies involving NMR and HRESIMS for the first time in this study.
Compounds 1~4 were preliminarily evaluated for their in vitro cytotoxicities against SF-268, MCF-7, HepG-2, and A549 cell lines with cisplatin as the positive control. Compound 3 showed significant cytotoxic activity against the tumor cell lines SF-268, MCF-7, HepG-2, and A549 with IC50 values of 1.9, 3.2, 2.5, and 5.0 μM, respectively. Compounds 1, 2, and 4 exhibited weak growth inhibitory activities against the four tumor cell lines with IC50 values in the range of 18.89~69.62 µmol·L-1 (Table 3).
下载:
导出CSV
| No. | 3 | Synthetic 3[22] | |||
| δH | δC | δH | δC | ||
| 1 | 183.3, C | 183.2, C | |||
| 2 | 151.0, C | 150.9, C | |||
| 3 | 7.05 (s) | 134.4, CH | 7.04 (s) | 134.4, C | |
| 4 | 185.5, C | 185.3, C | |||
| 4a | 117.8, C | 117.8, C | |||
| 5 | 7.78 (d, 7.7) | 134.5, CH | 7.76 (d, 7.6) | 134.5, CH | |
| 6 | 7.68 (t, 7.7) | 118.7, CH | 7.66 (t, 7.6) | 118.6, CH | |
| 7 | 7.32 (d, 7.7) | 134.5, CH | 7.30 (d, 7.6) | 134.5, CH | |
| 8 | 159.7, C | 159.6, CH | |||
| 8a | 121.2, C | 121.1, C | |||
| 1' | 146.4, C | 146.3, C | |||
| 2' | 114.9, C | 114.9, C | |||
| 3' | 6.72 (s) | 107.1, CH | 6.72 (s) | 107.1, CH | |
| 4' | 147.9, C | 147.8, C | |||
| 4a' | 129.0, C | 128.9, C | |||
| 5' | 7.87 (d, 8.5) | 116.0, CH | 7.86 (d, 8.5) | 115.9, CH | |
| 6' | 7.39 (t, 8.5) | 126.4, CH | 7.37 (t, 8.5) | 126.3, CH | |
| 7' | 6.87 (d, 8.5) | 105.6, CH | 6.86 (d, 8.5) | 105.6, CH | |
| 8' | 156.5, C | 156.4, C | |||
| 8a' | 115.4, C | 115.3, C | |||
| 8-OMe | 4.01 (s) | 56.6, CH3 | 3.99 (s) | 56.5, CH3 | |
| 4'-OMe | 3.96 (s) | 56.0, CH3 | 3.95 (s) | 56.1, CH3 | |
| 8'-OMe | 4.03 (s) | 56.2, CH3 | 4.00 (s) | 55.9, CH3 | |
下载:
导出CSV
| Compd. | IC50/(µmol•L-1) | |||
| SF-268 | MCF-7 | HepG-2 | A549 | |
| 1 | 43.01 | 35.58 | 22.75 | 27.93 |
| 2 | 33.24 | 65.04 | 69.62 | 66.25 |
| 3 | 1.9 | 3.2 | 2.5 | 5.0 |
| 4 | 31.38 | 35.16 | 18.89 | 25.44 |
| Cisplatin | 3.25 | 3.02 | 2.31 | 2.65 |
UV spectra were measured on a Shimadzu UV-2600 UV-visible spectrophotometer (Shimadzu, Kyoto, Japan). IR spectra were performed on a Shimadzu IR Affintiy-1 spectrometer (Shimadzu, Kyoto, Japan). 1D NMR and 2D NMR spectra were recorded on a Bruker Avance-500 spectrometer (Bruker, Fällanden, Switzerland) with TMS as internal standard. HRESIMS was measured on a Thermo MAT95XP high resolution mass spectrometer and ESIMS on a Thermo DSQ EI mass spectrometer (Thermo Scientific, Massachusetts, USA). All solvents were analytical grade (Guangzhou Chemical Plant, Guangzhou, China). Silica gel (200-300 mesh) was used for column chromatography, and precoated silica gel GF254 plates (Qingdao Marine Chemical Inc., Qingdao, China) were used for TLC spotting. C18 reversed-phase silica gel (40~63 µm, Merck, German) and Sephadex LH-20 gel (Pharmacia Fine Chemical Co. Ltd., Uppsala, Sweden) were also used for column chromatography. Thin-layer chromatography (TLC) spots were visualized under UV light and by dipping into 10% H2SO4 in alcohol followed by heating.
The strain FS521 was isolated from a sediment sample, which was collected at the depth of 4188 m in the South China Sea (115°34'45'' E, 15°58'38'' N), in October 2012. The strain was identified by sequence analysis of rDNA ITS (internal transcribed spacer) region. The sequence of ITS region of the fungal strain FS521 has been submitted to GenBank (Accession No. MN636334). By using BLAST (nucleotide sequence comparison program) to search the GenBank database, FS521 has 98.80% similarity to Hypoxylon rubiginosum STMA10237 (Accession No. KC968933). The strain is preserved at the Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Institute of Microbiology.
The fungal strain H. rubiginosum was grown on PDA medium at 28 ℃ for 4 d, and then inoculated into 20 L×3 Erlenmeyer flasks with rice solid medium (each flask contained 250 g of rice, 300 mL of natural filtered seawater) and incubated at room temperature for 24 d. All fermentation products were extracted three times with EtOAc and then concentrated under reduced pressure to obtain a crude extract (74.8 g). The crude extract was subjected to silica gel using gradient elution with petroleum ether/EtOAc (V:V=100:0→50:50) as eluent to give eight fractions (Fr.1~Fr.8). Fr.2 was chromatographed on silica gel eluting with n-hexane/EtOAc (V:V=10:1→1:2) to afford compound 3 (9.5 mg) and Fr.2-4. Fr.2-4 was further separated by reversed-phase HPLC using MeCN/H2O (V:V=50:50) with 0.05% formic acid at a flow rate of 3.0 mL/min to yield Fr.2-4-1 and Fr.2-4-2, which were purified with silica gel CC eluting with cyclohexane/EtOAc (V:V=30:1, 15:1, 10:1, 8:1, 5:1) to obtain compounds 2 (6.5 mg), 7 (5.0 mg) and 8 (4.6 mg), 11 (4.2 mg). Fr.5 was separated by RP-HPLC using MeOH/H2O (V:V=75:25) to obtain compounds 10 (3.0 mg) and 17 (3.5 mg). Fr.6 was fractioned by silica gel CC eluting with cyclohexane/EtOAc (V:V=5:1, 3:1, 2:1, 1:1, 1:2) to give two fractions Fr.6-1 and Fr.6-2. Subfraction Fr.6-2 was separated by RP-HPLC to give Fr.6-2-1 and Fr.6-2-2. Fr.6-2-2 was further purified by RP-HPLC with MeOH-H2O (V:V=80:20) to yield compounds 1 (6.5 mg), 6 (3.5 mg), and 9 (6.0 mg). Fr.6-2-1 was further purified by RP-HPLC with MeOH/ H2O (V:V=70:30) to give Fr.6-2-1-1, Fr.6.2-1-2, and Fr.6-2-1-5. All of them were sequentially purified by HPLC with n-hexane/iso-propanol (V:V=80:20) to yield compounds 12 (2.5mg), 14 (2.5 mg), 5 (3.0 mg) and 13 (3.3 mg). Fr.7 was separated by ODS CC eluting with MeOH/H2O (V:V=60:40, 70:30, 80:20, 90:10, 100:0) to give Fr.7-1 and Fr.7-6. Fr.7-1 was recrystallized from MeOH-H2O to afford compound 4 (10.0 mg). Fr.7-6 was purified over Sephadex LH-20 with CHCl3/ MeOH (V:V=50:50) and then separated by RP-HPLC using MeOH/H2O (V:V=70:30) to afford compounds 15 (5.0 mg) and 16 (6.0 mg).
Hypoxone A (1): Yellow powder, m.p. 136~137 ℃; [α]D20+22.5 (c 0.1, MeOH); UV (MeOH) λmax [log ε/(L·mol-1·cm-1)]: 216 (4.95), 253 (4.58), 316 (4.33) nm; 1H NMR and 13C NMR see Table 1; IR (KBr) νmax: 2926, 1732, 1682, 1593, 1472, 1373, 1277, 1233, 1196, 1032, 972, 800, 752 cm-1. HRESIMS calcd for C13H15O4Na [M+Na]+ 257.0784, found 257.0785.
4, 8-Dimethoxy-1-naphthol (2): Yellow needle, m.p. 116~117 ℃; 1H NMR and 13C NMR see Table 1. HRESIMS calcd for C12H13O3 [M+H]+ 205.0859, found 205.0854.
1'-Hydroxy-4', 8, 8'-trimethoxy[2, 2']binaphthalenyl-1, 4- dione (3): Black powder, m.p. 144~145 ℃; 1H NMR and 13C NMR see Table 2. HRESIMS calcd for C23H19O6 [M+H]+ 391.1176, found 391.1179.
Cytotoxicity of compounds 1~4 was assayed against four tumor cell lines including SF-268 (human glioma cell line), MCF-7 (human breast adenocarcinoma cell line), HepG-2 (human liver cell line), and A549 (human lung cancer cell line). Assays were performed by the SRB method[24].
Supporting Information 1D NMR and 2D NMR, HRESIMS spectra of compounds 1~4. These materials can be downloaded for free from the website (http://sioc-journal.cn/).
Koyama. K.; Kuramochi, D.; Kinoshiita, K.; Takahashi, T. J. Nat. Prod. 2002, 65, 1489. doi: 10.1021/np020140h
Gu, W.; Ge, H. M.; Song, Y. C.; Ding, H.; Zhu, L.; Zhao, X. A.; Tan, R. X. J. Nat. Prod. 2007, 70, 114. doi: 10.1021/np0604127
Quang, D. N.; Hashimoto, T.; Tanaka, M.; Stadler, M.; Asakawa, Y. Phytochemistry 2004, 65, 269.
Quang, D. N.; Hashimoto, T.; Stadler, M.; Radulovic, N.; Asakawa, Y. Planta Med. 2005, 71, 1058. doi: 10.1055/s-2005-873129
Edwards, R. L.; Maitland, D. J.; Whalley, A. J. S. J. Chem. Soc., Perkin Trans. 1 1989, 57.
Liu, H. X.; Tan, H. B.; Chen, K.; Zhao, L. Y.; Chen, Y. C.; Li, S. N.; Li, H. H.; Zhang, W. M. Org. Biomol. Chem. 2019, 17, 2346. doi: 10.1039/C8OB03223H
Liu, H. X.; Tan, H. B.; Wang, W. X.; Zhang, W. G.; Chen, Y. C.; Li, S. N.; Liu, Z. M.; Li, H. H.; Zhang, W. M. Org. Chem. Front. 2019, 6, 591. doi: 10.1039/C8QO01306C
Liu, H. X.; Tan, H. B.; Chen, Y. C.; Guo, X. Y.; Wang, W. X.; Guo, H.; Liu, Z. M.; Zhang, W. M. Org. Lett. 2019, 21, 1063. doi: 10.1021/acs.orglett.8b04107
Liu, H. X.; Tan, H. B.; Liu, Y.; Chen, Y. C.; Li, S. N.; Sun, Z. H.; Li, H. H.; Qiu, S. X.; Zhang, W. M. Fitoterapia 2017, 117, 1. doi: 10.1016/j.fitote.2016.12.005
Kögl F.; Becker H.; Detzel, A; de Voss, G. Justus Liebigs Ann. Chem. 1928, 465, 211. doi: 10.1002/jlac.19284650111
Rukachisirikul, V.; Sommart, U.; Phongpaichit, S. Chem. Pharm. Bull. 2007, 55, 1316. doi: 10.1248/cpb.55.1316
Talapatra, S. K.; Karmcharya, B.; De, S. C.; Talapatra, B. Phytochemistry 1988, 27, 3929. doi: 10.1016/0031-9422(88)83047-4
Chang, C. W.; Chang, H. S. Cheng, M. J.; Liu, T. W.; Hsieh, S. Y.; Yuan, G. F.; Chen, I. S. Chem. Biodiversity 2014, 11, 949. doi: 10.1002/cbdv.201300364
Chen, P.; Wu, J.; Dai, H. F.; Xie, X. C.; Mei, W. L. Chin. J. Med. Chem. 2008, 18, 279.
Kamisuki, S.; Ishimaru, C.; Onoda, K.; Kuriyama, I.; Ida, N.; Sugawara, F.; Yoshida, H.; Mizushina, Y. Bioorg. Med. Chem. 2007, 15, 3109. doi: 10.1016/j.bmc.2007.02.052
Allport, D. C.; Bu'Lock, J. D. J. Chem. Soc. 1960, 1960, 654.
Nadeau, A. K.; Sorensen, J. L. Tetrahedron Lett. 2011, 52, 1697. doi: 10.1016/j.tetlet.2011.01.150
Phan, D. H. T.; Kim, B.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 15608. doi: 10.1021/ja907711a
Li, Y. J.; Lv, S. W.; Wang, Y. H.; An F. T.; Liu, J.; Wang R.; Zhao, J. H. Inf. Trad. Chin. Med. 2003, 20, 25.
Zhao, Q.; Wang, C. X.; Yu, Y.; Wang, G. Q.; Zheng, Q. C.; Chen, G. D.; Lian, Y. Y.; Lin, F.; Guo, L. D.; Gao, H. J. Asian Nat. Prod. Res. 2015, 17, 567. doi: 10.1080/10286020.2015.1040776
Lakhrissi, M.; Chapleur, Y. J. Org. Chem. 1994, 59, 5752. doi: 10.1021/jo00098a039
Zhang, Q. J.; Dong, J. Y.; Cui, Q.; Li, S. S.; Cui, J. H. Synth. Commun. 2017, 47, 536. doi: 10.1080/00397911.2016.1199807
Takeya, T.; Kondo, H.; Otsuka, T.; Tomita, K.; Okamoto, I.; Tamura, O. Org. Lett. 2007, 9, 2807. doi: 10.1021/ol070951i
Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J. T.; Bokesch, H.; Kenney, S.; Boyd, M. R. J. Natl. Cancer Inst. 1990, 82, 1107. doi: 10.1093/jnci/82.13.1107
Table 1. 1H NMR (600 MHz) and 13C NMR (150 MHz) data for 1 and 2 in CDCl3
| No. | 1 | 2 | Synthetic 2[22] | |||||
| δH | δC | δH | δC | δH | δC | |||
| 1 | 196.1, C | 147.9, C | 147.9, C | |||||
| 2 | 2.90 (m), 2.64 (m) | 35.6, CH2 | 6.78 (d, 8.3) | 106.3, CH | 6.76 (d, 8.4) | 106.1, CH | ||
| 3 | 2.28 (m) | 27.8, CH2 | 6.78 (d, 8.3) | 106.3, CH | 6.76 (d, 8.4) | 106.1, CH | ||
| 4 | 6.05 (dd, 3.6, 5.7) | 69.8, CH | 148.1, C | 148.1, C | ||||
| 4a | 143.0, C | 127.9, C | 127.8, C | |||||
| 5 | 7.03 (d, 8.0) | 120.3, CH | 7.85 (d, 8.5) | 115.9, CH | 7.85 dd, 8.6, 0.8) | 115.5, CH | ||
| 6 | 7.50 (t, 8.0) | 134.6, CH | 7.34 (t, 8.5) | 125.2, CH | 7.32 (dd, 8.6, 7.7) | 125.2, CH | ||
| 7 | 7.00 (d, 8.0) | 112.6, CH | 6.85 (d, 8.5) | 104.9, CH | 6.84 (dd, 7.7, 0.8) | 104.9, CH | ||
| 8 | 160.0, C | 155.9, C | 155.9, C | |||||
| 8a | 124.4, C | |||||||
| 9 | 170.4, C | |||||||
| 10 | 2.10 (s) | 21.3, CH3 | ||||||
| 4-OMe | 3.93 (s) | 56.0, CH3 | 3.92 (s) | 56.0, CH3 | ||||
| 8-OMe | 3.93 (s) | 56.2, CH3 | 4.06 (s) | 56.1, CH3 | 4.03 (s) | 56.1, CH3 | ||
 下载: 导出CSV
下载: 导出CSV
Table 2. 1H NMR (600 MHz) and 13C NMR (150 MHz) data for 3 in CDCl3
| No. | 3 | Synthetic 3[22] | |||
| δH | δC | δH | δC | ||
| 1 | 183.3, C | 183.2, C | |||
| 2 | 151.0, C | 150.9, C | |||
| 3 | 7.05 (s) | 134.4, CH | 7.04 (s) | 134.4, C | |
| 4 | 185.5, C | 185.3, C | |||
| 4a | 117.8, C | 117.8, C | |||
| 5 | 7.78 (d, 7.7) | 134.5, CH | 7.76 (d, 7.6) | 134.5, CH | |
| 6 | 7.68 (t, 7.7) | 118.7, CH | 7.66 (t, 7.6) | 118.6, CH | |
| 7 | 7.32 (d, 7.7) | 134.5, CH | 7.30 (d, 7.6) | 134.5, CH | |
| 8 | 159.7, C | 159.6, CH | |||
| 8a | 121.2, C | 121.1, C | |||
| 1' | 146.4, C | 146.3, C | |||
| 2' | 114.9, C | 114.9, C | |||
| 3' | 6.72 (s) | 107.1, CH | 6.72 (s) | 107.1, CH | |
| 4' | 147.9, C | 147.8, C | |||
| 4a' | 129.0, C | 128.9, C | |||
| 5' | 7.87 (d, 8.5) | 116.0, CH | 7.86 (d, 8.5) | 115.9, CH | |
| 6' | 7.39 (t, 8.5) | 126.4, CH | 7.37 (t, 8.5) | 126.3, CH | |
| 7' | 6.87 (d, 8.5) | 105.6, CH | 6.86 (d, 8.5) | 105.6, CH | |
| 8' | 156.5, C | 156.4, C | |||
| 8a' | 115.4, C | 115.3, C | |||
| 8-OMe | 4.01 (s) | 56.6, CH3 | 3.99 (s) | 56.5, CH3 | |
| 4'-OMe | 3.96 (s) | 56.0, CH3 | 3.95 (s) | 56.1, CH3 | |
| 8'-OMe | 4.03 (s) | 56.2, CH3 | 4.00 (s) | 55.9, CH3 | |
下载: 导出CSV
Table 3. Cytotoxic activity of compounds 1~4
| Compd. | IC50/(µmol•L-1) | |||
| SF-268 | MCF-7 | HepG-2 | A549 | |
| 1 | 43.01 | 35.58 | 22.75 | 27.93 |
| 2 | 33.24 | 65.04 | 69.62 | 66.25 |
| 3 | 1.9 | 3.2 | 2.5 | 5.0 |
| 4 | 31.38 | 35.16 | 18.89 | 25.44 |
| Cisplatin | 3.25 | 3.02 | 2.31 | 2.65 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们