表 1
氯仿为三氯甲基源时反应条件优化a
Table 1.
Optimization of the reaction conditions while CHCl3 as reagent

α, β-不饱和羰基骨架通过区域或立体选择性的化学转化可构建多种多样的化合物[1], β, β-二氯-α, β-不饱和酮因β位氯原子的存在, 在后续转化中增添了更多的可能, 但至今其合成方法并不多[2].烯醇硅醚由于包含一个富电子的双键, 具有很好的与亲电试剂反应的活性, 等同于酮类似物, 可通过Aldol缩合[3]、Mannich反应[4]、烯丙基化反应[5]、Michael加成[6]、与多氟烷基亲电试剂反应[7]等在酮的α位引入各种基团.自由基具有反应活性高、来源广泛的特点, 自由基对烯醇硅醚双键加成同样具有很好的反应活性.氮自由基[8]、氧自由基[9]、硫自由基[10]、碳自由基[11]对烯醇硅醚双键的加成再经后继转化的文献大量涌现, 其中由卤代烃脱去卤素生成烃基自由基[12]的研究工作获得了广泛报道.
氯仿(CHCl3)和四氯化碳(CCl4)是常见溶剂, 同时也是很好的多氯甲基自由基源, 在自由基引发剂或光催化剂作用下产生多氯甲基自由基, 参与后续反应[13].设想以烯醇硅醚为反应底物, 氯仿或四氯化碳作为三氯甲基自由基源, 对烯醇硅醚双键进行加成, 脱去硅基得到α-三氯甲基酮, 进一步脱去一分子氯化氢得到β, β-二氯- α, β-不饱和酮化合物.实验证明, 这个研究策略可行, 多数烯醇硅醚能以良好收率获得目标产物. 1-芳基-1-三甲基硅氧基乙烯衍生物1的合成路线见Eq. (1).
|
|
(1) |
以1-苯基-1-三甲硅氧基乙烯(1a)为底物, 以氯仿为三氯甲基自由基源及溶剂, 反应温度110 ℃, 对反应进行了研究, 结果见表 1.
 下载:
导出CSV
下载:
导出CSV
 | |||
| Entry | Oxidant (equiv.) | CHCl3/mL | Yieldb/% |
| 1 | TBHP (2.5) | 3 | 12 |
| 2 | 30% H2O2 (2.5) | 3 | 16 |
| 3 | BPO (2.5) | 3 | 0 |
| 4 | ABIN (2.5) | 3 | 0 |
| 5 | DTBP (2.5) | 3 | 51 |
| 6c | DTBP (2.5) | 3 | Trace |
| 7d | DTBP (2.5) | 3 | Trace |
| 8 | DTBP (1.5) | 3 | 45 |
| 9 | DTBP (2.0) | 3 | 50 |
| 10 | DTBP (3.0) | 3 | 44 |
| 11e | DTBP (2.0) | 3 | 6 |
| 12f | DTBP (2.0) | 3 | 39 |
| 13 | DTBP (2.0) | 1 | 40 |
| 14 | DTBP (2.0) | 2 | 56 |
| 15g | DTBP (2.0) | 2 | 76 |
| 16h | DTBP (2.0) | 1.5 mmol | 20 |
| a Reaction conditions: 0.3 mmol of 1a, under N2, sealed tube. b Isolated yield. c 0.3 mmol of triethylamine (TEA) was used. d 0.3 mmol of acetic acid was used. e 90 ℃. f 130 ℃. g CHCl3 was dehydrated. h 2.0 mL of CH3CN as solvent. | |||
首先考察了氧化剂对反应的影响(Entries 1~5).其中以二叔丁基过氧化物(DTBP)为优选, 当选择2.5 equiv. DTBP时产物收率为51% (Entry 5).对酸碱添加剂的使用进行了考察, 反应发现添加碱(1.0 equiv.三乙胺)或添加酸(1.0 equiv.醋酸)时反应被显著抑制(Entries 6, 7). DTBP的用量实验表明2.0 equiv.最佳(Entries 8~10), 升高或降低反应温度没有带来正向的积极影响(Entries 11, 12);反应物浓度实验表明, 当选用2.0 mL CHCl3时产物收率稍有提高(Entry 14).令人惊喜的是, 当使用无水纯化氯仿时产物收率有了显著提高, 达到了76% (Entry 15);当选择CH3CN为溶剂, 氯仿用量为5.0 equiv.时, 并未能获得理想的结果(Entry 16).
综上所述, 确定了该反应的较优条件为: 0.3 mmol 1a, 2.0 mL无水纯化氯仿, 0.6 mmol DTBP, 110 ℃, 氮气保护下封管反应.
基于上述优化的反应条件, 对反应的底物适用性进行了考察, 结果见表 2.当氯仿作为三氯甲基自由基源时, 不同结构的烯醇硅醚均能以中等至良好收率转化为目标产物.实验结果表明:当R1为取代苯基时, 取代基的电子效应并不显著, 并未获得吸电子基有利或不利的结论; 同时苯环上邻位连有基团时并未表现出明显的位阻效应, 这和自由基对双键加成反应位点与芳基有一定距离有关. R1为噻吩-2-基时, 产物2o的收率为37%, 并不理想. R2为非氢原子时, 产物2m和2n的收率与2a相比有一定下降, 这与优化条件下, 后处理2m与2n时反应体系中存在一定量未转化为目标产物的中间产物α-三氯甲基酮有关.
当选择四氯化碳为三氯甲基自由基源及溶剂时, 在类似氯仿为试剂的优化条件下, 烯醇硅醚1a能以中等到良好的收率转化为目标产物2a, 稍有不同的是添加剂碱在反应中起到了相当关键的作用, 具体的优化结果见表 3.
下载:
导出CSV
 | |||||
| Entry | Oxidant (equiv.) |
CCl4/mL | Additive (equiv.) | T/℃ | Yieldb/% |
| 1 | BPO (2.0) | 3 | — | 110 | 18 |
| 2 | DTBP (2.0) | 3 | — | 110 | 38 |
| 3 | TBHP (2.0) | 3 | — | 110 | 0 |
| 4 | DCP (2.0) | 3 | — | 110 | 20 |
| 5 | DTBP (2.0) | 3 | TEA (2.0) | 110 | 66 |
| 6 | DTBP (2.0) | 3 | NaOAc (2.0) | 110 | 54 |
| 7 | DTBP (2.0) | 3 | KH2PO4 (2.0) | 110 | 45 |
| 8 | DTBP (2.0) | 3 | K2CO3 (2.0) | 110 | 49 |
| 9 | DTBP (2.0) | 3 | TEA (1.0) | 110 | 65 |
| 10 | DTBP (2.0) | 3 | TEA (1.5) | 110 | 72 |
| 11 | DTBP (1.5) | 3 | TEA (1.5) | 110 | 65 |
| 12 | DTBP (2.5) | 3 | TEA (1.5) | 110 | 67 |
| 13 | DTBP (2.0) | 3 | TEA (1.5) | 120 | 78 |
| 14 | DTBP (2.0) | 3 | TEA (1.5) | 130 | 73 |
| 15 | DTBP (2.0) | 2 | TEA (1.5) | 120 | 82 |
| 16c | DTBP (2.0) | 2 | TEA (1.5) | 120 | 83 |
| a Reaction conditions: 0.3 mmol 1a, under N2, sealed tube. b Isolated yield. c CCl4 was dehydrated. | |||||
首先考察了氧化剂对反应的影响(Entries 1~5), 其中以DTBP为优选, 当选择2.0 equiv. DTBP时产物收率为38% (Entry 2).对碱添加剂的使用进行了考察, 反应发现添加碱(醋酸钠、三乙胺、磷酸二氢钾、碳酸钾)时产物收率均有较显著提高, 其中以添加1.5 equiv.三乙胺为优选(Entries 5~10). DTBP的用量实验表明2.0 equiv.最佳(Entries 10~12), 当温度为120 ℃时产物收率有了一定的提高(Entry 13).当增大反应物浓度时产物收率达到82% (Entry 15);无水纯化四氯化碳代替试剂四氯化碳时收率稍有提高, 达到83% (Entry 16).
综上所述, 确定了该反应的较优条件为: 0.3 mmol 1a, 2.0 mL无水纯化CCl4, 0.6 mmol DTBP, 0.45 mmol TEA, 120 ℃, 氮气保护下封管反应.
基于上述优化的反应条件, 对反应底物的适用性进行了考察, 结果见表 4.当四氯化碳作为三氯甲基自由基源时, 与氯仿作对比, 大部分底物收率均有较明显的提高.实验结果表明:当R1为取代苯基时, 苯环上取代基的电子效应并不显著, 同时苯环上邻位连有基团时并未表现出明显的位阻效应.当R2为非氢原子时, 产物2n获得了理想的收率.当R1为噻吩-2-基时, 产物2o也获得了可接受的结果.
下载:
导出CSV

|
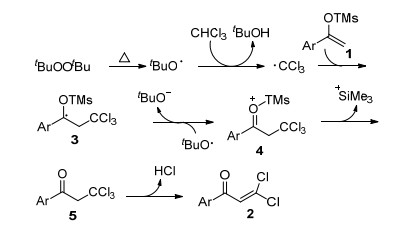
提出如Scheme 1所示可能的反应机理: DTBP受热分解生成叔丁基氧自由基, 氯仿在叔丁基氧自由基的氧化下生成三氯甲基自由基, 三氯甲基自由基对烯醇硅醚1双键进行加成生成中间体3, 3经叔丁基氧自由基氧化生成中间体4, 4脱去三甲基硅基生成α-三氯甲基芳酮5, 5进一步脱去一分子氯化氢得到目标产物2.四氯化碳为三氯甲基自由基源时机理类似.
以芳酮衍生的烯醇硅醚为底物, 在DTBP的作用下, 以氯仿和四氯化碳为三氯甲基自由基源及溶剂, 分别在110 ℃和120 ℃下建立了合成β, β-二氯-α, β-不饱和酮衍生物的新方法.该法操作简单, 无需金属催化, 对底物的基团容忍性强, 为合成β, β-二氯-α, β-不饱和酮衍生物提供了一个有效途径.
熔点在X-4型显微熔点仪上测定, 温度计未校正; 核磁共振谱在ANANCE III (500M)型核磁共振波谱仪上测定, TMS为内标, CDCl3为溶剂.所有试剂均为国产分析纯试剂, 柱层析硅胶为国产200~300目, 石油醚(PE)、乙酸乙酯(EA)为重蒸工业级试剂, 其中PE的沸程控制在60~90 ℃.氯仿无水纯化方法:氢化钙回流, 常压蒸馏; 四氯化碳无水纯化方法:五氧化二磷干燥, 常压蒸馏. 1-芳基-1-三甲基硅氧基乙烯衍生物1的合成参考文献[14]进行.
以2a化合物的合成为例.将0.5 mmol烯醇硅醚1a置于史莱克反应管中, 抽空换氮三次, 氮气氛围下加入3.4 mL无水纯化CHCl3, 1.0 mmol DTBP, 110 ℃封管反应4 h, 薄层色谱(TLC)跟踪反应, 反应完全后反应液经减压浓缩, 残余物经硅胶柱层析分离(展开剂为石油醚和乙酸乙酯混合液), 得淡黄色液体2a, 83.0 mg, 收率83%.其余产物的合成均与2a化合物方法相同.
当四氯化碳为三氯甲基源及溶剂时, 实验操作方法和上述类似.
1-苯基-3, 3-二氯-2-丙烯-1-酮(2a)[2c]:淡黄色油状物. CHCl3为试剂:产率76%; CCl4为试剂:产率83%. 1H NMR (500 MHz, CDCl3) δ: 7.29 (d, J=5.0 Hz, 1H), 7.51 (t, J=5.0 Hz, 2H), 7.62 (t, J=5.0 Hz, 1H), 7.94 (d, J=10.0 Hz, 2H).
1-(2-甲基苯基)-3, 3-二氯-2-丙烯-1-酮(2b):淡黄色油状物. CHCl3为试剂:产率79%; CCl4为试剂:产率66%. 1H NMR (500 MHz, CDCl3) δ: 7.61~7.58 (m, 1H), 7.43 (td, J=7.5, 1.3 Hz, 1H), 7.32~7.27 (m, 2H), 7.06 (s, 1H), 2.54 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 20.88, 125.89, 126.96, 129.12, 132.02, 134.98, 137.29, 138.82, 190.03. HRMS (ESI) calcd for C10H8Cl2O [M+H]+215.0030, found 215.0031.
1-(4-甲基苯基)-3, 3-二氯-2-丙烯-1-酮(2c)[2c]:淡黄色液体. CHCl3为试剂:产率74%; CCl4为试剂:产率64%. 1H NMR (500 MHz, CDCl3) δ: 7.86~7.82 (m, 2H), 7.31 (d, J=8.0 Hz, 2H), 7.26 (s, 1H), 2.44 (s, 3H).
1-(4-甲氧基苯基)-3, 3-二氯-2-丙烯-1-酮(2d)[2c]:淡黄色液体. CHCl3为试剂:产率51%; CCl4为试剂:产率82%. 1H NMR (500 MHz, CDCl3) δ: 7.93 (d, J=8.9 Hz, 2H), 7.22 (s, 1H), 6.98 (d, J=8.9 Hz, 2H), 3.90 (s, 3H).
1-(3-甲氧基苯基)-3, 3-二氯-2-丙烯-1-酮(2e):淡黄色液体. CHCl3为试剂:产率58%; CCl4为试剂:产率74%. 1H NMR (500 MHz, CDCl3) δ: 7.53~7.46 (m, 2H), 7.41 (t, J=7.9 Hz, 1H), 7.27 (d, J=1.7 Hz, 1H), 7.16 (ddd, J=8.2, 2.6, 0.9 Hz, 1H), 3.88 (s, 3H); 13C NMR (125 MHz, CDCl3) δ: 55.52, 112.62, 120.37, 121.09, 124.10, 129.83, 135.59, 138.32, 160.08, 186.41. HRMS (ESI) calcd for C10H8Cl2O2 [M+H]+ 230.9980, found 230.9976.
1-(4-氟苯基)-3, 3-二氯-2-丙烯-1-酮(2f)[2c]:淡黄色液体. CHCl3为试剂:产率64%; CCl4为试剂:产率91%. 1H NMR (500 MHz, CDCl3) δ: 8.01~7.94 (m, 2H), 7.24 (s, 1H), 7.21~7.15 (m, 2H).
1-(2-氟苯基)-3, 3-二氯-2-丙烯-1-酮(2g):淡黄色液体. CHCl3为试剂:产率50%; CCl4为试剂:产率85%. 1H NMR (500 MHz, CDCl3) δ: 7.84 (td, J=7.6, 1.8 Hz, 1H), 7.57 (dddd, J=8.3, 7.2, 5.1, 1.8 Hz, 1H), 7.30~7.25 (m, 2H), 7.16 (ddd, J=11.1, 8.3, 0.8 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ: 116.70(d, JC—F=225 Hz), 124.78 (d, JC—F=37.5 Hz), 126.18 (d, JC—F=125 Hz), 126.48 (d, JC—F=75.0 Hz), 131.08 (d, JC—F=25.0 Hz), 135.06 (d, JC—F=100.0 Hz), 136.49, 160.28, 162.31, 183.99 (d, JC—F=25.0 Hz); HRMS (ESI) calcd for C9H5Cl2FO [M+H]+ 218.9780, found 218.9777.
1-(4-氯苯基)-3, 3-二氯-2-丙烯-1-酮(2h)[2c]:淡黄色固体. CHCl3为试剂:产率49%; CCl4为试剂:产率74%. m.p. 61~62 ℃(文献值[2c]: 49~51 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.90~7.85 (m, 2H), 7.51~7.46 (m, 2H), 7.24 (s, 1H).
1-(2-氯苯基)-3, 3-二氯-2-丙烯-1-酮(2i):淡黄色液体. CHCl3为试剂:产率49%; CCl4为试剂:产率68%. 1H NMR (500 MHz, CDCl3) δ: 7.56~7.51 (m, 1H), 7.49~7.42 (m, 2H), 7.37 (ddd, J=7.7, 5.1, 3.5 Hz, 1H), 7.10 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 126.66, 127.21, 129.97, 130.55, 131.60, 132.57, 136.83, 138.35, 187.84. HRMS (ESI) calcd for C9H5Cl3O [M+H]+ 234.9484, found 234.9481.
1-(4-溴苯基)-3, 3-二氯-2-丙烯-1-酮(2j)[2d]:黄色固体. CHCl3为试剂:产率72%; CCl4为试剂:产率72%. m.p. 77~78 ℃(文献值[2d]72~73.5 ℃); 1H NMR (500 MHz, CDCl3) δ: 7.80 (d, J=8.6 Hz, 2H), 7.66 (d, J=8.7 Hz, 2H), 7.23 (s, 1H).
1-(4-硝基苯基)-3, 3-二氯-2-丙烯-1-酮(2k)[2c]:黄色固体. CHCl3为试剂:产率46%. m.p. 115~116 ℃(文献值[2c]113~114 ℃); 1H NMR (500 MHz, CDCl3) δ: 8.36 (d, J=8.8 Hz, 2H), 8.09 (d, J=8.9 Hz, 2H), 7.31 (s, 1H).
1-(1-萘基)-3, 3-二氯-2-丙烯-1-酮(2l):淡黄色油状物. CHCl3为试剂:产率64%; CCl4为试剂:产率42%. 1H NMR (500 MHz, CDCl3) δ: 8.64 (d, J=8.6 Hz, 1H), 8.05 (d, J=8.2 Hz, 1H), 7.90 (dd, J=21.9, 7.6 Hz, 2H), 7.68~7.50 (m, 3H), 7.21 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 124.46, 125.45, 126.79, 127.33, 128.33, 128.62, 129.06, 130.31, 133.59, 133.96, 135.09, 135.33, 189.71. HRMS (ESI) calcd for C13H8Cl2O [M+H]+ 251.0030, found 251.0028.
1, 2-二苯基-3, 3-二氯-2-丙烯-1-酮(2m)[2e]:淡黄色液体. CHCl3为试剂:产率58%. 1H NMR (500 MHz, CDCl3) δ: 8.08~8.02 (m, 2H), 7.65~7.59 (m, 1H), 7.50~7.55 (m, 4H), 7.42~7.33 (m, 3H).
2-二氯亚甲基-1-四氢萘酮(2n):淡黄色液体. CHCl3为试剂:产率57%; CCl4为试剂:产率64%. 1H NMR (500 MHz, CDCl3) δ: 8.13 (dd, J=7.9, 1.1 Hz, 1H), 7.52 (td, J=7.5, 1.4 Hz, 1H), 7.38 (t, J=7.5 Hz, 1H), 7.26 (d, 1H), 3.14~3.05 (m, 4H); 13C NMR (125 MHz, CDCl3) δ: 28.74, 31.26, 127.24, 128.35, 128.42, 128.45, 133.03, 133.30, 133.69, 142.76, 184.64. HRMS (ESI) calcd for C11H8Cl2O [M+H]+ 227.0030, found 227.0025.
1-(2-噻吩基)-3, 3-二氯-2-丙烯-1-酮(2o)[15]:淡黄色液体. CHCl3为试剂:产率37%; CCl4为试剂:产率49%); 1H NMR (500 MHz, CDCl3) δ: 7.75~7.71 (m, 2H), 7.25 (s, 1H), 7.18 (dd, J=4.9, 3.9 Hz, 1H).
1-(3-硝基苯基)-3, 3-二氯-2-丙烯-1-酮(2p)[2d]:黄色固体. CCl4为试剂:产率41%. m.p. 102~103 ℃(文献值[2d] 96~97 ℃). 1H NMR (500 MHz, CDCl3) δ: 8.74 (t, J=1.9 Hz, 1H), 8.48 (ddd, J=8.2, 2.2, 1.0 Hz, 1H), 8.31~8.24 (m, 1H), 7.74 (t, J=8.0 Hz, 1H), 7.34 (s, 1H).
1-(4-三氟甲基苯基)-3, 3-二氯-2-丙烯-1-酮(2q):黄色固体. CCl4为试剂:产率66%. m.p. 56~57 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.04 (d, J=8.2 Hz, 2H), 7.78 (d, J=8.2 Hz, 2H), 7.29 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 122.39, 123.34, 124.56, 125.95(t, JC—F=37.5 Hz), 128.78, 134.78, 135.04, 137.41, 139.75, 185.57; HRMS (ESI) calcd for C10H5Cl2F3O [M+H]+ 268.9748, found 268.9745.
1-(3-氯苯基)-3, 3-二氯-2-丙烯-1-酮(2r):淡黄色液体. CCl4为试剂:产率81%. 1H NMR (500 MHz, CDCl3) δ: 7.90 (t, J=1.8 Hz, 1H), 7.83~7.78 (m, 1H), 7.59 (ddd, J=8.0, 2.0, 1.0 Hz, 1H), 7.46 (t, J=7.9 Hz, 1H), 7.25 (s, 1H); 13C NMR (125 MHz, CDCl3) δ: 123.45, 126.54, 128.52, 130.20, 133.62, 135.28, 136.82, 138.57, 185.28. HRMS (ESI) calcd for C9H5Cl3O [M+H]+ 234.9484, found 234.9480.
(a) Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2002, 41, 1668.
(b) Melchiorre, P. Angew. Chem., Int. Ed. 2012, 51, 9748.
(a) Galina, G. L.; Valentina, A. K.; Elena, V. R.; Khanh, Q. H.; Dmitrii, O. S.; Igor, B. R. Tetrahedron. 2011, 67, 1844.
(b) Ian, W. J. S.; Sudhir, B. A. J. Org. Chem. 1982, 47, 374.
(c) Nobumasa, K.; Kumiko, U.; Manabu, Y.; Toshio, S. J. Organomet. Chem. 1998, 552, 39.
(d) Shinji, M.; Yoshiaki, K.; Tomoyuki, A.; Noboru, S.; Shireru, T. J. Chem. Soc., Chem. Commun. 1972, 741.
(e) Pavel, S. L.; Ivan, V. S.; Victor, N. K.; Vitaly, A. R.; Alexander, V. P.; Valentina, A. K.; Igor, B. R.; Valentine, G. N. Org. Chem. Front. 2019, 6, 335.
(f) Somai, M.; Krishna, B.; Lee, Y. R. Adv. Synth. Catal. 2014, 356(16), 3422
(a) Lefebvre, O.; Brigaud, T.; Portella, C. J. Org. Chem. 2001, 66, 1941.
(b) Itsuno, S.; Arima, S.; Haraguchi, N. Tetrahedron 2005, 61, 12074.
(c) Dell'Amico, L.; Zanardi, F. Angew. Chem., Int. Ed. 2019, 58, 3264.
(d) Li, J. D.; Li, Y. A.; Sun, J. A.; Gui, Y.; Huang, Y. K.; Zha, Z. G.; Wang, Z. Y. Chem. Commun. 2019, 55, 630.
(a) Kashikura, W.; Mori, K.; Akiyama, T. Org. Lett. 2011, 13, 1860.
(b) Li, J. S.; Liu, Y. J.; Zhang, G. W.; Ma, J. A. Org. Lett. 2017, 19, 6364.
Étienne, B.; Katy, C.; Olivier, M.; Mélanie, T.; Jean-Francois, P. J. Am. Chem. Soc. 2007, 129, 1034. doi: 10.1021/ja067501q
(a) Yu, J. S.; Liao, F. M.; Gao, W. M.; Liao, K.; Zuo, R. L.; Zhou, J. Angew. Chem., Int. Ed. 2015, 54, 7381.
(b) Hao, Y. J.; Hu, X. S.; Yu, J. S.; Zhou, F.; Zhou, Y.; Zhou. J. Tetrahedron 2018, 74, 7395.
(a) Huo, H. H.; Wang, C. Y.; Harms, K.; Meggers, E. J. Am. Chem. Soc. 2015, 137, 9551.
(b) Katayev, D.; Vaxclavík, J.; Brüning, F.; Commare, B.; Togni, A. Chem. Commun. 2016, 52, 4049.
(c) Jérémy, J.; Khaled, C.; Ren, Y. F.; Hervé, V.; Maylis, O.; Sébastien, B.; Louis, F.; Marine, D. M. Chem.-Eur. J. 2017, 23, 15030.
(d) Xu, C.; Song, X. N.; Guo, J.; Chen, S. B.; Gao, J.; Jiang, J.; Gao, F. Y.; Li, Y. X. Org. Lett. 2018, 20, 3933.
(a) Rathore, R.; Kochi. J. K. J. Org. Chem. 1996, 61, 627.
(b) Zlotorzynska, M.; Zhai, H.; Sammis, G. M. J. Org. Chem. 2010, 75, 864.
(a) Zlotorzynska, M.; Zhai, H.; Sammis, G. M. Org. Lett. 2008, 10, 5083.
(b) Montserrat, R.; Joe, C. T. L.; Christine, R. D.; Glenn, M. S. J. Org. Chem. 2011, 76, 7720.
(a) Kamigata, N.; Udodaira, K.; Shimizu, T. J. Chem. Soc., Perkin Trans. 1 1997, 783.
(b) Liu, T. S.; Zheng, D. Q.; Ding, Y. C.; Fan, X. N.; Wu, J. Chem. Asian J. 2017, 12, 465.
(a) Sakakura, T.; Haraa, M.; Tanaka. M. J. Chem. Soc., Perkin Trans. 1 1994, 283.
(b) Diego, F. B.; Jose, C. G. G. Adv. Synth. Catal. 2018, 360, 2773.
(c) Kong, W. G.; Yu, C. J.; An, H. J.; Song, Q. L. Org. Lett. 2018, 20, 349.
(a) Mikami, K.; Tomita, Y.; Ichikawa, Y.; Amikura, K.; Itoh, Y. Org. Lett. 2006, 8, 4671.
(b) Pham, P. V.; Nagib, D. A.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2011, 50, 6119.
(c) Huang, M. W.; Li, L.; Zhao, Z. G.; Chen, Q. Y.; Guo, Y. Synthesis 2015, 47, 3891.
(d) Zhang, X.; Qin, J.; Huang, X. Q.; Meggers, E. Eur. J. Org. Chem. 2018, 571.
Chloroform as dichloromethyl radical source, see:
(a) Chen, C.; Tan, H.; Liu, B. F.; Yue, C. C.; Liu, W. B. Org. Chem. Front. 2018, 5, 3143.
(b) Robynne, K. N.; Su, Y. L.; Liu, S. Q.; Melina, R.; Zhang, X. H.; Michael, P. D. J. Am. Chem. Soc. 2019, 141, 16643.
Chloroform or carbon tetrachloride as trichloromethyl radical source, see:
(c) Dowbenko, R. Org. Synth. Coll. 1973, 5, 93.
(d) Mitani, M.; Takuya, K.; Kuratate, T. J. Org. Chem. 1994, 59, 1279.
(e) Laurent, Q.; Katrin, T.; Kay, S. J. Am. Chem. Soc. 2006, 128, 7440.
(f) Zhou, Y. H.; Wu, C. P.; Dong, X. L.; Qu, J. P. J. Org. Chem. 2016, 81, 5202.
Khan, I.; Reed-Berendt, B. G.; Melen, R. L.; Morrill, L. C. Angew. Chem., Int. Ed. 2018, 57, 12356. doi: 10.1002/anie.201808800
Burkhardt, U.; Johne, S. J. Prakt. Chem. 1987, 329, 332. doi: 10.1002/prac.19873290225
表 1 氯仿为三氯甲基源时反应条件优化a
Table 1. Optimization of the reaction conditions while CHCl3 as reagent
| | |||
| Entry | Oxidant (equiv.) | CHCl3/mL | Yieldb/% |
| 1 | TBHP (2.5) | 3 | 12 |
| 2 | 30% H2O2 (2.5) | 3 | 16 |
| 3 | BPO (2.5) | 3 | 0 |
| 4 | ABIN (2.5) | 3 | 0 |
| 5 | DTBP (2.5) | 3 | 51 |
| 6c | DTBP (2.5) | 3 | Trace |
| 7d | DTBP (2.5) | 3 | Trace |
| 8 | DTBP (1.5) | 3 | 45 |
| 9 | DTBP (2.0) | 3 | 50 |
| 10 | DTBP (3.0) | 3 | 44 |
| 11e | DTBP (2.0) | 3 | 6 |
| 12f | DTBP (2.0) | 3 | 39 |
| 13 | DTBP (2.0) | 1 | 40 |
| 14 | DTBP (2.0) | 2 | 56 |
| 15g | DTBP (2.0) | 2 | 76 |
| 16h | DTBP (2.0) | 1.5 mmol | 20 |
| a Reaction conditions: 0.3 mmol of 1a, under N2, sealed tube. b Isolated yield. c 0.3 mmol of triethylamine (TEA) was used. d 0.3 mmol of acetic acid was used. e 90 ℃. f 130 ℃. g CHCl3 was dehydrated. h 2.0 mL of CH3CN as solvent. | |||
 下载: 导出CSV
下载: 导出CSV
表 3 CCl4为试剂时反应条件优化a
Table 3. Optimization of the reaction conditions while CCl4 as reagent
| | |||||
| Entry | Oxidant (equiv.) |
CCl4/mL | Additive (equiv.) | T/℃ | Yieldb/% |
| 1 | BPO (2.0) | 3 | — | 110 | 18 |
| 2 | DTBP (2.0) | 3 | — | 110 | 38 |
| 3 | TBHP (2.0) | 3 | — | 110 | 0 |
| 4 | DCP (2.0) | 3 | — | 110 | 20 |
| 5 | DTBP (2.0) | 3 | TEA (2.0) | 110 | 66 |
| 6 | DTBP (2.0) | 3 | NaOAc (2.0) | 110 | 54 |
| 7 | DTBP (2.0) | 3 | KH2PO4 (2.0) | 110 | 45 |
| 8 | DTBP (2.0) | 3 | K2CO3 (2.0) | 110 | 49 |
| 9 | DTBP (2.0) | 3 | TEA (1.0) | 110 | 65 |
| 10 | DTBP (2.0) | 3 | TEA (1.5) | 110 | 72 |
| 11 | DTBP (1.5) | 3 | TEA (1.5) | 110 | 65 |
| 12 | DTBP (2.5) | 3 | TEA (1.5) | 110 | 67 |
| 13 | DTBP (2.0) | 3 | TEA (1.5) | 120 | 78 |
| 14 | DTBP (2.0) | 3 | TEA (1.5) | 130 | 73 |
| 15 | DTBP (2.0) | 2 | TEA (1.5) | 120 | 82 |
| 16c | DTBP (2.0) | 2 | TEA (1.5) | 120 | 83 |
| a Reaction conditions: 0.3 mmol 1a, under N2, sealed tube. b Isolated yield. c CCl4 was dehydrated. | |||||
下载: 导出CSV
表 4 CCl4为试剂烯醇硅醚结构对反应的影响a
Table 4. Effect of enol silyl ethers structure to the reaction while CCl4 as reagent
|
|
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们