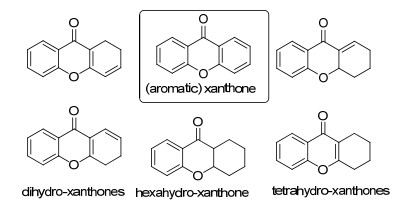
图 1.
Xanthone的结构
Figure 1.
Structures of xanthone

Xanthone的文名为呫吨酮或氧杂蒽酮, 是一类由细菌、真菌、地衣等微生物和高等植物产生的次级代谢产物.根据这类天然产物的xanthone三环结构不饱和度不同[1], 又可以分为aromaticxanthone、dihydroxanthone、tetrahydroxanthone以及hexahydroxanthone(图 1).该类化合物具有广泛、良好的生物活性, 包括能作用于心血管、肾脏、肝脏、神经元和免疫系统以及抗肿瘤[2], 抗病毒、抗细菌、抗真菌和抗疟疾[3]活性, 还有抗炎和抗球虫[4]等活性, 所以此类骨架结构属于一类“优势结构”, 吸引了大量药物化学家的关注[5].
多环型xanthone是一类具有高度氧化态的六环骨架结构的天然产物, 目前分离得到的此类化合物主要来源于放线菌属, 包括Actinomadura、Actinoplanes、Amycolatopsis、Kibdelosporangium和Streptomyces[6]等.这类化合物有着非常广谱但程度各异的抗细菌、抗真菌、抗寄生虫活性, 包括耐甲氧西林金黄色葡萄球菌(MRSA)和耐万古霉素粪肠球菌[7], 对多个肿瘤细胞系也有明显的抑制活性.正是因为它们独特的化学结构和良好的生物活性, 近一二十年来, 这类天然产物的分离、生物及化学合成和药理及药化研究显著增加.
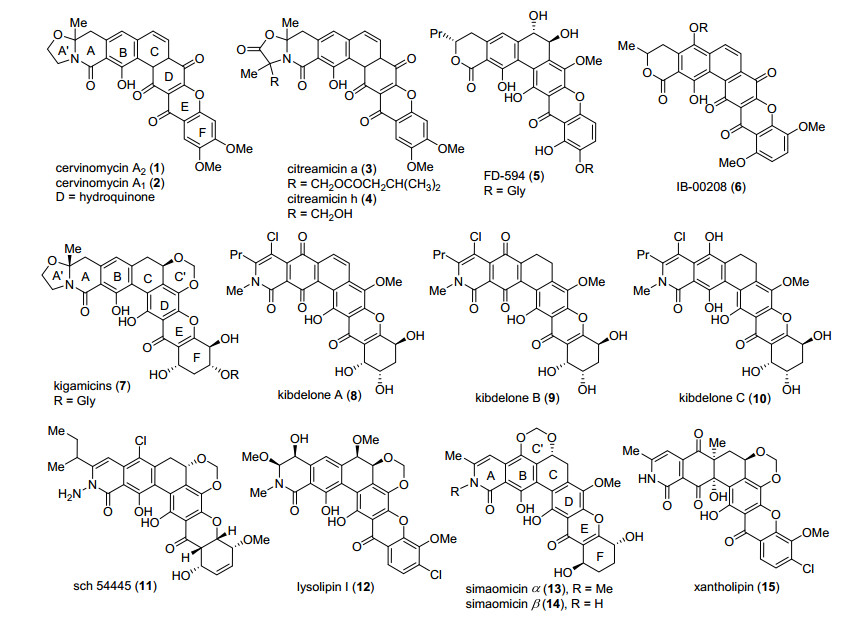
图 2中罗列了部分多环型xanthone天然产物, 这些化合物除了具有代表性的xanthone环和高氧化态的六环核心骨架外, 大多含有在聚酮类天然产物中较为少见的环状内酰胺, 如kibdelones (8~10)[8]、simaomicins (13, 14)[9]、kigamicins (7)[10, 11]和xantholipin (15)[12]等, 也有A环以内酯形式存在的, 如FD-594 (5)[13, 14]和IB-00208 (6)[15, 16]等.除了A环的差异, 有些在A环左侧还含有一个取代的四氢噁唑环, 如kigamicins (7)、cervinomycins (1, 2)[17, 18]和citreamicins (3, 4)[19, 20]等.除了上述差异, 它们主要的不同还在于BCD和F环氧化态, 取代官能团的数量不同, 特别是C环和F环, 例如C环可以有饱和、不饱和、单羟基取代、双羟基取代等多种存在形式.在此基础上, kigamicins (7)、simaomicins (13, 14)和xantholipin (15)等多个天然产物还含Chin. J.Org.Chem. 2020, 40, 563~574有较为少见的缩醛六元环C'. F环除了不饱和度不同之外, 还有羟基数目和构型上的差异.在kigamicins (7)和FD-594 (5)中, F环的羟基还连接有糖基片段.
早在1989年, Kelly小组[21]就以汇聚式路线完成了(±)-cervinomycin A1 (2)和A2 (1)全合成.由于缺乏对多取代、高氧化态F环的有效构建方法, 此后的20年多环型xanthone天然产物的合成研究几乎都是针对F环为苯环的一类天然产物, 包括1991年Rao小组[22]报道了(±)-cervinomycin A1 (2)和A2 (1)的全合成工作和2009年Suzuki小组[23]完成了FD-594 (5)糖苷配基的不对称合成. 2011年Porco小组[24]和Ready小组[25]分别报道了kibdelone C (10)的不对称全合成工作.2013年Ready小组[26]完成了(-)-simaomicin α (13)的合成工作. 2018年高栓虎课题组[27]也完成了kibdelone C (10)的不对称形式全合成.
Cervinomycins A1 (2)和A2 (1)是Omure小组[17]1982年从链霉菌属Streptomyces cervinus sp.的培养液中分离得到的, 并于1986年确定了其化学结构.经过大量的活性测试[18], 发现cervinomycins A1 (2)和A2 (1)对细菌具有较为广谱的抗菌活性, 其中它们对Clostridium perfringens和Eubacterium limosum的最低抑菌浓度(MIC)值相同, 分别可达为12, 0.2 nmol/L; 对Clostridium perfringens、Eubacterium limosum的MIC值均低至24 nmol/L; 对许多厌氧细菌和支原体也有明显的抑制活性.从结构上来说, cervinomycins A1 (2)和A2(1)的差别在于D环的氧化态不同, 两者也能通过氧化/还原相互转化.Cervinomycins属于芳香型多环xanthone, 且含有xanthone天然产物中并不常见的四氢噁唑环, 这点与kigamicins相同.
Kelly小组[21]以Heck反应和6π电环化为关键反应, 实现了(±)-cervinomycins A1 (2)和A2 (1)全合成(Scheme 1).首先从二碘化物16开始, 与17经过加成/消除得到中间体18, 对苯二醌经过还原得到对苯二酚, 在酸性条件下环化, 对两个酚羟基进行MOM保护得到xanthone三环DEF片段19.片段23从原料20开始, 经过三步在苯环上引入两个取代基得到中间体21, 经过两步转化得到烯基内酯中间22, 随后发生胺解和Mitsunobu反应, 构建了四氢噁唑环, 得到A'AB片段23.
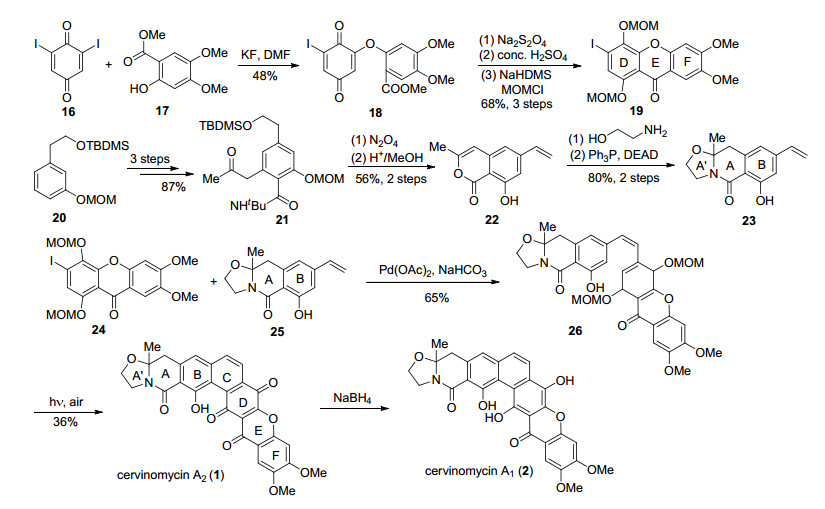
片段19和23通过Heck反应进行偶联, 得到光反应前体26, 随后以高压汞灯作为光源, 发生6π电环化, 同时伴随着MOM的脱除和对苯二酚氧化直接得到了(±)-cervinomycin A2 (1), 经过还原得到(±)-cervino- mycin A1 (2).
1991年, Rao课题组[22]以ABCD四环中间体31为关键化合物完成了(±)-cervinomycin A1 (2)和A2 (1)的全合成工作(Scheme 2).首先从多取代萘27开始, 经过10步转化得到BCD三环中间体28, 4步对官能团的转化得到了四环内酯31, 通过D环的氧化, 再与32发生加成/消除, 最后经过还原、保护、傅克酰基化得到六环中间体34.水解内酯, 并将羧基保护、羟基氧化为酮得到35, 随后碱性条件下发生缩合构建出四氢噁唑环, 接着选择性地将D环氧化, 得到(±)-cervinomycin A2(1), 再将D环对苯二醌结构还原可得到(±)-cervinomycin A1 (2).相比较Kelly小组而言, Rao小组的合成效率并不太高, 特别是构建ABCD四环中间体31, 作者使用了大量的步骤来构建A环, 后续步骤与Kelly小组较为类似.

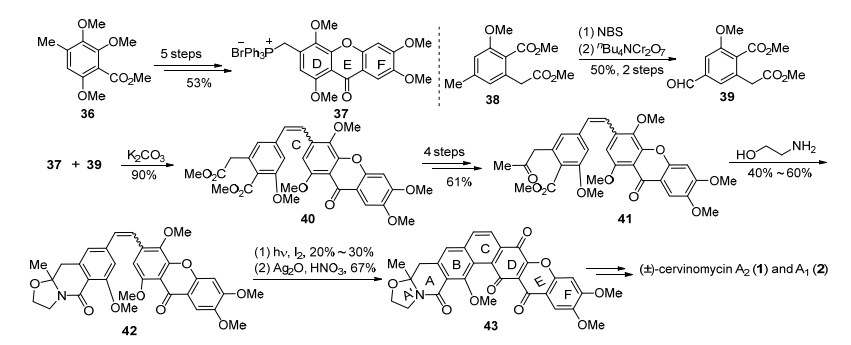
1994年, Mehta小组以傅克酰基化为关键反应, 5步制备了xanthone三环DEF片段37, 同时利用38制备了醛基化合物39 (Scheme 3).通过Wittig反应连接两个片段, 得到顺反混合物中间体40, 再经过4步的官能团调整, 构建出酮基侧链.随后41再与乙醇胺发生缩合, 得到四氢噁唑环, 这一步收率并不稳定.虽然Mehta小组同样使用了6π电环化来构建C环, 但是与Kelly小组不同的是, 他们使用了紫外光作为光源, 碘作为催化剂, 收率相对不高, 除了底物的原因外, 还由于四氢噁唑环在酸性条件下不够稳定, 容易开环.最终通过D环的氧化和脱保护得到(±)-cervinomycin A2 (1), 再经过还原得到了(±)-cervinomycin A1 (2).
三个小组对(±)-cervinomycins A1 (2)和A2 (1)的全合成工作虽然在效率上有高低之分, 不过也各有特色.
FD-594 (5)是1998年由Kakinuma小组[13, 14]从链霉菌属Streptomyces sp. TA-0256.的培养液中分离得到的, 并通过测定发现其具有优秀的抗细菌活性, 例如对Sta- phylococcus aureus 209-Pj、Staphylococcus epidermidis、Bacillussubtilis ATCC 633的MIC值分别仅为0.10, 0.39和0.39 μg/mL, 同时对HL-60, P388和L1210等多个肿瘤细胞株均表现出了较好的抑制活性, IC50值在0.10~0.25 μg/mL之间.从结构上来说, 不同于大多数多环xanthone天然产物, FD-594 (5)的A环为一个手性内酯环, C环含有2个手性的反式羟基, 整个六环骨架与三糖片段以C—O糖苷键相连.反式邻二醇和糖基片段的存在都使得FD-594 (5)的合成难度相对较高.值得一提的是, FD-594 (5)六环骨架存在溶剂依赖的阻旋异构现象, 使其和糖苷配基以及含有六环骨架的一系列简单衍生物在氯仿和甲醇中的比旋光度正负相反.
2009年, Suzuki小组[23]利用手性传递作为关键策略完成了FD-594 aglycon (60)的不对称全合成, 这是第一例含有六环骨架的多环型xanthone天然产物不对称全合成(Scheme 4).首先, 作者从44开始, 通过溴代、酚保护、醛保护得到了加成前体45, 卤锂交换后对手性环氧化合物46进行开环, 从而引入了手性羟基, 得到47.经过钯催化的插羰酯化反应为关键步骤, 构建了手性内酯, 对官能团和保护基进行调整, 得到了片段49.对于多取代xanthone片段55的制备, 从50开始, 通过芳构化、酯基转化为醛、溴代等8步反应, 得到53, 随后正丁基锂作用过的54对53加成, 再经过4步的官能团转化和氧化态调整得到55.片段49和55发生酯化反应连接2个片段, 并发生分子内的Heck反应形成C环的C—C键连接.使用(S)-缬氨醇[28]对新形成的内酯进行不对称开环反应, 引入了轴手性, 得到手性传递的关键中间体57.经9步转化得到二醛化合物58, 随后在二碘化钐的作用下发生pinacol偶联[29], 从而顺利地构建出了手性的反式邻二醇, 得到C环关环的关键中间体59.最后, 脱除所有保护基, 完成了FD-594aglycon (60)的合成.虽然作者并未完成FD-594 (5)的全合成, 但是通过对苷元的合成, 提供了一种构建C环反式邻二醇结构的解决方案, 即使过程看起来并不是很高效.这对于合成C环具有一个或者两个手性羟基的一类多环xanthone天然产物来说, 具有重要意义.
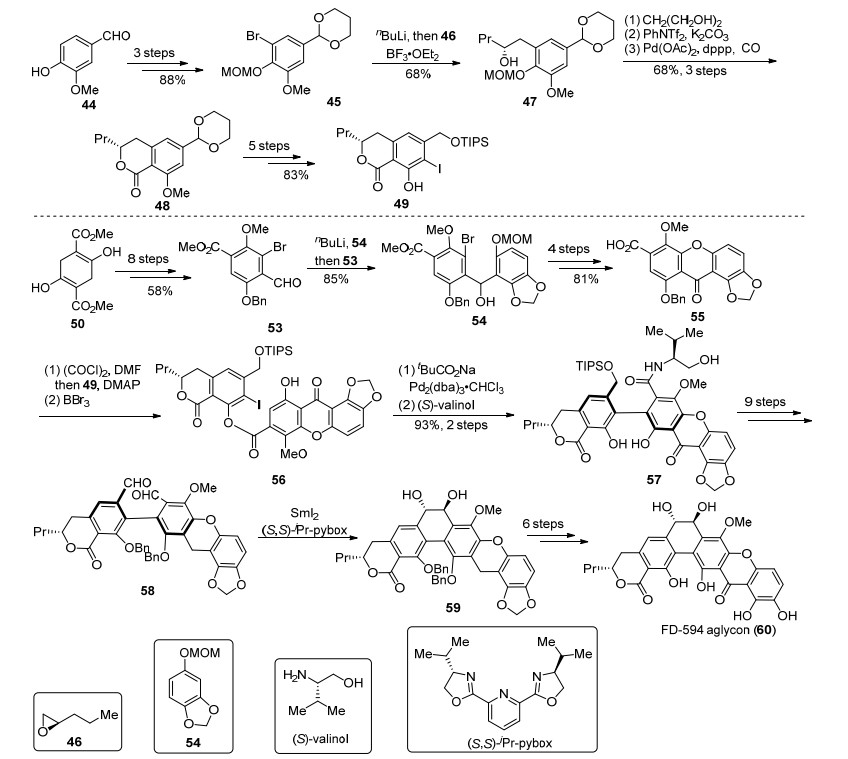
2003年, Canedo和Romero等[15, 16]在对采自西班牙海岸的一系列微生物进行培养和次级代谢产物分离过程中, 从放线菌属Actinomadura sp.的培养液中分离得到了IB-00208.随后他们对IB-00208 (6)进行了活性测试和结构鉴定.IB-00208(6)显示了非常好的对革兰氏阳性菌的抗菌活性, 其中对Staphylococcus aureus、Bacillus subtilis和Micrococcus luteus的MIC值能分别低至1.4, 1.4和0.09nmol/L, 同时对多个肿瘤细胞株也有很好的抑制活性, 其中对P388D1, A-549, HT-29和SKMEL-28的MIC值都能低至1 nmol/L.结构上来说, IB-00208(6)由一个单糖片段和六环核心骨架通过C—O糖苷键连接而成, 其中六环核心骨架含有D环被氧化的全芳环型xanthone单元.
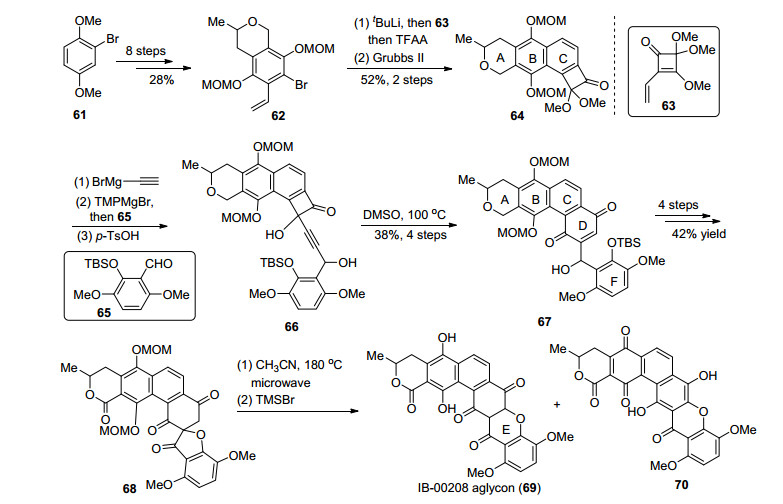
2015年, Martin小组[37]利用Moore重排反应[30]作为关键步骤, 完成了IB-00208 aglycon(69)的合成(Scheme 5).从61出发, 经过8步取代基引入和官能团调整得到了溴代片段62, 卤锂交换之后对63进行加成, 再发生烯烃复分解得到含有四元环的ABC三环中间体64, 通过引入炔基, 并对65加成构建了关键前体66.在DMSO中, 高温加热下发生Moore重排, 能够单一性地得到67.经过4步反应得到螺环中间体68, 在微波作用下再次发生重排得到xanthone结构, 脱去保护基从而完成了IB-00208 aglycon (69)的全合成.不过由于69和70容易异构化, 在脱保护过程中得到的69会不断向70转化, 最终只能制得混合物, 并不能分离.
Kibdelones A~C (图 2)是由Capon小组[8]于2006年从土壤放线菌Kibdelosporangium sp. (MST-108465)的培养液中分离得到的.同时被分离到的还有一系列相应连有Rhamnose单糖的糖苷产物和六环骨架上进一步发生氧化的产物. 2006年, 该小组[31]又分离得到了六环骨架异构的产物isokibdelones A~C及其糖苷化产物. Kibde- lones A~C被分离得到后, 该小组测试了它们的生物活性, 发现它们对革兰氏阳性菌Bacillus subtilis的LD99低至0.1 nmol/L, 线虫Haemonchus contortus的LD99均不大于10 nmol/L, 其中以kibdelone A (8)的活性最高. Kibdelone A (8)还对革兰氏阴性菌Escherichia coli也有很好的抑制活性, LD99为2.7 nmol/L.同时, 它们的抗肿瘤活性测试显示, kibdelones A和B对小细胞肺癌、结肠癌、黑色素瘤、卵巢癌、肾癌和乳腺癌等多个肿瘤细胞株的GI50值都处于1~4 nmol/之间, kibdelone C (10)的活性最高, 均低于1 nmol/L.
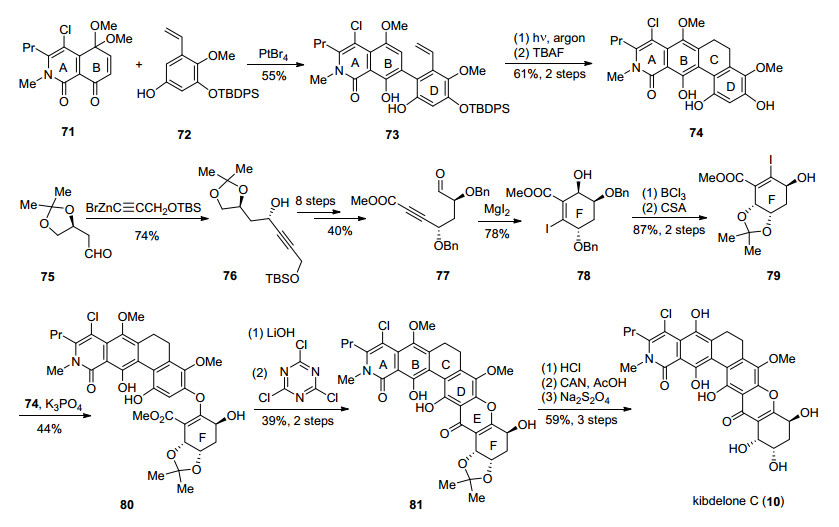
从结构上看, kibdelones A~C的差别在于BC环的氧化态不同, kibdelones A (8)和B (9)的B环为对苯二醌结构, kibdelone C (10)为对苯二酚, 对于C环而言, kibdelone A (8)的C-19和C-20之间为双键, B、C则为单键.这样的结构导致了kibdelones B (9)和C (10)在甲醇溶液中、40 ℃下会缓慢地发生氧化和异构化, 经过18 h之后溶液组分均稳定在A:B:C=3:1:2.除此之外, kibdelones A~C的DEF环属于hexahydroxanthone, F环含有三个手性羟基, 这是合成这类天然产物的主要难点.针对kibdelone系列天然产物, 迄今为止, Porco小组分别于2011年[24]和2013年[32]完成了kibdelones C (10)和A (8)的全合成, Ready小组[25]于2011年完成了kibdelone C (10)对映体的全合成, 高栓虎课题组[27]于2018年完成了kibdelone C (10)的形式合成.
2011年, Porco小组[34]利用Pt(IV)催化的芳构化[33]、光促进的6π电环化以及二碘化镁促进的halo-Michael/ aldol等关键反应制备了合成片段, 通过片段的连接和B环的选择性氧化率先完成了kibdelone C (10)的全合成, 并确证了其绝对构型(Scheme 6).首先, 预制备片段71和72, 通过Pt(IV)催化的芳构化得到ABD三环中间体73, 通过高压汞灯的照射, 发生6π电环化反应成功得到了ABCD四环化合物74. F环的合成是由75开始, 在锌的螯合作用下对醛加成得到手性羟基化合物76, 再经过8步的转化得到关键中间体77, 再通过二碘化镁促进的halo-Michael/aldol反应构建出F环, 得到碘代物78.保护基调整得到79之后, 在碱性条件下74对其加成, 再消除HI, 从而连接了两个片段, 得到oxa-Michael产物80, 通过傅克反应顺利地得到了六环骨架81.在通过氧化/还原得到B环对苯二酚结构时, 作者发现中性条件下硝酸铈铵氧化时, B环和D环不存在选择性, 只能得到混合物; 当在乙酸下时可以选择性地对B环进行氧化得到对苯二醌, 最终经过还原完成了kibdelone C (10)的全合成
2011年, Porco小组[32]在kibdelone C (10)的合成基础上, 通过对合成中的底物和诸多反应进行了优化改良, 高效简洁地实现了kibdelone A (8)的全合成(Scheme 7).首先使用三氯化铟、六氟异丙醇和乙腈混合溶液的反应体系替换了四溴化铂, 使得71和72的芳构化反应收率得到大幅提升.由于kibdelones A (8)和C (10)在环的不饱和度上不同, 在光反应时需要加入碘作为氧化剂[36]来引入C环的碳碳双键, 得到四环化合物82.对于加成/消除底物84的制备则是通过采用并改进了Hudlicky小组[35]发展的酶催化的不对称去芳构化作为关键反应来制备.同时由于C环双键的引入, 导致了底物82相对74反应活性变低, 最终作者通过使用六氟异丙醇酯提高了底物85的反应活性, 顺利的在原有的碱性条件下发生加成/消除, 连接了片段82和85.随后通过傅克酰基化反应关环, 硝酸铈铵选择性氧化B环, 完成了kibdelone A (8)的全合成.
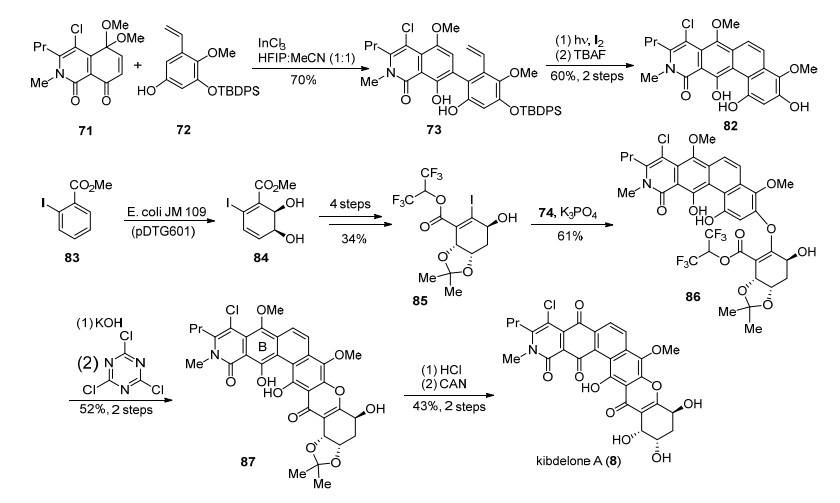
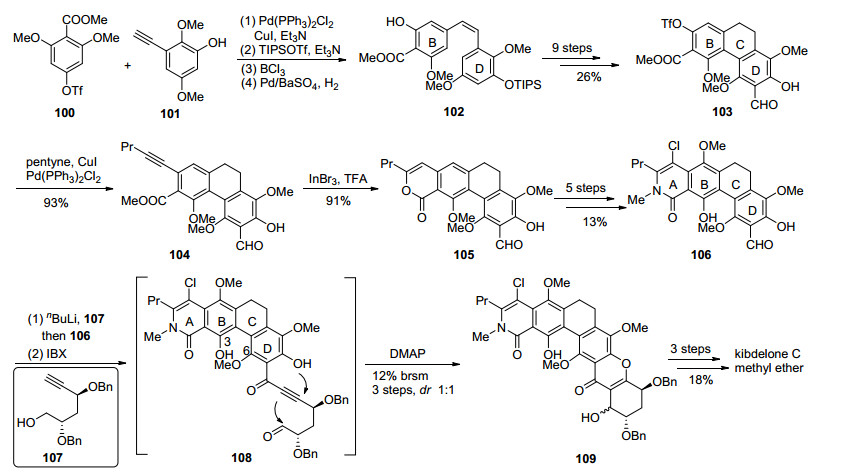
2011年Ready小组[25]与Porco小组同时报道了对kibdelone C (10)的全合成工作, 由于分离小组并未测定kibdelones的绝对构型, 最终Ready小组合成得到是天然kibdelone C (10)的对映体(Scheme 8). Ready小组首先从多取代苯甲酸88开始, 选择了首先构建A环的反式来得到AB双环片段91, 引入的炔基用以连接DEF环.溴代xanthone三环制备则是从92开始, 首先92经过不对称环氧化及其开环[38], 引入F环的所有手性羟基, 得到93, 经过4步转化构建出F环. 94发生卤锂交换, 并对多取代苯甲醛95加成得到96, 沿用Nicolaou小组[39]合成diversonol过程中的构建xanthone三环的方法, 顺利地得到了DEF三环化合物98.片段91和98经过Sonogashira反应偶联、碳碳叁键完全氢化得到的产物在铜(II)络合物催化下发生自由基碘代[40], 并使用Boc保护碘邻位的酚羟基得到99.作者发现游离的酚羟基并不能很好地进行Heck反应, 引入Boc除了能够保护酚羟基, 还能参与金属钯的配位.得到的化合物99经过Heck偶联、氯代、脱保护等步骤最终得到了(-)- kibdelone C (10).
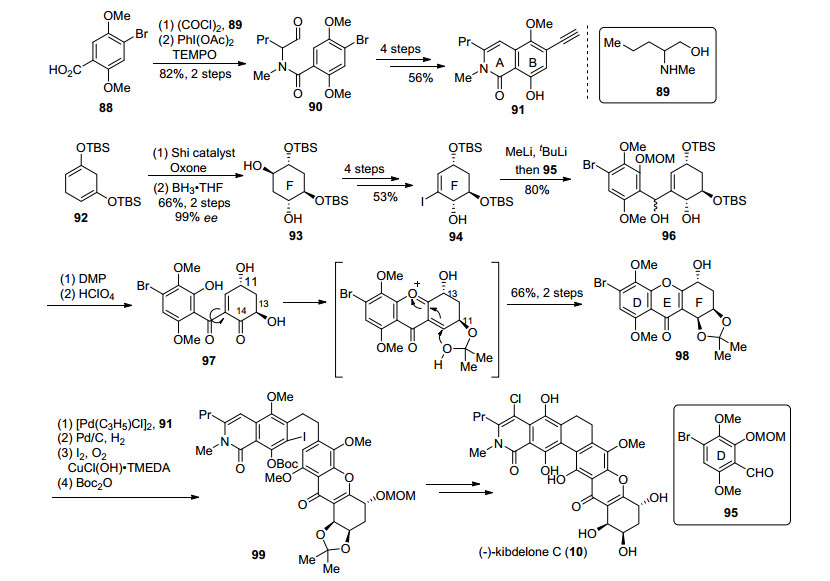
2018年, 本课题组[41]以光促进的6π电环化反应和DMAP催化的oxa-Michael/aldol串联环化反应为关键步骤, 完成了kibdelone C (10)的形式合成(Scheme 9).从简单片段100和101出发, 经过Sonogashira偶联、保护基调整和选择性氢化制得顺式双键中间体102.经过光促进的6π电环化, 构建了BCD三环体系, 对三环取代基进行调整, 得到103, 再经过Sonogashira偶联引入炔基, 三溴化铟催化的内酯化等步骤最终得到加成前体106[42]. 107在过量正丁基锂作用下形成双负离子, 并对106加成, IBX对产物进行氧化, 得到醛基中间体108. Dake小组[41]于2014年发展了DMAP促进的串联环化反应构建xanthone三环体系的方法.利用此方法, 108在DMAP作用下发生串联环化并得到了预期产物109, 不过由于底物结构的原因, 收率较低.在后续的研究中发现, 此串联环化反应效率主要依赖于底物性质和位阻效应, 特别是6位酚羟基保护带来的位阻效应对反应影响尤为严重, 因此109的较低收率可能正是由C环关环后3位羟基和6位甲氧基保护带来的位阻效应所致.最终109经过3步转化得到了kibdelone C (10)的甲氧基醚, 通过与Porco和Ready小组中间体的比对确认, 完成了kibdelone C (10)的形式合成.
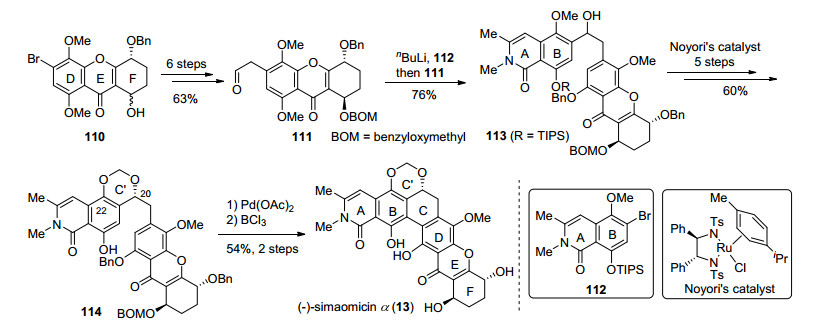
simaomicin α (13)是由美国氰胺公司科学家于1990年从一种马杜拉放线菌的培养液中分离得到的[9].其具有非常优良的抗球虫活性, 被分离组誉为“已报道的天然产物中最有潜力的抗球虫药”, 对病鸡饲喂1 mg/kg即可达到治疗效果.同时其对多种革兰氏阳性菌Staphylococcus aureus、S. epidermidis、Streptococcus faecalis、Streptococcus (Enterococcus sp.)、S. mutans和S. sanguis都有良好的抗菌活性, MIC值在0.06 μg/mL以下, 但对革兰氏阴性菌的抗菌活性并不明显.从结构上来说, simaomicin α (13)与kibdelone C (10)差异较大, 前者C环具有一个手性羟基, 并与B环酚羟基共同形成了缩醛六元环C', A环侧链为甲基, F环少一个羟基.
2013年Ready小组在kibdelone C (10)的合成工作基础上完成了(-)-simaomicin α (13)的全合成(Scheme 10).首先利用已有方法制备了DEF三环片段110, 并通过6步反应将其转化为醛111.片段112经过卤锂交换后, 对醛基片段111进行加成, 顺利的实现了片段的连接, 使用Noyori催化剂[43]对羰基的还原构建了C环的手性羟基, 并在碱性条件下发生烷基化, 构建了六元缩醛环, 得到114.最后通过钯(II)促进的脱氢偶联实现了C环的关环, 最后脱去保护基从而完成了(-)-simao- micin α (13)的全合成. Ready小组沿用了诸多(-)- kibdelone C (10)合成工作中的方法, 快速高效地实现了片段的合成, 同时C环手性羟基和六元缩醛环的构建方法也为多环xanthone其它成员的合成, 提供了很高的参考价值.
kigamicins (7)是2003年由Kunimoto小组[10, 11]于链霉菌属Amycolatopsis sp. ML630-mF1的培养液中分离得到的, 包含A~E一共5个成员, 它们的不同仅仅在于糖片段的种类和数目差异. 2005年, 通过kigamicin A的X单晶衍射确定了kigamicins A、C和D相对构型, 再通过降解得到的糖与D-amicetose比对, 确定了其绝对构型.由于kigamicins B和E的含量较低, 未分离得到足够的样品, 不能确定它们的绝对构型, 不过依据相同的生化途径, kigamicins A~E在绝对构型上可能是保持一致的. Kigamicins (7)具有良好而独特的抗肿瘤活性, 对PANC-1细胞具有选择性的细胞毒性, 营养饥饿相较正常营养条件下要强100倍.除此之外, kigamicins A~E对许多革兰氏阳性菌具有广泛而强烈的抗菌活性, 包括耐甲氧西林的金黄色葡萄球菌, 对某些细菌的MIC能达到0.025 μg/mL.从结构上来说, kigamicins (7)具有一个高氧化态hexahydroxanthone在内的8环骨架, 具有5个手性中心, 8环骨架中还包含有独特的四氢噁唑环A'和六元缩甲醛环C', 合成难度相对较大.
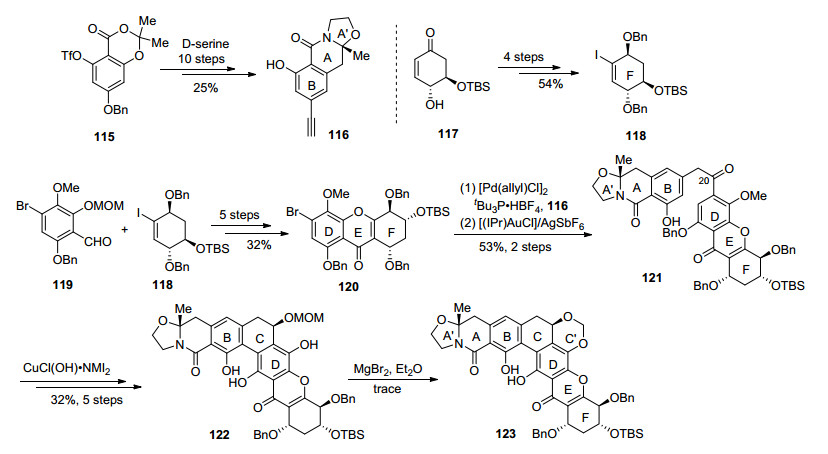
2019年, Ready小组[44]报道了对kigamicins (7)的合成研究工作(Scheme 11).首先依据Hosokawa小组合成TMC-66的方法[45], 从115开始, 经过与D-丝氨酸缩合、脱羧等关键步骤, 10步制备了炔基片段116.从奎宁酸出发[46]制备得到的117, 经过4步制得碘代多羟基化合物118, 完成了对三个手性羟基的构建, 卤锂交换后对119加成, 5步转化后得到了DEF三环片段120. Sonogashira偶联连接116和120片段, 金催化的区域选择性的炔基水合反应[47], C-20位转化为羰基, 使用Noyori催化剂[43]立体选择性地还原构建出C-20的手性羟基, 使用铜(II)复合物促进的氧化偶联[45, 48], 顺利地实现了C环的关环得到122.在尝试碱性条件下通过烷基化构建C'环失败后, 作者希望利用溴化镁和保护基MOM作用, 通过路易斯酸促进的氧鎓途径实现C'的关环, 不过最后只得到了痕量产物, 并不能进行充分的结构鉴定, 使得对kigamicins (7)的合成折戟于此.虽然作者最终并未完成kigamicins (7)或其糖苷配基的全合成, 但是制备了大量片段和高级中间体, 对以后完成kigamicins (7)的合成工作有着重要的参考价值.
介绍了多环型xanthone类天然产物的分离、活性以及截止目前已发表的相关全合成工作.由于它们良好的生物活性和独特的化学结构, 不断地吸引着大量的学者对这一类天然产物进行分离鉴定、活性研究以及合成研究.虽然目前对这类天然产物已经取得了丰富的研究成果, 但是仍然有很多的未知需要我们去探索, 而且在如今这个抗生素研发乏力和“超级细菌”不断涌现的时代, 具有良好抗菌活性的多环型xanthone类天然产物被赋予了更多的潜在价值.
Masters, K. S.; Brase, S. Chem. Rev. 2012, 112, 3717. doi: 10.1021/cr100446h
(a) Cheng, G.; Sun, J.; Fridlender, Z. G.; Ching, L. C. S.; Wang, L. M.; Albelda, S. M. J. Biol. Chem. 2010, 285, 10553.
(b) Gobbi, S.; Zimmer, C.; Belluti, F.; Hartmann, R. W.; Rampa, A.; Recanatini, M.; Bisi, A. J. Med. Chem. 2010, 53, 5347.
(c) Palmeira, A.; Paiva, A.; Sousa, E.; Seca, H.; Almeida, G. M.; Lima, R. T.; Fernandezs, M. X.; Pinto, M.; Vasconcelos, M. H. Chem. Biol. Drug Des. 2010, 76, 43.
Honda, N. K.; Pavan, F. R.; Coelho, R. G.; Micheletti, A. C.; de Andrade Leite, S. R.; Lopes, T. I. B.; Beatriz, A.; Mitsutsu, M. Y.; Brum, R. L.; Leite, C. Q. F. Phytomedicine 2010, 17, 328. doi: 10.1016/j.phymed.2009.07.018
Pinto, M. M.M.; Sousa, M. E.; Nascimento, M. S. Curr. Med. Chem. 2005, 12, 2517. doi: 10.2174/092986705774370691
Lesch, B.; Bräse, S. Angew. Chem., Int. Ed. 2004, 43, 115. doi: 10.1002/anie.200352154
Winter, D. K.; Sloman, D. L.; Porco, J. Nat. Prod. Rep. 2013, 30, 382. doi: 10.1039/c3np20122h
Peoples, A. J.; Zhang, Q.; Millet, W. P.; Rothfeder, M. T.; Pescatore, B. C.; Madden, A. A.; Ling, L.; Moore, C. M. J. Antibiot. 2008, 61, 457. doi: 10.1038/ja.2008.62
(a) Ratnayake, R.; Lacey, E.; Tennant, S.; Gill, J. H.; Capon, R. J. Org. Lett. 2006, 8, 5267.
(b) Ratnayake, R.; Lacey, E.; Tennant, S.; Gill, J. H.; Capon, R. J. Chem.-Eur. J. 2007, 13, 1610.
(a) Lee, T. M.; Carter, G. T.; Borders, D. B. J. Chem. Soc., Chem. Commun. 1989, 22, 1771.
(b) Korshalla, J.; Maiese, W. M.; Goodman, J.; Torrey, M. J.; Kantor, S.; Labeda, D. P.; Greenstein, M. J. Antibiot. 1990, 43, 1059.
Kunimoto, S.; Lu, J.; Esumi, H.; Yamazaki, Y.; Kinoshita, N.; Honma, Y.; Hamada, M.; Ohsono, M.; Ishizuka, M.; Takeuchi, T. J. Antibiot. 2003, 56, 1004. doi: 10.7164/antibiotics.56.1004
Kunimoto, S.; Someno, T.; Yamazaki, Y.; Lu, J.; Esumi, H.; Naganawa, H. J. Antibiot. 2003, 56, 1012. doi: 10.7164/antibiotics.56.1012
Terui, Y.; Chu, Y.; Li, J.-Y.; Ando, T.; Yamamoto, H.; Kawamura, Y.; Tomishima, Y.; Uchida, S.; Okazaki, T.; Munetomo, E.; Seki, T.; Yamamoto, K.; Murakami, S.; Kawashima, A. Tetrahedron Lett. 2003, 44, 5427. doi: 10.1016/S0040-4039(03)01318-2
Qiao, Y.-F.; Okazaki, T.; Ando, T.; Mizoue, K.; Kondo, K.; Eguchi, T.; Kakinuma, K. J. Antibiot. 1998, 51, 282. doi: 10.7164/antibiotics.51.282
Kondo, K.; Eguchi, T.; Kakinuma, K.; Mizoue, K.; Qiao, Y.-F. J. Antibiot. 1998, 51, 288. doi: 10.7164/antibiotics.51.288
Malet-Cascon, L.; Romero, F.; Espliego-Vazquez, F.; Gravalos, D.; Fernandez-Puentes, J. L. J. Antibiot. 2003, 56, 219. doi: 10.7164/antibiotics.56.219
Rodriguez, J. C.; Puentes, J. L. F.; Baz, J. P.; Canedo, L. M. J. Antibiot. 2003, 56, 318. doi: 10.7164/antibiotics.56.318
Omura, S.; Iwai, Y.; Hinotozawa, K.; Takahashi, Y.; Kato, J.; Nakagawa, A.; Hirano, A.; Shimizu, H.; Haneda, K. J. Antibiot. 1982, 35, 645. doi: 10.7164/antibiotics.35.645
Omura, S.; Nakagawa, A.; Kushida, K.; Lukacs, G. J. Am. Chem. Soc. 1986, 108, 6088. doi: 10.1021/ja00279a095
Maiese, W. M.; Lechevalier, M. P.; Lechevalier, H. A.; Korshalla, J.; Goodman, J.; Wildey, M. J.; Kuck, N.; Greenstein, M. J. Antibiot. 1989, 42, 846. doi: 10.7164/antibiotics.42.846
Carter, G. T.; Nietsche, J. A.; Williams, D. R.; Borders, D. B. J. Antibiot. 1990, 43, 504. doi: 10.7164/antibiotics.43.504
Kelly, T. R.; Jagoe, C. T.; Li, Q. J. Am. Chem. Soc. 1989, 111, 4522. doi: 10.1021/ja00194a071
(a) Rao, A. V. R.; Yadav, J. S.; Reddy, K. K.; V., U. Tetrahedron Lett. 1991, 32, 5199.
(b) Mehta, G.; Shah, S. R.; Venkateswarlu, Y. Tetrahedron 1994, 50, 11729.
Masuo, R.; Ohmori, K.; Hintermann, L.; Yoshida, S.; Suzuki, K. Angew. Chem., Int. Ed. 2009, 48, 3462. doi: 10.1002/anie.200806338
Sloman, D. L.; Bacon, J. W.; Porco, J. A. J. Am. Chem. Soc. 2011, 133, 9952. doi: 10.1021/ja203642n
Butler, J. R.; Wang, C.; Bian, J.; Ready, J. M. J. Am. Chem. Soc. 2011, 133, 9956. doi: 10.1021/ja204040k
Wang, Y.; Wang, C.; Butler, J. R.; Ready, J. M. Angew. Chem., Int. Ed. 2013, 52, 10796. doi: 10.1002/anie.201304812
Dai, Y.; Ma, F.; Shen, Y.; Xie, T.; Gao, S. Org. Lett. 2018, 20, 2872. doi: 10.1021/acs.orglett.8b00901
(a) Bringmann, G.; Breuning, M.; Tasler, S. Synthesis 1999, 525.
(b) Bringmann, G.; Mortimer, J. P.; Keller, P. A.; Gresser, M. J.; Garner, J.; Breuning, M. Angew. Chem., Int. Ed. 2005, 44, 5384.
Tamiya, M.; Ohmori, K.; Kitamura, M.; Kato, H.; Arai, T.; Oorui, M.; Suzuki, K. Chem.-Eur. J. 2007, 13, 9791. doi: 10.1002/chem.200700863
(a) Karlsson, J. O.; Nguyen, N. V.; Foland, L. D.; Moore, H. W. J. Am. Chem. Soc. 1985, 107, 3392.
(b) Foland, L. D.; Karlsson, J. O.; Perri, S. T.; Schwabe, R.; Xu, S.; Patil, S.; Moore, H. W. J. Am. Chem. Soc. 1989, 111, 975.
Ratnayake, R.; Lacey, E.; Tennant, S.; Gill, J. H.; Capon, R. J. Org. Lett. 2006, 8, 5267. doi: 10.1021/ol062113e
Winter, D. K.; Endoma-Arias, M. A.; Hudlicky, T.; Beutler, J. A.; Porco, J. A. J. Org. Chem. 2013, 78, 7617. doi: 10.1021/jo401169z
Sloman, D. L.; Mitasev, B.; Scully, S. S.; Beutler, J. A.; Porco, J. A. Angew. Chem., Int. Ed. 2011, 50, 2511. doi: 10.1002/anie.201007613
(a) Wei, H.-X.; Timmons, C.; Farag, M. A.; Pare, P. W.; Li, G. Org. Biomol. Chem. 2004, 2, 2893.
(b) Wei, H.-X.; Hu, J.; Jasoni, R. L.; Li, G.; Pare, P. W. Helv. Chim. Acta 2004, 87, 2359.
(c) Dai, Y.; Shen, Y.; Gao, S. Chin. J. Org. Chem. 2018, 38, 1608(in Chinese).
(代义华, 申艳芳, 高栓虎, 有机化学, 2018, 38, 1608.)
Endoma, M. A. A.; Bai, V. P.; Hansen, J.; Hudlicky, T. Org. Process Res. Dev. 2002, 6, 525. doi: 10.1021/op020013s
(a) Liu, L.; Yang, B.; Katz, T. J.; Poindexter, M. K. J. Org. Chem. 1991, 56, 3769.
(b) Talele, H. R.; Gohil, J.; Bedekar, A. V. Bull. Chem. Soc. Jpn. 2009, 82, 1182.
Yang, J.; Knueppel, D.; Cheng, B.; Mans, D.; Martin, S. F. Org. Lett. 2015, 17, 114. doi: 10.1021/ol503336t
Shi, Y. Acc. Chem. Res. 2004, 37, 488. doi: 10.1021/ar030063x
Nicolaou, K. C.; Li, A. Angew. Chem., Int. Ed. 2008, 47, 6579. doi: 10.1002/anie.200802632
(a) Li, X.; Hewgley, J. B.; Mulrooney, C. A.; Yang, J.; Kozlowski, M. C. J. Org. Chem. 2003, 68, 5500.
(b) Nakajima, M.; Miyoshi, I.; Kanayama, K.; Hashimoto, S.; Noji, M.; Koga, K. J. Org. Chem. 1999, 64, 2264.
Castillo-Contreras, E. B.; Dake, G. R. Org. Lett. 2014, 16, 1642. doi: 10.1021/ol5002945
(a) Sakai, N.; Annaka, K.; Konakahara, T. J. Org. Chem. 2006, 71, 3653.
(b) Bianchi, G.; Chiarini, M.; Marinelli, F.; Rossi, L.; Arcadi, A. Adv. Synth. Catal. 2010, 352, 136.
Noyori, R.; Hashiguchi, S. Acc. Chem. Res. 1997, 30, 97. doi: 10.1021/ar9502341
Ma, A. J.; Ready, J. M. Org. Lett. 2019, 21, 1148. doi: 10.1021/acs.orglett.9b00098
Hosokawa, S.; Fumiyama, H.; Fukuda, H.; Fukuda, T.; Seki, M.; Tatsuta, K. Tetrahedron Lett. 2007, 48, 7305. doi: 10.1016/j.tetlet.2007.08.037
Barros, M. T.; Maycock, C. D.; Ventura, M. R. Chem.-Eur. J. 2000, 6, 3991. doi: 10.1002/1521-3765(20001103)6:21<3991::AID-CHEM3991>3.3.CO;2-M
Marion, N.; Ramon, R. S.; Nolan, S. P. J. Am. Chem. Soc. 2009, 131, 448. doi: 10.1021/ja809403e
Noji, M.; Nakajima, M.; Koga, K. Tetrahedron Lett. 1994, 35, 7983. doi: 10.1016/S0040-4039(00)78402-4

图式 1 Kelly小组对(±)-cervinomycins A1和A2的全合成
Scheme 1 Total synthesis of (±)-cervinomycins A1 and A2 by Kelly's group

图式 2 Rao小组对(±)-cervinomycins A1和A2的全合成
Scheme 2 Total synthesis of (±)-cervinomycins A1 and A2 by Rao's group

图式 3 Mehta小组对(±)-cervinomycins A1和A2的全合成
Scheme 3 Total synthesis of (±)-cervinomycins A1 and A2 by Mehta's group

图式 4 Suzuki小组对FD-594 aglycon的全合成
Scheme 4 Total synthesis of FD-594 aglycon by Suzuki's group

图式 5 Martin小组对IB-00208 aglycon的全合成
Scheme 5 Total synthesis of IB-00208 aglycon by Martin's group

图式 10 Ready小组对(-)-simaomicin α的全合成
Scheme 10 Total synthesis of (-)-simaomicin α by Ready's group

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:





