图 1.
SAM、SAH以及DOT1L抑制剂结构
Figure 1.
Structures of SAM, SAH, and DOT1L inhibitors

DOT1L(类端粒沉默干扰体1)是一种不含SET (histone lysine methyltransferases)结构域的组蛋白赖氨酸甲基转移酶(HKMTs), 它特异性催化位于核小体中的组蛋白H3上赖氨酸79 (H3K79)的单、双和三甲基化. S-腺苷-L-甲硫氨酸(SAM)作为甲基供体, 在HKMTs作用下将其甲基转移至底物, 生成相应的酶促反应产物S-腺苷-L-高半光氨酸(SAH)(图 1)[1-2]. DOT1L的活性异常, 可以导致HOXA9与MEIS1高表达, 诱发混合谱系白血病(Mixed Lineage Leukemia, MLL)的发生, 是这种恶性肿瘤白血病发生的必需驱动因子[3-4].因此, 通过抑制DOT1L有望实现混合谱系白血病MLL的治疗[5-7].
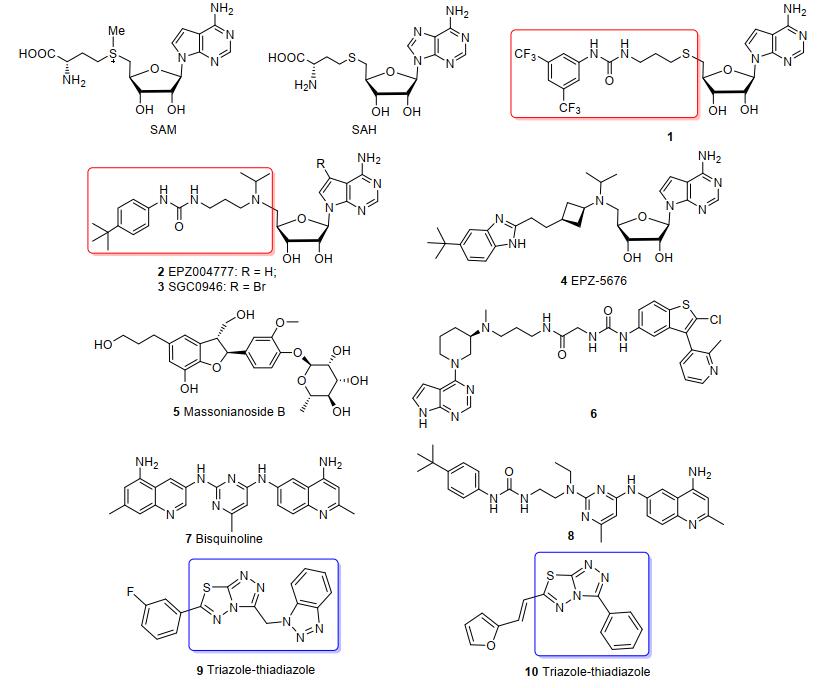
目前, 已报道的DOT1L抑制剂按结构分为两类, 一类是基于SAM及其相应的酶促反应产物SAH结构设计的核苷类DOT1L抑制剂[8-15], 如化合物1~4(图 1); 另一类则是非核苷类DOT1L抑制剂[16-24], 如化合物5~10(图 1).
Epizyme公司报道的化合物1~3分子中含有脲基侧链, 该链较SAM中的半胱氨酸链能更好地与DOT1L催化域口袋匹配, 使分子表现出更高效的DOT1L抑制活性.化合物4和6[19]中也均含有类似的氨基侧链.化合物7[21]、9[22]和10[23]是中国科学院上海药物研究所近年来报道的双喹啉类和三氮唑并噻二唑类DOT1L抑制剂.分子模拟结果表明, 化合物7的喹啉连嘧啶部分可以有效占据SAM的结合位点, 从而表现出良好的DOT1L抑制活性.依据优势片段结合的药物设计方法, 实验室设计合成了新的连接有氨基脲链的喹啉连嘧啶化合物8[24], 亦表现了良好的DOT1L抑制活性, 并显著地提高了化合物的抗肿瘤细胞活性.
在此工作基础上, 因化合物9和10中的三氮唑并噻二唑部分主要也是与SAM结合位点作用, 因此, 本文依据优势片段结合的药物设计方法, 拟设计合成新的连接有氨基脲链的三氮唑并噻二唑结构衍生物, 并测试化合物的DOT1L酶抑制活性, 以期发现高效的DOT1L抑制剂.
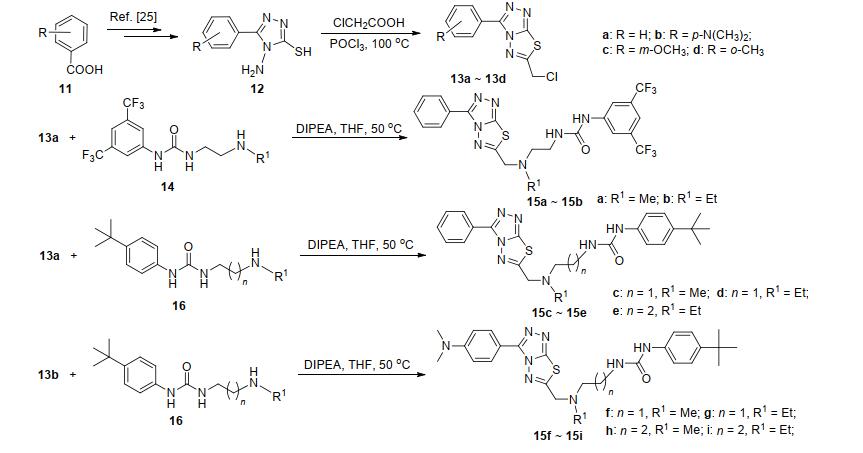
参照文献方法[25], 以芳香酸11为起始原料, 经酯化、肼解、二硫化碳(CS2)成盐及水合肼关环四步反应, 得到4-氨基-5-巯基-1, 2, 4-三唑衍生物12.在三氯氧磷条件下, 化合物12与氯乙酸脱水环合得到含苄氯的三氮唑并噻二唑关键中间体13.弱碱性条件下, 化合物13a和13b分别与已有的氨基脲链14和16[24], 经亲核取代反应合成了目标化合物15a~15i (Scheme 1).
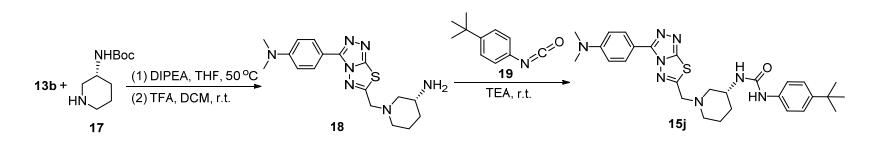
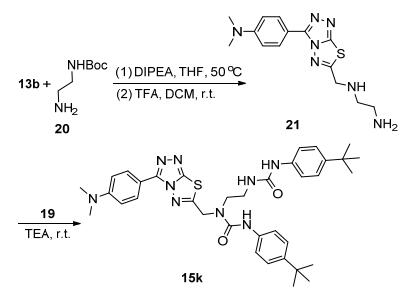
为拓展氨基上的R1取代基团, 叔丁氧羰基(Boc)保护的3-氨基哌啶17与13b发生亲核取代反应, 酸性条件下脱除Boc后得到化合物18, 再与4-叔丁基苯基异氰酸酯19反应, 合成了目标化合物15j (Scheme 2).此外, 以Boc保护的乙二胺和13b为原料, 经上述同样的步骤合成了化合物21, 继而与19反应得到含双脲基链的化合物15k (Scheme 3).由于氨基与异氰酸酯反应活性高, 调整试剂的比例、缚酸剂和反应温度, 均没有得到含单脲基链的产物.
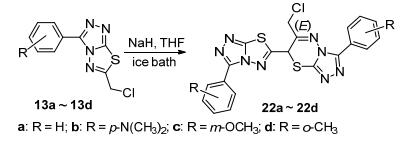
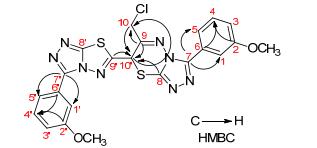
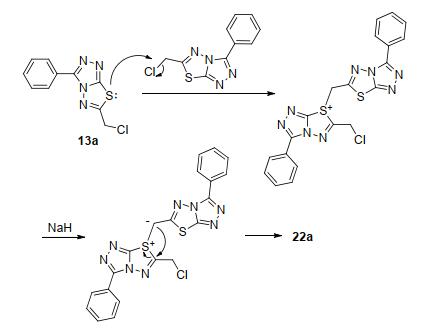
在合成化合物15a时, 强碱如氢化钠(NaH)条件下, 化合物13a和氨基脲链14不能反应制备目标化合物 15a, 而是生成了13a的二聚类似物22a (Scheme 4).随后, 直接以13a为原料, 室温NaH条件下进行反应1 h, 除生成22a和一个极性大难以展开分离的副产物外, 无其它杂质生成, 但反应收率不高, 只有31.7%.经简单条件摸索, 在冰浴条件下反应时间延长至3 h, 收率可达70.5%.在相同的条件下, 分别以13b~13d为原料, 高产率地合成了相应的二聚类似物22b~22d.该类化合物结构经波谱分析鉴定为三氮唑并噻二唑连三氮唑并噻二嗪类衍生物.以化合物22c的结构鉴定为例, 高分辨质谱确定该化合物的分子式为C22H18ClN8O2S2 ([M+H]+), 即两分子的原料减去一个氯化氢.通过1H, 1H-COSY和HSQC二维核磁共振(NMR)谱确定C、H信号归属后, 氢谱中δ 6.26处的单峰和碳谱中δ 36.8处的碳信号显示噻二嗪环的存在.进一步通过HMBC谱和一些关键的相关信号峰(图 2), 如C-9'、C-8、C-9、C-10/10'-H以及C-10'、C-9/10-H等, 确证化合物结构如图 2所示.
推测可能的反应机理如Scheme 5所示: 13a中的硫原子亲核性强, 取代另一分子中较活泼的相当于苄位的氯原子, 给出硫鎓; 硫正离子的形成, 活化了与其直接相连的“苄位”, 使得该位置的氢酸度加大, 更易被NaH夺去形成硫叶立德; 后者重排扩环得到三氮唑并噻二唑连三氮唑并噻二嗪类化合物22a.
对所合成的化合物15a~15k和22a~22d进行了DOT1L酶抑制活性测试[24], 化合物10为阳性对照. 50 µmol•L-1浓度时抑制率75%以上的化合物, 进一步测试了其IC50值, 结果见表 1.由表 1可知, 所测试化合物50 µmol•L-1时大多表现出中等或较弱的DOT1L抑制活性, 均低于阳性对照化合物10的酶抑制活性, 其半数抑制浓度(IC50)值为7.25 µmol•L-1.初步的构效关系分析表明, 三氮唑并噻二唑部分所连接的氨基脲侧链的体积较大, 可能不利于分子与酶的结合, 导致所合成的化合物活性要低于阳性对照化合物.含双脲基链的化合物15k, 在50 µmol•L-1时其DOT1L抑制活性为79.62%, IC50值为25.92 µmol•L-1, 活性相对较好, 这可能是15k中两个叔丁基的疏水性较强, 使其更有利于与DOT1L结合, 导致活性略有增强.二聚类似物22a~22d在50 µmol•L-1时也表现出一定的DOT1L抑制活性, 其中22a表现出了较好的酶抑制活性, IC50为10.59 µmol•L-1, 是所测四个二聚类似物中活性最好的, 提示这可能是一类新结构的DOT1L抑制剂.进一步的合成活性研究正在进行之中.
 下载:
导出CSV
下载:
导出CSV
| Compd. | Inhibition rate/% | IC50/(μmol"L-1) |
| 15a | 31.35 | —a |
| 15c | 0.32 | — |
| 15e | 11.35 | — |
| 15g | 22.02 | — |
| 15i | 62.35 | — |
| 15k | 79.62 | 25.92 |
| 22b | 69.86 | — |
| 22d | 39.36 | — |
| 15b | 11.04 | — |
| 15d | 3.21 | — |
| 15f | 50.27 | — |
| 15h | 37.43 | — |
| 15j | 55.47 | — |
| 22a | 86.16 | 10.59 |
| 22c | 47.12 | — |
| 10 | 86.71 | 7.25 |
| a Tested concentration at 50 µmol•L-1, “—” undertermined. | ||
为了更直观地分析化合物10、15k、15i和22a的构效关系, 通过分子对接测试了以上4个化合物与DOT1L的可能结合模式[26-28].因化合物15k和15i结构中含有与EPZ004777 (2, 图 1)相似的氨基脲侧链, 所以对接的大分子选用了DOT1L与EPZ004777的复合物结构(PDB ID:4EKI)[13], 结果如图 3所示.结合模式分析显示:化合物10, 15k, 15i和22a均能较好地占据DOT1L的SAM结合口袋.与其他化合物相比, 阳性对照化合物10的三氮唑并噻二唑连接的丙烯呋喃基团更小、更容易进入口袋, 同时苯基与F243具有Pi-Pi堆积相互作用, 从而使得化合物10能更好地占据DOT1L的SAM结合口袋.而化合物22a的三氮唑并噻二唑连接的三氮唑并噻二嗪基团较大, 不易进入延伸结合口袋致其活性减小, 这与实验测得的10活性优于22a的结果一致.对于连接有氨基脲链的衍生物15i和15k而言, 其氨基脲侧链空间位阻较大, 亦使其不容易进入SAM的延伸结合口袋; 并且三氮唑环上体积突出, 且易带正电的N, N-二甲基苯胺基团, 使得15i和15k的活性更弱于10和22a; 但化合物15k中两个叔丁基的疏水性较强, 有利于其与DOT1L结合, 使得15k的活性要优于化合物15i的.
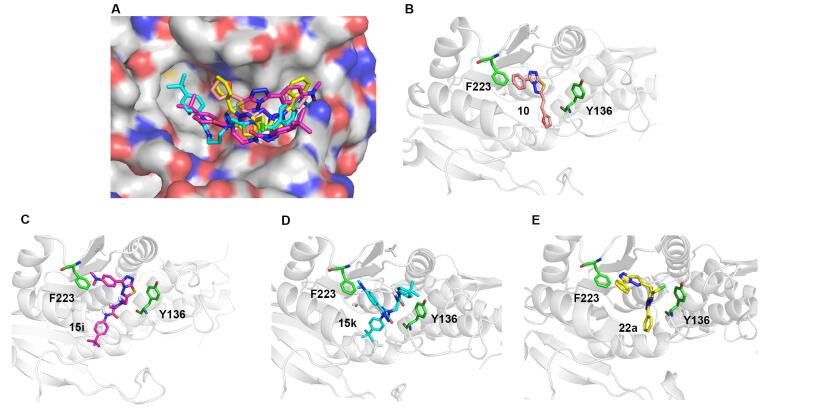
(A) Compounds 10, 15i, 15k and 22a were aligned with SAM (salmon) in the SAM binding pocket of DOT1L (4EKI); (B) 10; (C) 15i; (D) 15k; (E) 22a
依据优势片段结合的药物设计理念, 设计合成了新的连接有氨基脲链的三氮唑并噻二唑衍生物15a~15k, 以及三氮唑并噻二唑连三氮唑并噻二嗪类结构衍生物22a~22d.化合物在50 µmol•L-1时, 表现出中等或较弱的DOT1L抑制活性, 其中化合物15k和二聚类似物22a是所测化合物中, 两个活性最好的衍生物, IC50值分别为25.92和10.59 µmol•L-1.分子对接实验提示, 氨基脲侧链的空间位阻可能阻碍了分子与酶的结合, 致使其活性降低.
熔点由SGW® X-4显微熔点仪(温度计未校正)测定; 核磁共振谱用BRUKER AC-P600 (600 MHz)型核磁共振仪、Brucker advance II 400 FT-NMR型核磁共振仪, 内标为TMS; 质谱(ESI)用Agilent G6300离子阱液相质谱联用仪; 高分辨质谱(ESI)用FTICR-MS (Ionspec 7.0T)型质谱仪测定; 层析用硅胶(200~300目)为青岛海洋化工厂产品.本工作所用其他试剂均为分析纯, 无水试剂均按常规方法处理, 水为二次蒸馏水.
化合物12a[25] (3.8 g, 20 mmol)和60 mL三氯氧磷置于250 mL圆底烧瓶中, 加入1.2 equiv.氯乙酸升温至100 ℃反应10 h.待薄层色谱(TLC)检测反应完全, 加50 mL水淬灭三氯氧磷, 然后用40%氢氧化钠溶液中和溶液至中性, 析出棕黄色固体, 减压抽滤.滤饼用水(10 mL×2)洗涤, 然后柱层析分离[V(乙酸乙酯):V(二氯甲烷)=1:10]得到化合物13a.
按相同的方法, 分别以12b~12d为原料, 得到了相应的产物13b~13d.
6-(氯甲基)-3-苯基-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑(13a):无色油状, 产率53.2%. 1H NMR (600 MHz, CDCl3) δ: 8.22 (d, J=8.4 Hz, 2H), 7.46 (d, J=6.6 Hz, 3H), 4.84 (s, 2H); 13C NMR (100 MHz, CDCl3) δ: 166.0, 154.6, 146.5, 130.5, 128.9, 126.3, 125.2, 39.6; HR-ESI- MS calcd for C10H8ClN4S [M+H]+ 251.0158, found 251.0153.
4-{6-(氯甲基)-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-3-基}-N, N-二甲基苯胺(13b):黄色固体, 产率51.0%. m.p. 166.5~167.7 ℃; 1H NMR (600 MHz, CDCl3+CD3OH) δ: 8.01 (d, J=9.0 Hz, 2H), 6.71 (d, J=8.4 Hz, 2H), 4.86 (s, 2H), 2.99 (s, 6H); 13C NMR (100 MHz, CDCl3+CD3OH) δ: 169.8, 157.7, 155.7, 151.2, 131.5, 115.7, 115.6, 115.4, 43.7, 43.4; HR-ESI-MS calcd for C12H13ClN5S [M+H]+ 294.0580, found 294.0586.
6-(氯甲基)-3-(3-甲氧基苯基)-[1, 2, 4]三唑并[3, 4-b]- [1, 3, 4]噻二唑(13c):白色固体, 产率49.7%. m.p. 124.7~125.5 ℃; 1H NMR (600 MHz, CDCl3) δ: 7.84 (d, J=24.0 Hz, 2H), 7.40~7.38 (m, 1H), 7.00 (d, J=6.0 Hz, 1H), 4.85 (s, 2H), 3.86 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 166.0, 159.9, 154.6, 146.4, 130.0, 126.4, 118.7, 116.8, 111.4, 55.5, 39.6; HR-ESI-MS calcd for C11H10ClN4OS [M+H]+ 281.0264, found 281.0272.
6-(氯甲基)-3-(邻甲苯基)-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑(13d):无色油状, 产率57.8%. 1H NMR (600 MHz, CDCl3) δ: 7.76 (d, J=7.8 Hz, 1H), 7.39 (t, J=7.8 Hz, 1H), 7.35 (d, J=7.2 Hz, 1H), 7.31 (t, J=7.8 Hz, 1H), 4.80 (s, 2H), 2.53 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 165.7, 154.1, 147.1, 138.1, 131.3, 130.5, 129.4, 125.9, 124.4, 39.6; HR-ESI-MS calcd for C11H10ClN4S [M+H]+ 265.0315, found 265.0322.
于50 mL圆底烧瓶中, 称取13a (0.1 g, 0.5 mmol)溶于15 mL四氢呋喃(THF)中, 加入N, N-二异丙基乙胺(DIPEA, 348 µL, 4.0 equiv.)和氨基脲链14[24] (0.2 g, 1.2 equiv), 升温至50 ℃反应36 h. TLC监测反应完全后, 旋蒸除去THF.残留物用10 mL二氯甲烷溶解, 依次用0.1 mol/L盐酸(5 mL×3)和水(5 mL×3)洗, 以无水硫酸钠干燥, 浓缩的残留物经硅胶色谱柱分离[V(乙酸乙酯):V(二氯甲烷)=2:1]得到白色固体化合物15a.
按相同的方法, 分别以13a和13b, 以及氨基脲链14和16[24]为原料, 得到相应的产物15b~15i.
1-[3, 5-二(三氟甲基)苯基]-3-{2-{甲基[(3-苯基- [1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基]氨基}乙基}脲(15a):白色固体, 产率33.4%. m.p. 146.3~147.8 ℃; 1H NMR (600 MHz, CDCl3) δ: 9.41 (s, 1H), 8.12 (d, J=7.8 Hz, 2H), 7.97 (s, 2H), 7.49~7.44 (m, 3H), 7.38 (s, 1H), 6.92 (s, 1H), 3.92 (s, 2H), 3.55 (m, 2H), 2.80 (t, J=5.4 Hz, 2H), 2.43 (s, 3H); 13C NMR (150 MHz, CDCl3) δ: 174.6, 155.6, 155.0, 146.5, 141.8, 132.0, 131.8, 131.1, 129.2, 126.3, 124.5, 124.2, 122.4, 117.9, 114.8, 57.2, 57.1, 43.2, 38.2; HR-ESI-MS calcd for C22H20F6N7- OS [M+H]+, 544.1354, found 544.1352.
1-[3, 5-二(三氟甲基)苯基]-3-{3-{甲基[(3-苯基- [1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基]氨基}丙基}脲(15b):白色固体, 产率35.8%. m.p. 178.3~179.3 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.05 (d, J=7.8 Hz, 2H), 7.96 (s, 2H), 7.42 (d, J=7.2 Hz, 1H), 7.40~7.36 (m, 3H), 6.99 (s, 1H), 4.05 (s, 2H), 3.54 (d, J=4.2 Hz, 2H), 2.94 (s, 2H), 2.78 (q, J=6.6 Hz, 2H), 1.10 (t, J=6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 176.0, 155.7, 154.9, 146.3, 141.9, 132.0, 131.7, 130.9, 129.1, 126.2, 124.4, 124.2, 122.4, 117.9, 114.7, 53.9, 53.8, 39.1, 29.7, 11.6; HR-ESI-MS calcd for C23H22F6N7OS [M+H]+ 558.1510, found 558.1505.
1-[4-(叔丁基)苯基]-3-{2-{甲基[(3-苯基-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基]氨基}乙基}脲(15c):白色固体, 产率58.0%. m.p. 101.9~103.7 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.27 (d, J=12.0 Hz, 1H), 8.19~8.151 (m, 2H), 7.45~7.44 (m, 3H), 7.33 (d, J=8.4 Hz, 2H), 7.25 (d, J=9.0 Hz, 2H), 6.31 (s, 1H), 3.83 (s, 2H), 3.46 (dd, J=6.0 Hz, J=10.8 Hz, 2H), 2.70 (t, J=5.4 Hz, 2H), 2.36 (s, 3H), 1.26 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 174.0, 156.6, 145.6, 137.0, 130.5, 128.9, 126.3, 125.8, 125.2, 119.6, 57.4, 57.1, 42.8, 37.9, 34.2, 31.4; HR-ESI-MS calcd for C24H30N7OS [M+H]+ 464.2232, found 464.2238.
1-[4-(叔丁基)苯基]-3-{2-{乙基[(3-苯基-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基]氨基}乙基}脲(15d):白色固体, 产率47.6%. m.p. 178.0~181.3 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.36~8.28 (m, 1H), 8.15 (dd, J=2.4 Hz, J=6.0 Hz, 2H), 7.42~7.41 (m, 3H), 7.31 (d, J=9.0 Hz, 2H), 7.24 (d, J=8.4 Hz, 2H), 6.37 (d, J=4.2 Hz, 1H), 3.91 (s, 2H), 3.42 (q, J=5.4 Hz, 2H), 2.77 (t, J=6.0 Hz, 2H), 2.66 (q, J=7.2 Hz, 2H), 1.26 (s, 9H), 1.02 (t, J=7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 175.6, 156.6, 154.9, 146.0, 145.5, 137.0, 130.4, 128.9, 126.3, 125.7, 125.2, 119.6, 53.7, 53.6, 48.7, 38.4, 31.4, 11.7; HR-ESI- MS calcd for C25H32N7OS [M+H]+ 478.2388, found 478.2391.
1-[4-(叔丁基)苯基]-3-{3-{乙基[(3-苯基-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基]氨基}丙基}脲(15e):白色油状, 产率38.8%. 1H NMR (600 MHz, CDCl3) δ: 8.18 (t, J=4.8 Hz, 2H), 7.99 (s, 1H), 7.46~7.44 (m, 3H), 7.27 (d, J=1.8 Hz, 1H), 7.26 (s, 1H), 7.22 (d, J=8.4 Hz, 2H), 6.31 (t, J=5.4 Hz, 1H), 3.83 (s, 2H), 3.33 (dd, J=6.0 Hz, J=6.6 Hz, 2H), 2.69 (t, J=7.2 Hz, 2H), 2.65 (q, J=7.2 Hz, 2H), 1.78~1.73 (m, 2H), 1.25 (s, 9H), 1.04 (t, J=7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 175.8, 156.6, 154.8, 146.0, 145.4, 136.9, 130.4, 128.9, 126.3, 125.3, 119.5, 53.4, 51.4, 48.1, 37.8, 34.1, 28.0, 11.7; HR-ESI-MS calcd for C26H34N7OS [M+H]+ 492.2545, found 492.2538.
1-[4-(叔丁基)苯基]-3-{2-{[(3-(4-(二甲基氨基)苯基)-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基](甲基)氨基}乙基}脲(15f):白色固体, 产率64.0%. m.p. 125.7~127.3 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.42 (s, 1H), 8.03 (d, J=9.0 Hz, 2H), 7.36 (d, J=9.0 Hz, 2H), 7.25 (d, J=9.0 Hz, 2H), 6.70 (d, J=9.0 Hz, 2H), 6.46 (t, J=5.4 Hz, 1H), 3.82 (s, 2H), 3.48 (dd, J=5.4, 10.8 Hz, 2 H), 3.00 (s, 6H), 2.71 (t, J=5.4 Hz, 2H), 2.36 (s, 3H), 1.27 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 172.8, 156.8, 151.6, 146.8, 137.2, 127.6, 125.7 119.5, 119.5, 112.6, 111.7, 57.3, 57.1, 42.8, 40.1, 37.9, 34.2, 31.5, 29.7; HR- ESI-MS calcd for C26H35N8OS [M+H]+ 507.2654, found 507.2655.
1-[4-(叔丁基)苯基]-3-{2-{[(3-(4-(二甲基氨基)苯基)-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基](乙基)氨基}乙基}脲(15g):白色固体, 产率63.6%. m.p. 179.8~181.3 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.25 (s, 1H), 8.02 (d, J=8.4 Hz, 2H), 7.33 (d, J=7.8 Hz, 2H), 7.25 (d, J=7.8 Hz, 2H), 6.69 (d, J=8.4 Hz, 2H), 6.40 (s, 1H), 3.92 (s, 2H), 3.46 (d, J=4.8 Hz, 2H), 3.00 (s, 6H), 2.80 (s, 2H), 2.68 (q, J=6.6 Hz, 2H), 1.27 (s, 9H), 1.04 (t, J=6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 174.1, 156.5, 151.5, 146.7, 145.8, 136.8, 127.6, 125.8, 125.7, 120.2, 112.7, 111.7, 53.7, 53.6, 48.8, 40.1, 38.5, 34.2, 31.4, 29.7, 11.8; HR-ESI-MS calcd for C27H37N8OS [M+H]+ 521.2811, found 521.2816.
1-[4-(叔丁基)苯基]-3-{3-{[(3-(4-(二甲基氨基)苯基)-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基](甲基)氨基}丙基}脲(15h):白色固体, 产率38.5%. m.p. 205.4~206.7 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.07~8.04 (m, 3H), 7.29 (d, J=8.4 Hz, 2H), 7.21 (d, J=8.4 Hz, 2H), 6.72 (d, J=9.0 Hz, 2H), 6.37 (t, J=4.8 Hz, 2H), 3.73 (s, 2H), 3.35 (dd, J=6.0, 12.0 Hz, 2H), 3.01 (s, 6H), 2.64 (t, J=6.6 Hz, 2H), 2.30 (s, 3H), 1.78~1.74 (m, 2H), 1.44 (s, 2H), 1.24 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 173.1, 156.7, 153.8, 151.6, 146.8, 145.1, 137.2, 127.6, 125.6, 119.4, 112.6, 111.7, 57.1, 55.1, 42.4, 40.1, 37.6, 34.1, 31.4, 28.4, 27.9; HR-ESI-MS calcd for C27H37- N8OS [M+H]+ 521.2811, found 521.2813.
1-[4-(叔丁基)苯基]-3-{3-{[(3-(4-(二甲基氨基)苯基)-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基)甲基](乙基)氨基}丙基}脲(15i):白色固体, 产率33.7%. m.p. 174.5~175.8 ℃; 1H NMR (600 MHz, CDCl3) δ: 8.34 (s, 1H), 8.03 (d, J=9.0 Hz, 2H), 7.30 (d, J=2.4 Hz, 2H), 7.20 (d, J=8.4 Hz, 2H), 6.69 (d, J=9.0 Hz, 2 H), 6.59 (s, 1H), 3.78 (s, 2H), 3.31 (d, J=4.8 Hz, 2H), 2.98 (s, 6H), 2.67 (t, J=7.2 Hz, 2H), 2.60 (q, J=7.2 Hz, 2H), 1.75~1.70 (m, 2H), 1.24 (s, 9H), 1.00 (t, J=7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ: 174.7, 156.8, 153.8, 151.5, 146.7, 144.8, 137.3, 127.6, 125.6, 119.1, 112.6, 111.7, 77.4, 77.3, 77.2, 77.0, 53.3, 51.3, 48.1, 40.1, 37.8, 315, 29.7, 27.9, 11.7; HR-ESI-MS calcd for C28H39N8OS [M+H]+ 535.2967, found 535.2970.
于50 mL圆底烧瓶中, 称取13b (0.3 g, 1.0 mmol)溶于15 mL四氢呋喃(THF)中, 加入DIPEA (661 µL, 4.0 equiv.)为缚酸剂, 以及R-3-(叔丁氧羰基氨基)哌啶(17) (0.2 g, 1.2 equiv.), 升温至50 ℃反应36 h. TLC监测反应完全后, 旋蒸除去THF.残留物用10 mL乙酸乙酯溶解, 依次用0.1 mol/L盐酸(5 mL×3)和水(5 mL×3)洗, 以无水硫酸钠干燥, 浓缩得残留物经硅胶色谱柱分离[V(乙酸乙酯):V(二氯甲烷)=1:6], 得到哌啶环上氨基叔丁氧羰基氨基(Boc)保护的白色固体化合物.
于25 mL圆底烧瓶中, 称取上述化合物(0.5 g, 1 mmol)溶于4 mL二氯甲烷, 加入2 mL三氟醋酸, 室温下搅拌. TLC监测反应完全后, 旋蒸除去二氯甲烷及三氟醋酸.经硅胶色谱柱分离[V(甲醇):V(二氯甲烷)=1:10]得到无色油状化合物18.
称取化合物18 (0.4 g, 1 mmol)于25 mL圆底烧瓶中, 加入5 mL三乙胺(TEA)和1.7 mmol/mL的对叔丁基异氰酸酯19 (720 μL, 1.2 equiv.), 室温反应0.5 h, TLC检测反应结束.用等体积的乙酸乙酯和水的混合溶液(5 mL×3)萃取, 收集乙酸乙酯相.用无水硫酸钠干燥有机相后, 浓缩得到的残留物经硅胶色谱柱分离[V(乙酸乙酯):V(二氯甲烷)=1:1], 得到白色油状物15j.
按相同的方法, 以13b和Boc保护的乙二胺20为起始原料, 经中间体化合物21, 得到了相应的目标产物15k.
(R)-1-[4-(叔丁基)苯基]-3-{1-{{3-[4-(二甲基氨基)苯基]-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基}甲基}哌啶- 3-基}脲(15j):白色油状, 产率60.0%. 1H NMR (600 MHz, CDCl3) δ: 8.40 (s, 1H), 8.09 (d, J=8.4 Hz, 2H), 7.31 (d, J=8.4 Hz, 2H), 7.22 (d, J=8.4 Hz, 2H), 6.72 (d, J=8.4 Hz, 2H), 6.32 (s, 1H), 4.03 (s, 1H), 3.70 (s, 2H), 2.98 (s, 6H), 2.92 (s, 1H), 2.85 (s, 1H), 2.43 (s, 3H), 1.68 (s, 2H), 1.49 (d, J=28.2 Hz, 2H), 1.23 (s, 9H); 13C NMR (150 MHz, CDCl3) δ: 172.0, 155.9, 153.9, 151.6, 146.9, 145.4, 137.0, 127.6, 125.7, 119.7, 112.7, 111.8, 59.2, 57.6, 53.9, 45.7, 40.1, 34.2, 31.4, 29.4, 22.4; HR-ESI-MS calcd for C28H37N8OS [M+H]+ 533.2810, found 533.2804.
3-[4-(叔丁基)苯基]-1-{2-{3-[4-(叔丁基)苯基]脲基}乙基}-1-{{3-[4-(二甲基氨基)苯基]-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基}甲基}脲(15k):白色固体, 产率23.7%. m.p. 155.3~155.9 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.64 (d, J=8.4 Hz, 2H), 7.52 (s, 1H), 7.32 (d, J=2.8 Hz, 4H), 7.14 (s, 4H), 6.62 (d, J=8.4 Hz, 2H), 5.81 (s, 1H), 4.81 (s, 1H), 4.30 (s, 1H), 3.57~3.19 (m, 5H), 2.89 (s, 6H), 1.21 (d, J=6.0 Hz, 18H); 13C NMR (100 MHz, CDCl3) δ: 167.7, 161.9, 156.7, 156.2, 151.6, 151.5, 145.7, 136.2, 129.8, 128.8, 126.7, 126.1, 125.7, 120.0, 112.3, 111.6, 40.0, 34.6, 34.2, 31.4, 31.2; HR-ESI-MS calcd for C36H46N9O2S [M+H]+ 668.3495, found 668.3496.
称取氢化钠(NaH, 0.2 g, 4.0 equiv.)于25 mL反应瓶中, 加入5 mL THF, 冰浴下搅拌10 min, 滴加化合物13a (0.3 g, 1 mmol)的四氢呋喃溶液5 mL, 继续冰浴条件下反应3 h, TLC检测反应结束.加甲醇淬灭NaH, 浓缩得到粗产物经柱层析分离[V(甲醇):V(二氯甲烷)=1:10), 得到白色固体22a.
按相同的方法, 分别以13b~13d为原料, 得到了相应的目标产物22b~22d.
6-(氯甲基)-3-苯基-7-(3-苯基-[1, 2, 4]三唑并[3, 4-b]- [1, 3, 4]噻二唑-6-基)-7H-[11, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二嗪(22a):白色固体, 产率70.5%. m.p. 131.4~132.6 ℃; 1H NMR (600 MHz, DMSO-d6) δ: 8.15~8.13 (m, 2H), 7.81 (d, J=7.8 Hz, 2H), 7.65~7.64 (m, 3H), 7.36 (t, J=7.2 Hz, 1H), 7.10 (t, J=7.8 Hz, 2H), 6.28 (s, 1H), 5.05 (s, 2H); 13C NMR (150 MHz, DMSO-d6) δ: 164.6, 154.9, 154.0, 150.8, 144.8, 139.3, 130.8, 130.3, 129.0, 128.8, 127.7, 125.3, 125.2, 124.9, 44.9, 36.1; HR-ESI-MS calcd for C20H14ClN8S2 [M+H]+ 465.0471, found 465.0476.
4-{6-{6-(氯甲基)-3-[4-(二甲基氨基)苯基]-7H- [1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二嗪-7-基}-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-3-基}-N, N-二甲基苯胺(22b):黄色固体, 产率68.2%. m.p. 178.2~180.3 ℃; 1H NMR (600 MHz, DMSO-d6) δ: 8.13 (d, J=8.4 Hz, 2H), 7.57 (d, J=8.4 Hz, 2H), 6.92 (d, J=9.0 Hz, 2H), 6.2 (d, J=8.4 Hz, 2H), 5.05 (s, 2H), 3.02 (s, 6H), 2.80 (s, 6H); 13C NMR (150 MHz, CDCl3+CD3OD) δ: 162.9, 153.4, 152.4, 152.0, 151.7, 138.4, 129.5, 127.2, 111.7, 111.6, 111.2, 45.2, 39.7, 39.6; HR-ESI-MS calcd for C24H24ClN10S2 [M+H]+ 551.1315, found 551.1312.
6-(氯甲基)-3-(3-甲氧基苯基)-7-[3-(3-甲氧基苯基)- [1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基]-7H-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二嗪(22c):黄色固体, 产率77.4%. m.p. 128.0~130.8 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 7.67~7.63 (m, 2H), 7.54~7.50 (m, 1H), 7.32 (d, J=11.4 Hz, 1H), 7.20 (dd, J=3.0 Hz, J=9.0 Hz, 1H), 6.98 (dd, J=3.0 Hz, J=12.6 Hz, 1H), 6.90 (t, J=12.0 Hz, 1H), 6.26 (s, 1H), 5.06 (d, J=1.8 Hz, 2H), 3.82 (s, 3H), 3.71 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 165.4, 159.9, 159.3, 154.2, 151.2, 145.3, 139.7, 130.6, 130.3, 126.8, 126.6, 120.6, 118.0, 116.9, 116.8, 113.5, 110.1, 55.8, 55.6, 45.3, 36.8; HR-ESI-MS calcd for C22H18ClN8O2S2 [M+H]+ 525.0682, found 525.0689.
6-(氯甲基)-3-(邻甲苯基)-7-[3-(邻甲苯基)-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二唑-6-基]-7H-[1, 2, 4]三唑并[3, 4-b][1, 3, 4]噻二嗪(22d):黄色固体, 产率75.4%. m.p. 205.4~206.7 ℃; 1H NMR (400 MHz, DMSO-d6) δ: 7.48~7.40 (m, 3H), 7.35 (d, J=11.4 Hz, 1H), 7.30 (d, J=11.4 Hz, 1H), 7.23 (t, J=11.4 Hz, 1H), 7.17 (t, J=12.0 Hz, 1H), 7.08 (d, J=10.8 Hz, 1H), 6.24 (s, 1H), 4.86 (d, J=6.4 Hz, 2H), 2.21 (s, 3H), 2.05 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ: 165.4, 154.6, 153.4, 152.4, 146.4, 138.7, 138.3, 137.7, 131.4, 131.1, 131.0, 129.9, 126.5, 126.0, 125.2, 124.9, 45.4, 37.0, 20.5, 20.0; HR-ESI-MS calcd for C22H18ClN8S2 [M+H]+ 493.0784, found 493.0787.
DOT1L酶抑制活性通过AlphaLISA酶活实验进行检测.实验Buffer (50 mmol•L-1 Tris-HCl, pH 8.0, 150 mmol•L-1 NaCl, 3 mmol•L-1 MgCl2, 0.1% bovine serum albumin, BSA)稀释后的化合物15a~15k和22a~22d (50 μmol•L-1)与纯化得到的DOT1L蛋白(80 nmol•L-1)先加入到384孔板(white opaque OptiPlate-384, PerkinElmer)中, 室温孵育10 min.向反应体系中加入1 μmol•L-1 SAM (AdoMet)和0.25 ng核小体(Reaction Biology), 室温孵育30 min.然后向反应体系中加入高盐Buffer (50 mmol•L-1 Tris-HCl, pH 7.4, 1 mol•L-1 NaCl, 0.1% Tween20, 0.3% poly-L-lysine), 室温孵育15 min终止酶活反应.向实验体系中加入组蛋白H3受体磁珠和H3K79me2抗体的混合物, 室温孵育1 h.最后向实验体系中加入链酶亲和素供体磁珠, 并室温孵育30 min, 通过PerkinElmer的酶标仪进行信号检测.
用实验Buffer梯度稀释化合物15k和22a, 按相同的方法检测DOT1L酶抑制活性, 并通过GraphPad Prism5进行IC50曲线拟合.
化合物10、15i、15k和22a与DOT1L (PDB ID:4EKI)的对接通过Maestro[26]完成.首先通过Maestro相应模块对蛋白和小分子化合物进行处理与准备, 同时预测小分子质子化状态及生成的立体异构体.然后采用Maestro中Glide模块的SP模式进行Glide对接[27-30].对接结果用于结合模式的分析.
辅助材料(Supporting Information) 化合物15a~15k和22a~22d的核磁共振氢谱和碳谱, 化合物22c的二维核磁谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Min, J. R.; Feng, Q.; Li, Z. Z.; Zhang, Y.; Xu, R. M. Cell 2003, 112, 711. doi: 10.1016/S0092-8674(03)00114-4
Kim, W.; Choi, M.; Kim, J. E. Cell Cycle 2014, 13, 726. doi: 10.4161/cc.28104
Bernt, K. M.; Zhu, N.; Sinha, A. U.; Vempati, S.; Faber, J.; Krivtsov, A. V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; Pollock, R. M.; Richon, V. M.; Kung, A. L.; Armstrong, S. A. Cancer Cell 2011, 20, 66. doi: 10.1016/j.ccr.2011.06.010
Lillico, R.; Lawrence, C. K. Lakowski, T. M. J. Proteome Res. 2018, 17, 2657. doi: 10.1021/acs.jproteome.8b00118
Anglin, J. L.; Song, Y. J. Med. Chem. 2013, 56, 8972. doi: 10.1021/jm4007752
周仕海, 孙朋举, 赵勇强, 张岩, 余聂芳, 药学学报, 2018, 53, 500.Zhou, S. H.; Sun, P. J.; Zhao, Y. Q.; Zhang, Y.; Yu, N. F. Acta Pharm. Sin. 2018, 53, 500(in Chinese).
Ümit Kaniskan, H.; Martini, M. L.; Jin, J. Chem. Rev. 2018, 118, 989. doi: 10.1021/acs.chemrev.6b00801
Basavapathruni, A.; Jin, L.; Daigle, S. R.; Majer, C. R.; Therkelsen, C. A.; Wigle, T. J.; Kuntz, K. W.; Chesworth, R.; Pollock, R. M.; Scott, M. P. Moyer, M. P.; Richon, V. M.; Copeland, R. A.; Olhava, E. J. Chem. Biol. Drug Des. 2012, 80, 971. doi: 10.1111/cbdd.12050
Daigle, S. R.; Olhava, E. J.; Therkelsen, C. A.; Majer, C. R.; Sneeringer, C. J.; Song, J.; Johnston, L. D.; Scott, M. P.; Smith, J. J.; Xiao, Y.; Jin, L.; Kuntz, K. W.; Chesworth, R.; Moyer, M. P.; Bernt, K. M.; Tseng, J. C.; Kung, A. L.; Copeland, R. A.; Richon, V. M.; Pollock, R. M. Cancer Cell 2011, 20, 53. doi: 10.1016/j.ccr.2011.06.009
Stein, E. M.; Garcia-Manero, G.; Rizzieri, D. A.; Savona, M.; Tibes, R.; Altman, J. K.; Jongen-Lavrencic, M.; Dohner, H.; Armstrong, S.; Pollock, R. M.; Waters, N. J.; Legler, M.; Thomson, B.; Daigle, S.; McDonald, A.; Campbell, C.; Olhava, E.; Hedrick, E. E.; Lowenberg, B.; Copeland, R. A.; Tallman, M. S. Blood 2014, 124, 387. doi: 10.1182/blood.V124.21.387.387
Yao, Y.; Chen, P.; Diao, J.; Cheng, G.; Deng, L.; Anglin, J. L.; Prasad, B. V.; Song, Y. J. Am. Chem. Soc. 2011, 133, 16746. doi: 10.1021/ja206312b
Anglin, J. L.; Deng, L.; Yao, Y.; Cai, G.; Liu, Z.; Jiang, H.; Cheng, G.; Chen, P.; Dong, S.; Song, Y. J. Med. Chem. 2012, 55, 8066.
Yu, W.; Chory, E. J.; Wernimont, A. K.; Tempel, W.; Scopton, A.; Federation, A.; Marineau, J. J.; Qi, J.; Barsyte-Lovejoy, D.; Yi, J.; Marcellus, R.; Iacob, R. E.; Engen, J. R.; Griffin, C.; Aman, A.; Wienholds, E.; Li, F.; Pineda, J.; Estiu, G.; Shatseva, T.; Hajian, T.; Al-Awar, R.; Dick, J. E.; Vedadi, M.; Brown, P. J.; Arrowsmith, C. H.; Bradner, J. E.; Schapira, M. Nat. Commun. 2012, 3, 1288. doi: 10.1038/ncomms2304
Yu, W.; Smil, D.; Li, F.; Tempel, W.; Fedorov, O.; Nguyen, K. T.; Bolshan, Y.; Al-Awar, R.; Knapp, S.; Arrowsmith, C. H.; Vedadi, M.; Brown, P. J.; Schapira, M. Bioorg. Med. Chem. 2013, 21, 1787. doi: 10.1016/j.bmc.2013.01.049
Spurr, S. S.; Bayle, E. D.; Yu, W. Y.; Li, F. L.; Tempel, W.; Vedadi, M.; Schapira, M.; Fish, P. V. Bioorg. Med. Chem. Lett. 2016, 26, 4518. doi: 10.1016/j.bmcl.2016.07.041
Chen, J.; Park, H. J. ACS Chem. Biol. 2019, 14, 873. doi: 10.1021/acschembio.8b00933
Scheufler, C.; Möbitz, H.; Gaul, C.; Ragot, C.; Be, C.; Fernández, C.; Beyer, K. M.; Tiedt, R.; Stauffer, F. ACS Med. Chem. Lett. 2016, 7, 730. doi: 10.1021/acsmedchemlett.6b00168
Chao, C.; Zhu, H.; Stauffer, F.; Caravatti, G.; Vollmer, S.; Machauer, R.; Holzer, P.; Möbitz, H.; Scheufler, C.; Klumpp, M.; Tiedt, R.; Beyer, K. S.; Calkins, K.; Guthy, D.; Kiffe, M.; Zhang, J.; Gaul, C. ACS Med. Chem. Lett. 2016, 7, 735. doi: 10.1021/acsmedchemlett.6b00167
Möbitz, H.; Machauer, R.; Holzer, P.; Vaupel, A.; Stauffer, F.; Ragot, C.; Caravatti, G.; Scheufler, C.; Fernández, C.; Hommel, U.; Tiedt, R.; Beyer, K. S.; Chao, C.; Zhu, H.; Gaul, C. ACS Med. Chem. Lett. 2017, 8, 338. doi: 10.1021/acsmedchemlett.6b00519
Chen, S. J.; Wang, Y. L.; Zhou, W.; Li, S. S.; Peng, J. L.; Shi, Z.; Hu, J. C.; Liu, Y. C.; Ding, H.; Lin, Y. Y.; Li, L. J.; Cheng, S. F.; Liu, J. Q.; Lu, T.; Jiang, H. L.; Liu, B.; Zheng, M. Y.; Luo, C. J. Med. Chem. 2014, 57, 9028. doi: 10.1021/jm501134e
Chen, S.; Li, L.; Chen, Y.; Hu, J.; Liu, J.; Liu, Y. C.; Liu, R.; Zhang, Y.; Meng, F.; Zhu, K.; Lu, J.; Zheng, M.; Chen, K.; Zhang, J.; Jiang, H.; Yao, Z.; Luo, C. J. Chem. Inf. Model. 2016, 56, 527. doi: 10.1021/acs.jcim.5b00738
Wang, Y. L.; Li, L. J.; Zhang, B. D.; Xing, J.; Chen, S. J.; Wan, W.; Song, Y. K.; Jiang, H.; Jiang, H. L.; Luo, C.; Zheng, M. Y. J. Med. Chem. 2017, 60, 2026. doi: 10.1021/acs.jmedchem.6b01785
Song, Y. K.; Li, L. J.; Chen, Y. T.; Liu, J. Q.; Xiao, S. H.; Lian, F. L.; Zhang, N. X.; Ding, H.; Zhang, Y. Y.; Chen, K. X.; Jiang, H. L.; Zhang, C. H.; Liu, Y. C.; Chen, S. J.; Luo, C. Bioorg. Med. Chem. 2018, 26, 1751. doi: 10.1016/j.bmc.2018.02.020
Zhang, L.; Chen, Y. T.; Liu, N.; Li, L. J.; Xiao, S. H.; Li, X. L.; Chen, K. X.; Luo, C.; Chen, S. J.; Chen, H. Bioorg. Chem. 2018, 80, 649. doi: 10.1016/j.bioorg.2018.07.022
Cui, P. L.; Li, X. L.; Zhu, M. Y.; Wang, B. H.; Liu, J.; Chen, H. Eur. J. Med. Chem. 2017, 127, 159. doi: 10.1016/j.ejmech.2016.12.053
Maestro, Version 10.4, Schrödinger, LLC, New York, 2015.
Friesner, R. A.; Banks, J. L.; Murphy, R. B.; Halgren, T. A.; Klicic, J. J.; Mainz, D. T.; Repasky, M. P.; Knoll, E. H.; Shelley, M.; Perry, J. K.; Shaw, D. E.; Francis, P.; Shenkin, P. S. J. Med. Chem. 2004, 47, 1739. doi: 10.1021/jm0306430
Friesner, R. A.; Murphy, R. B.; Repasky, M. P.; Frye, L. L.; Greenwood, J. R.; Halgren, T. A.; Sanschagrin, P. C.; Mainz, D. T. J. Med. Chem. 2006, 49, 6177. doi: 10.1021/jm051256o
Halgren, T. A.; Murphy, R. B.; Friesner, R. A.; Beard, H. S.; Frye, L. L.; Thomas Pollard, W.; Banks, J. L. J. Med. Chem. 2004, 47, 1750. doi: 10.1021/jm030644s
Glide, Version 6.9, Schrödinger, LLC, New York, 2015.

图 3 活性化合物与DOT1L的结合模式
Figure 3 Putative binding poses of the active compounds
(A) Compounds 10, 15i, 15k and 22a were aligned with SAM (salmon) in the SAM binding pocket of DOT1L (4EKI); (B) 10; (C) 15i; (D) 15k; (E) 22a
表 1 化合物15a~15k和22a~22d的DOT1L抑制活性a
Table 1. DOT1L Inhibitory activity of compounds 15a~15k and 22a~22d
| Compd. | Inhibition rate/% | IC50/(μmol"L-1) |
| 15a | 31.35 | —a |
| 15c | 0.32 | — |
| 15e | 11.35 | — |
| 15g | 22.02 | — |
| 15i | 62.35 | — |
| 15k | 79.62 | 25.92 |
| 22b | 69.86 | — |
| 22d | 39.36 | — |
| 15b | 11.04 | — |
| 15d | 3.21 | — |
| 15f | 50.27 | — |
| 15h | 37.43 | — |
| 15j | 55.47 | — |
| 22a | 86.16 | 10.59 |
| 22c | 47.12 | — |
| 10 | 86.71 | 7.25 |
| a Tested concentration at 50 µmol•L-1, “—” undertermined. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们