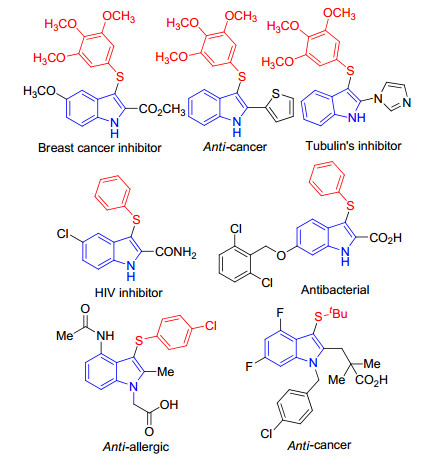
图 1.
Representative examples of bioactive indole-based organosulfur compounds

Indoles and their derivatives are prevalent in natural products and biologically active compounds.[1] Among the various indole derivatives, sulfur-bearing indoles represent a class of very important organosulfur heterocyclic compounds, as they are present in many biologically and phar-maceutically important molecules (Figure 1).[2] which display a broad spectrum of biological and pharmaceutical activities, including anti-HIV, [3] anti-cancer, [4] anti- obesity, [5] anti-allergies, [6] etc.
As a result, much effort has been made towards development of efficient route to access indole thioethers. Traditionally, metal-catalyzed C—S bond formation between C—X/C—H and different sulfur sources such as thiols, sulfonyl chlorides, disulfides, sulfonyl hydrazides, and other S-containing reagents is well known.[7] In spite of some great advantages, some of these processes could suffer from certain drawbacks, including pre-functionalization of reactants, harsh reaction conditions, and toxic metal catalysts. In recent years, transition metal-free C—S bond formation by direct C—H sulfenylation of indoles has attracted much attention because the metal-free procedures are greener and cost-effective, and more and more excellent and significant works have been presented.[8] The recent five-year progress in direct C—H bond sulfenylation of indoles under transition metal-free conditions is discussed and their mechanisms in detail are described
Because of its electronic properties, indoles could be easily functionalized at the C(3) position through reaction with electrophiles. Thus, thioethers can be easily introduced at C(3) by using a number of sulfenylating reagents. However, 2-sulfenylindoles is hard to be acquired by direct C—H sulfenylation of C(3)-unsubstituted non-protected indoles due to the weak reactivity of the C—H bond at the C(2) position.
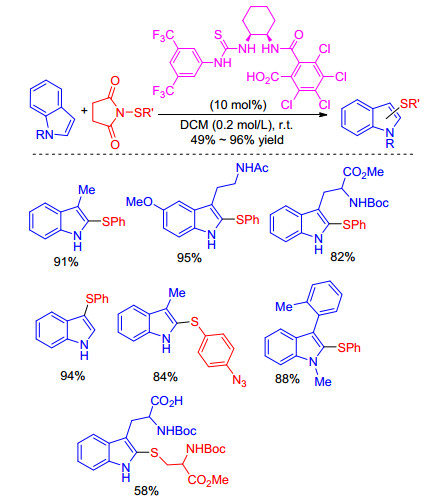
If using C(3)-substituted indoles as substrate, C(2)—H bond sulfenylation of indoles can easily take place under transition metal-free conditions with various sulfenylating reagents. In 2017, Gustafson group[9] demonstrated that a conjugate Lewis base-Brønsted acid catalyzed sulfenylation at C(2) position of C(3)-unsubstituted indoles with N-(thio)- succinimides under mild conditions (Scheme 1). An efficient catalyst that contains a thiourea tethered to carboxylic acid was found to sulfenylate electron rich heterocycles at room temperature. Obviously, the C—H sulfenylation will happen at C(3) position with C(3)-unsubstituted indoles as substrate.
In most of the literature about the C(3)—H bond sulfenyaltion of indoles, special C(3)-substituted substrates were also employed to be sulfenylated utilizing the same strategy and 2-sulfenylindoles could be obtained in moderate to high yields with different sulfenylating reagents including thiols, disulfides, sulfonyl chlorides, sulfonyl hydrazides, N-thioimides, etc. under transition metal-free conditions.[18, 29~31, 34~36, 42, 46]
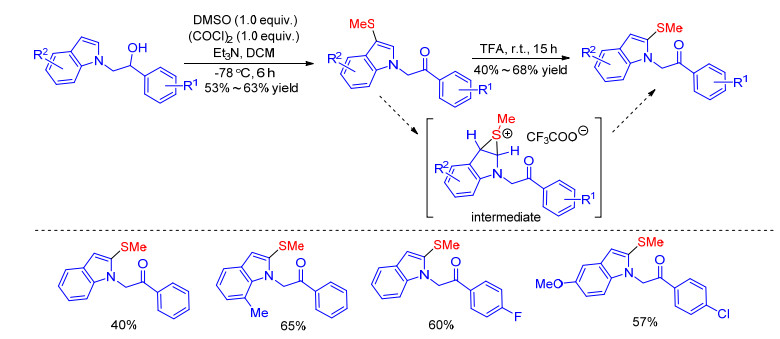
In 2015, the stepwise construction of 2-sulfenyl indoles could be successfully achieved by Xu group[10] through combination of the C(3)—H sulfenylation and acid- catalyzed rearrangement of 3-sulfenylindoles. Firstly, an unprecedented sulfenylation of N-substituted indoles was achieved using DMSO as sulfenylating source via metal-free oxidation and methylthiolation. Subsequently, the rearrangement of 3-methylthioether-substituted indoles promoted by trifluoroacetic acid led to the facile synthesis of the corresponding 2-sulfenyl indoles (Scheme 2). All the desired 2-methylthioether indoles were isolated in moderate to good yields. Notably, the novel and general sulfenylation of indoles might be very valuable and attractive protocol in medicinal and organosulfur chemistry.
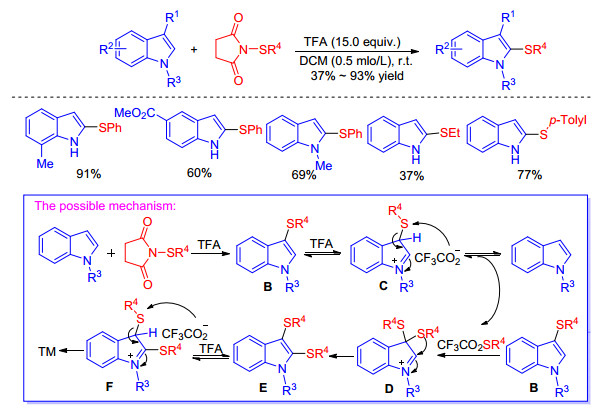
In addition, Cossy group[11] presented an efficient direct C—H sulfenylation reaction at the C(2) position of C(3)-unsubstituted N-unprotected indoles using N-(thio)succinic-mides as the sulfur sources. Experimental results confirmed that the desired product 2-sulfenylindoles could be produced through the rearrangement of C(3)-sulfenylated indoles rather than direct C—H sulfenylation at the C(2) position (Scheme 3). Firstly, the C(3)-sulfenylated indole B was formed, which can be protonated by trifluoroacetic acid (TFA) giving the intermediate C, then it proceed the trifluoroacetate anion induced desulfenylation to generate the starting indole as well as CF3CO2SR4. As the strong electrophilic sulfenylating reagent, CF3CO2SR4 can react with compound B to afford the 3, 3-bis-sulfide indolenium D. One of the sulfide groups migrate to the C(2) position leading to the 2, 3-disubstituted indole E which underwent protonation and desulfenylation to produce the expected 2-sulfenylindole as well as the acyloxysulfide. The acyloxysulfide can be trapped by the unsubstituted indole to regenerate the C(3)-sulfenylated indole B to enter catalytic cycle. But it’s worthy to be pointed out that when an electron-withdrawing group is present on the nitrogen or sulfenylated reagents, the C(3)-sulfenylated indoles were isolated in high yields.
As we all know, the direct C—H bond sulfenylation of indoles usually occurred at C(3) position due to its electronic properties. In the past years, great efforts also have been devoted to create new protocols for the construction of C—S bonds. At present, various sulfenylating reagents such as thiols, disulfides, sulfonyl hydrazides, sulfinic acids, and so on have been employed for the C(3)—H bond sulfenylation of indoles.[12]
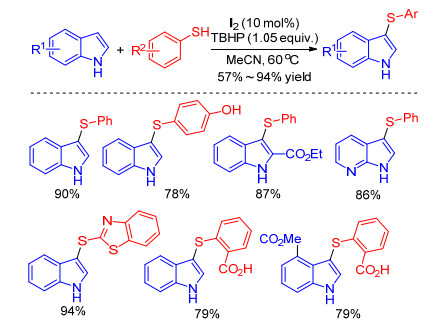
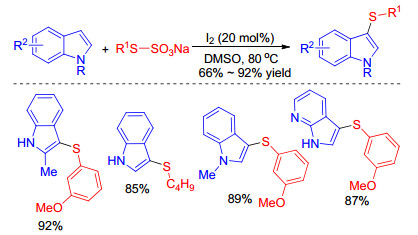
Iodine-catalyzed sulfenylation of indoles with various thiophenols in the presence of different oxidants such as TBHP, [13] H2O2, [14] and DMSO[15] were described by Wang, Huang, Shen et al. The 3-sulfenylindoles could be obtained with good to excellent yields under environmental mild conditions (Scheme 4). The notable features of these protocols include the extensive scope of substrates, the mild reaction conditions and the high selectivity of reaction site delivery promising applications in drug discovery and functional materials.
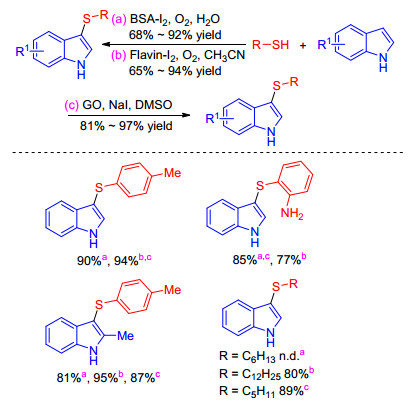
The green combinational oxidants of BSA-O2, Flavin-O2 or GO-DMSO were also employed to promote the sulfenylation of indoles under iodine-catalyzed conditions. In 2016, Sinha and co-workers[16] coalesced biocatalyst and chemocatalyst for direct sulfenylation of indoles as well as hydroxyaryls with readily available thiophenols using BSA- O2 as cooperative oxidant in aqueous medium. Subse-quently, Basu group[17] disclosed the sulfenylation of indoles with thiols in the presence of catalytic grapheme oxide and NaI using DMSO as both solvent and oxidant, Iida group[18] presented a flavin-iodine system realizing the novel direct sulfenylation of indoles with thiols under O2 condition (Scheme 5).
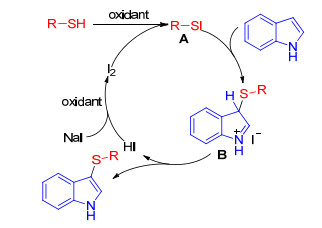
The mechanism of all iodine-catalyzed sulfenylation of indoles with additional oxidants proposed by these groups is coincident and presented in Scheme 6. Firstly, RSH reacts with I2 which is added directly or produces by oxidation of NaI to form an electrophilic species RSI A, which can provide the electrophile RS+ and react with indole moiety to produce intermediate B. Then deprotonation of B gives the desired products and HI that is oxidized by different oxidants to regenerate I2 which again enters the catalytic cycle.
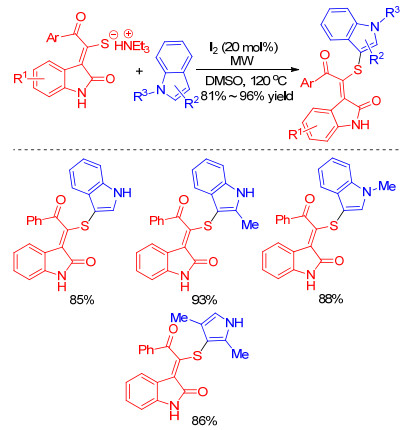
Using I2/DMSO catalytic system with aid of microwave irradiation, readily available triethylammonium thiolates were proven to be new and eco-friendly sulfenylating agents for the synthesis of sulfenylated indoles and pyrroles by Li and co-workers in 2017 (Scheme 7).[19] In this protocol, various functional groups on substrates are tolerated to generate the desired products in good yields.
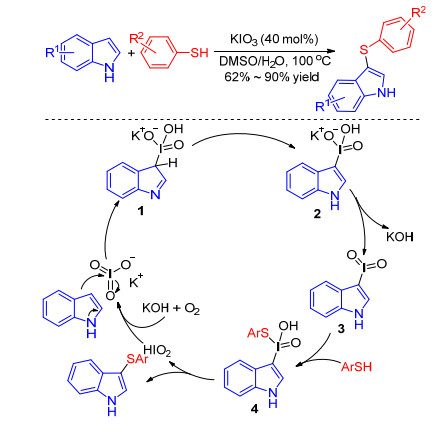
Liu, [20] Lin[21] et al. found by using simple inorganic iodine salt KIO3 as the sole catalyst, the sulfenylated indole products were easily synthesized with good tolerance and satisfactory yield in DMSO/H2O. Initially, indoles react with KIO3 to produce polyvalent iodine intermediate 2 through intermediate 1, which releases KOH to yield intermediate 3. The addition of thiophenol to the I=O bond generates intermediate 4. The reductive elimination of 4 provides the target product and iodous acid, which is then oxidized to regenerate potassium iodate for further catalysis in the presence of KOH and air (Scheme 8).
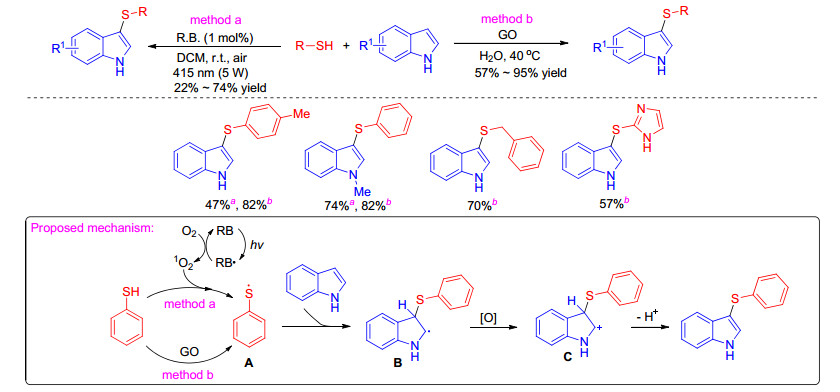
Indoles and their derivatives also can be sulfenylated with thiophenols through free radical mechanism catalyzed by rose bengal or grapheme oxide. In 2017, Guo group[22] developed an efficient photocatalytic direct sulfenylation of indoles employing thiophenols as the sulfenylating agents, inexpensive rose bengal as the photocatalyst (Scheme 9). This protocol shows mild conditions, readily available starting materials, high atom and step economy, and environmental advantages. In 2019, Wu group[23] presented the graphene oxide mediated sulfenylation of indoles using thiols as sulfenylating reagent under iodine and transition metal free conditions (Scheme 9). The two proposed reaction mechanisms are in common which rose bengal or graphene oxide might act as a radical initiator transforming benzenethiol into phenylthiophenol radical A. Then the thioyl radical A interacted with indoles to produce the radical intermediate B. After that, B was oxidized to the intermediate C. Finally, deprotonation of intermediate C led to the formation of 3-sulfenylindoles.
Disulfides are commonly used organic sulfur reagents, and they can react with iodine to afford electrophilic species RSI which can provide the electrophile RS+ and react with various substrates to produce sulfenylated products.[24]
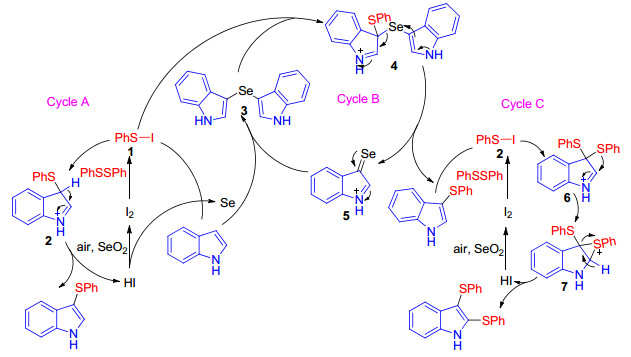
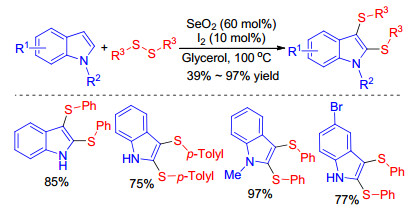
In 2018, Lenardao group described the synthesis of sulfenylindoles through the C—H sulfenylation of indoles with diaryl disulfides using the unprecedent I2/SeO2 catalytic system (Scheme 10).[25] As depicted in Scheme 10, a possible mechanism which contains three catalytic cycles was proposed by authors. In Cycle A, disulfide firstly react with I2 to form PhSI, which then reacts with indole to form products and HI; this cycle is completed by the reaction of HI with SeO2 to form I2. The in situ formed selenium by-product facilitates Cycle B by reacting with indole to form intermediate 3. Intermediate 4 is formed after reaction with PhSI, which is generated in situ from PhSSPh and I2, and then suffers disproportionation to deliver the expected product and selenone 5. Nucleophilic attack of 5 by indole forms more 3 and HI, which is oxidized back to I2 by SeO2, giving more Se (cycle B). This efficient method with good substituent tolerance on both the disulfide and the indoles can furnish diverse sulfenylindoles.
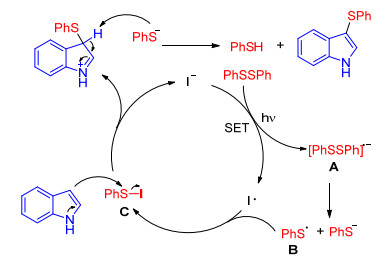
Yan and co-workers[26] successfully used catalytic amount of sodium iodine instead of iodine as promoter to synthesize 3-arylthioindoles in good yields via the photoirradiation of indoles and disulfides. This protocol has good functional group tolerance and eco-friendly mild conditions, but only using aryl disulfides as sulfenylating reagents. A possible reaction mechanism is outlined in Scheme 11. The diphenyl disulfide is firstly activated under the photoirradiation to provide a diphenyl disulfide anion radical A via single electron transfer. The decomposition of A generates thiophenol anion and phenylthiyl radical B, which reacts with the iodine radical to afford the arylsulfenyl iodine C. Finally, the subsequent electrophilic substitution with the indoles provides the desired product 3-arylthioindole (Scheme 11).
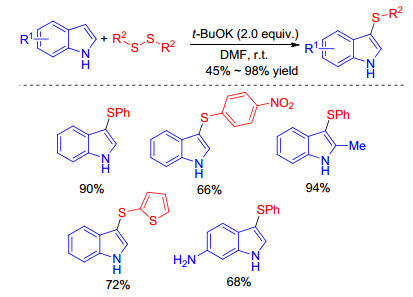
Under iodine-free conditions, Song and co-workers[27] achieved an efficient t-BuOK promoted C(3)-sulfenylation of indoles with disulfides (Scheme 12). The presented protocol featuring mild reaction conditions and short reaction times exhibits a broad functional group tolerance affording structurally diverse sulfenylated indoles with or without protecting groups.
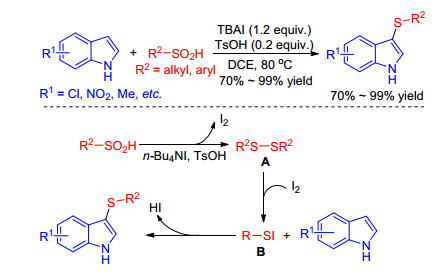
In 2015, Liu and co-workers[28] described an unprecedented by-product I2 promoted sulfenylation of indoles in which sulfinic acids were used as sulfenylating reagents. In the presence of TsOH and Bu4NI, lots of aryl- and alkylsulfinic acids underwent well sulfenylation with substituted indoles to give structurally diverse desired products in high yields (Scheme 13). They proposed the reaction pathway depicted in Scheme 13. Initially, in the presence of TsOH and n-Bu4NI, the sulfinic acid is transformed to disulfides A, which reacts with I2 to give the sulfenyl iodide B. Then the intermediate B is attacked by indole to give products and HI. Iodine regenerated from the reaction of HI and sulfinic acid could promote the reaction continually.
The sulfenylation of indoles with sulfinic acids in the presence of TsOH and n-Bu4NI was also reported by Barman and coworkers.[29] Although the reaction mechanism is alike, however, these reactions proceed under microwave irradiation with excellent yield, shorter reaction time and solvent-free conditions.
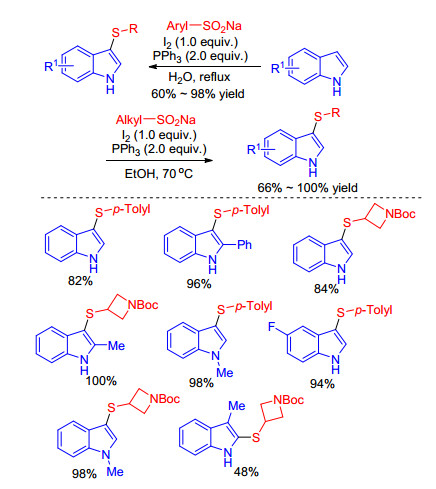
Harrity, [30] Xie[31] et al. successively described an iodine- PPh3 mediated sulfenylation of indoles with sodium sulfinates as the sulfur source. They used different sodium (aryl or alkyl) sulfinates as sulfenylating reagents affording the relevant sulfenylindoles in moderate to excellent yields under mild conditions (Scheme 14). The reaction mechanism is the same as above Barman’s work, except that PPh3 is used instead of diethyl phosphite as reductant.
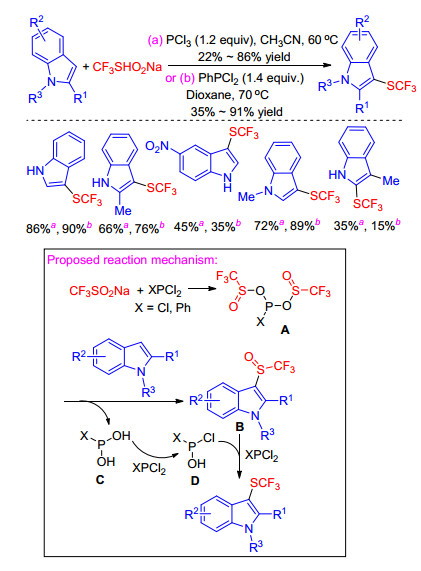
In 2017, Zhao and co-workers[32, 33] continuously published two articles on the trifluoromethylthiolation of indoles using CF3SO2Na in the presence of PCl3 or PhPCl2. First of all, they achieved the direct trifluoromethylthiolation of electron-rich aromatics with CF3SO2Na in the presence of PCl3 used as a reducing and chlorination reagent. The transition metal-free protocol utilized cheap and readily available reagents and exhibited good atom economy and functional group compatibility. However, due to reacting rapidly with indole, the slow addition of PCl3 was carried out using a syringe pump leading to complicated operation and long reaction time. Thus, they replaced PCl3 with PhPCl2 which is less electrophilic than PCl3 and reacts less rapidly with indoles (Scheme 15).
A plausible reaction mechanism for this transformation was depicted in Scheme 15. Initially, CF3SO2Na may react with PCl3 to form intermediate A, which is attacked by indole twice to give intermediate B and C. Intermediate B reacts with PCl3 to generate P(OH)Cl2 (D). Reduction of B by PCl3 and D gives the corresponding indole trifluoromethylthioethers.
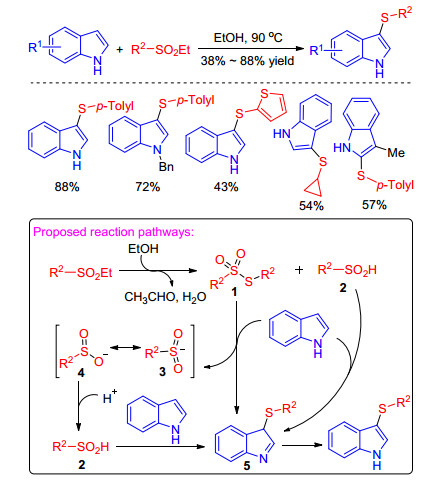
In 2018, Yang et al.[34] disclosed a catalyst-free sulfenylation of indoles with sulfinic esters as new sulfur electrophiles in ethanol. The protocol afforded a series of structurally diverse sulfenylated indoles in high yields under catalyst-free conditions. The possible reaction pathway proposed is that the sulfinic ester is reduced by ethanol to form sulfonothioate 1 and sulfinic acid 2, which proceed a electrophilic reaction with indoles to generate intermediates 3 and 5. Then the sulfinic acid 2 was formed from anion 4, resonance of intermediate 3, which can also react with indole to provide the intermediate 5. Finally, the intermediate 5 transforms into the desired sulfenylated indoles (Scheme 16).
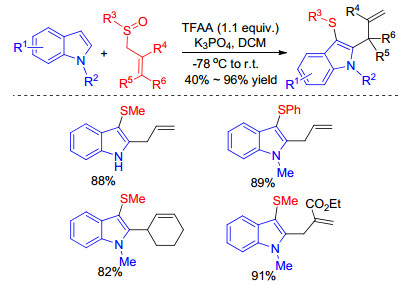
Indoles can also be sulfenylated by various sulfoxides under transition metal-free conditions. For example, Xu group established an unexpected and unprecedented methylthiolation of substituted indoles using dimethyl sulfoxide as the sulfur source under the Swern oxidation conditions, in which ketone-containing 3-sulfenylindoles was directly synthesized from readily available starting materials in good yields.[10] In 2018, Procter and co-workers developed a cascade vicinal bifunctionalization of indoles that provides an efficient method to the C(3) sulfenyl, C(2) carbon indole derivatives. The present strategy underwent an interrupted Pummerer coupling of activated allyl sulfoxides and a charge accelerated [3, 3]-sigmatropic rearrangement of allyl heteroaryl sulfonium salts (Scheme 17).[35] The handy method is not only applicable to indoles but also to other heterocyclic compounds affording C(3) and C(2) dual functionalized products in moderate to good yields under mild conditions.
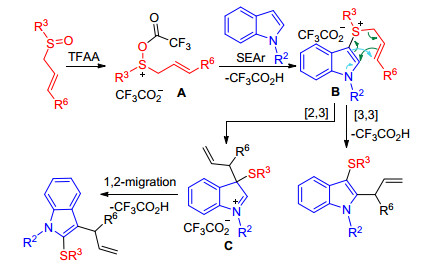
However, when γ-substituted allyl sulfoxides were employed, the C(2) thio, C(3) allyl indoles were also formed showing inferior regioselectivity, upon utilizing the more hindered N-iPr indole, C(2) sulfenyl C(3) allyl regioisomer was obtained as the sole regioisomeric product. Authors proposed the following mechanism for the dual functionalization cascade and reaction regioselectivity (Scheme 18). Activation of the allylic sulfoxide with TFAA generates sulfoxonium salt A, which is trapped by indole through the inherently nucleophilic C(3) position to form sulfonium salt B. Subsequent charge accelerated [3, 3]-sigmatropic rearrangement B accomplishes the second functionalization event and delivers the desired C(3) sulfenyl, C(2) allyl indoles. When the more hindered group was incorporated in γ position or nitrogen, the [3, 3]-sigmatropic rearrangement is disfavored on steric grounds and instead a [2, 3]-sigma- tropic rearrangement of allyl sulfonium salt B forms intermediate C, which then undergoes facile sulfur migration to C(2), likely via an episulfonium ion intermediate.
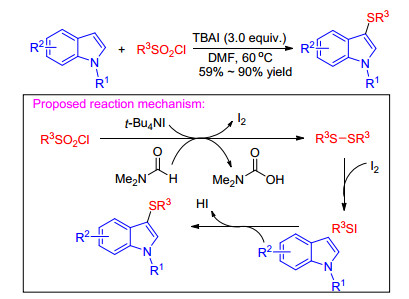
The iodine-mediated sulfenylation of indoles with low- cost, and readily available, stable aryl or alkyl sulfonyl chlorides using different reducing agents such as iodine, PPh3 and DMF has been demonstrated.[36] In 2017, Xiang group[37] found a new TBAI-mediated sulfenylation of indole derivatives and other electron-rich aromatics with various sulfonyl chlorides. This protocol featured easy operation, highly regioselective, and showed a broad functional group tolerance. A possible reaction mechanism was proposed in Scheme 19. Initially, in the presence of reducing reagent TBAI and solvent DMF, the sulfonyl chloride is transformed into disulfides, which reacts with iodine to give R3SI. Then R3SI is attacked by indoles generating the desired products 3-sulfenylindoles and iodide that reacts with sulfonyl chloride to enter the next catalytic cycle again. By means of this kind of protocol, a range of C(3) arylthiolated, alkylthiolated, trifluoromethylthiolated and difluoromethylthiolated indoles and other electron rich aromatics are generated in high to excellent yields under mild conditions.
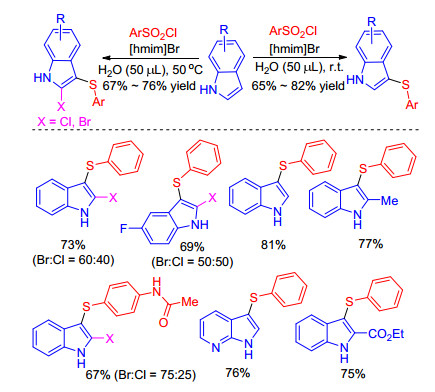
Subsequently, Sinha and co-workers[38] developed eco- friendly and temperature tunable sulfenylation of indoles, in which the synergistic effect between [hmim]Br-ArSO2Cl exclusively leaded to the formation of 3-arylthio indoles at room temperature, while at 50 ℃ this reaction provided an unexpected 2-halo-3-arylthioindoles without addition of any external halogenating agent (Scheme 20). The insightful mechanism research shows that this procedure also includes the formation of RSX and then nucleophilic reaction with indoles. Moreover, this tunable protocol is mild, efficient and has a broad substrate scope.
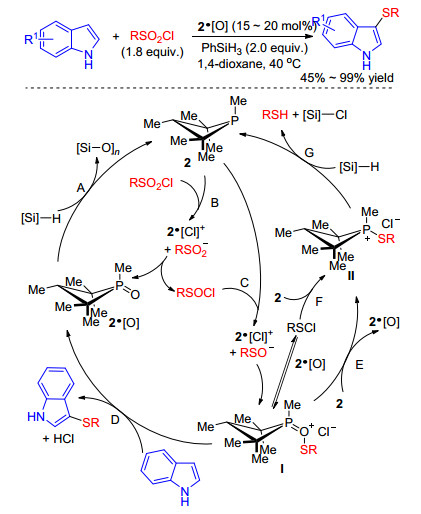
Recently, Radosevich group[39] reported an efficient organophosphorus-catalyzed sulfenylation of indoles. This progress was catalyzed by a readily available phosphine (phosphetane) in conjunction with a hydrosilane terminal reductant to afford the desired products. Based on the experimental results, authors proposed a plausible reaction mechanism for this phosphacatalytic deoxygenation/sulfa- nylation of indoles as outlined in Scheme 21. Firstly, the precatalyst 2·[O] is reduced to the active phosphetane 2 by phenylsilane (step A). Subsequently, 2 operates on RSO2Cl to the formation of active sulfenyl electrophiles I by twofold deoxygenation of the sulfonyl chlorides (steps B, C). In effect, intermediate I may be viewed as a phosphine oxide Lewis base adduct of sulfenium fragment, which can enhance the reactivity of electrophilic reagents.[40] Finally, reaction of I with indoles formed the target product and regenerated 2·[O] to complete the catalytic cycle (step D). Alternatively, formation of intermediate II may proceed directly from I (step E) or via sulfenyl chloride RSCl (step F), upon which II can rejoin the catalytic cycle by reduction with terminal phenylsilane (step G) (Scheme 21).
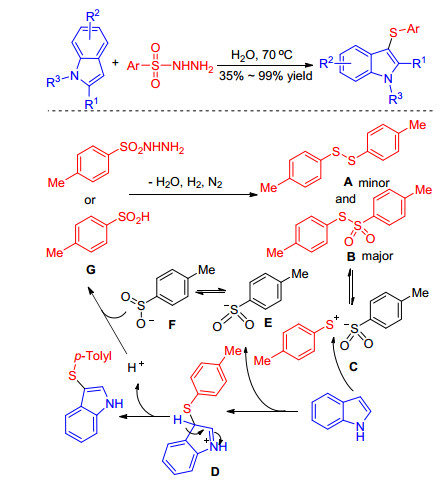
Sulfonyl hydrazide is a high atom-economical, odorless, non-toxic sulfenylating reagent, and has been already devoted to construct 3-sulfenylindoles. Wang and co-work- ers[41] achieved the catalyst-free synthesis of 3-sulfen- ylindoles with sulfonyl hydrazides in water without ligand or additive. Besides, a variety of functional groups could tolerate the reaction conditions well and the by-products were only nitrogen and water. A possible reaction mechanism was proposed and depicted in Scheme 22. Firstly, sulfonylhydrazides transform into minor A and major intermediate B which can dissociate into C in the presence of water. Subsequently, the intermediate C is nucleophilic attacked by indoles generating intermediate D and E. The intermediate E resonates with sulfur-centered anion F, which can form sulfinic acids G in the presence of water. Meanwhile, the intermediate D quickly transforms into desired product with the release of proton (Scheme 22).
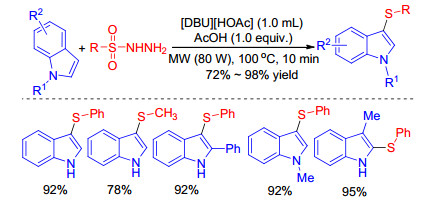
Barman et al.[42] found that under microwave irradiation, using organic ionic base-Brønsted acid could improve sulfenylation efficiency of indoles with sulfonyl hydrazides as thiol surrogate. A variety of aryl- and alkyl-sulfonyl hydrazides react with indoles well to give diverse 3-sulfenyl- indoles in moderate to excellent yields in 10 min (Scheme 23). The process could be a greener alternative due to the short reaction time, high yield, broad substrates scope and use of a recyclable ionic liquid.
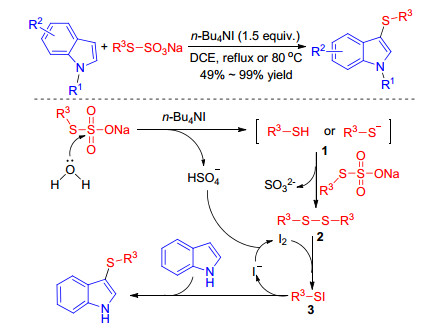
Bunte salts have been developed and well applied in organic synthesis due to their easy preparation and stabilities without unpleasant smell. It was also reported that Bunte salts reacted with iodine to generate electrophilic sulfenyl iodide (RSI) species which was known to undergo sulfenylation reaction of indoles with concomitant formation of HI.[43] In 2016, Luo group[44] developed an efficient catalytic method for the synthesis of 3-sulfenylindoles under metal-free conditions using readily available odorless crystalline Bunte salts as the sulfenylating agents and DMSO as both the oxidant (Scheme 24). This reaction is practical and environmentally benign, selective at C(3) position of indoles, and compatible with a wide range of substrates giving the desired products in excellent yields.
Subsequently Ji and co-workers[45] achieved the TBAI catalyzed 3-sulfenylation of indoles with Bunte salts. Authors hypothesized that the Bunte salt reacts with water in the presence of TBAI to produce the thiol or the thiol anion and the bisulfate anion, which further oxidizes iodide ion to furnish iodine. Another Bunte salt is nucleophilic attacked by the thiol anion to afford disulfide, which reacts with I2 to form an electrophilic species RSI. The desired product and iodide ion are obtained via the reaction of electrophilic species RSI with indole and deprotonation (Scheme 25). This protocol deftly used in situ generated bisulfate anion to oxidate iodine ion without extra-oxidant, and is more environmentally friendly therefore.
Notably, the indoles is an electron-rich aromatic compound that can undergo nucleophilic reactions at both C(2) and C(3) positions. Although many methods have been successfully established to construct structurally diverse mono-sulfenyl indoles, protocols that can accomplish double C—H sulfenylation of indoles at 2- and 3-positions still remained scarce.
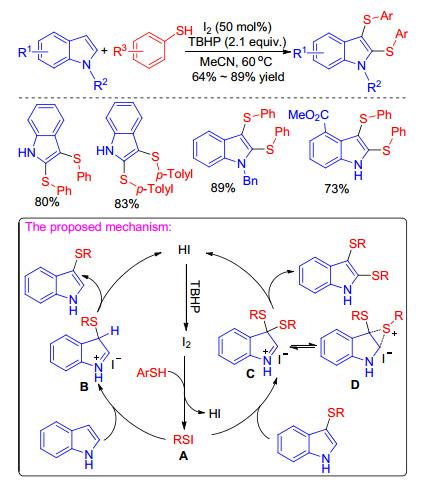
In 2015, Wang group[13] described the synthesis of 2, 3-bissulfenyl indoles under transition metal-free conditions and using readily available thiols as sulfur source when they performed the C(3)—H bond sulfenylation of indoles. This efficient method has good substituent tolerance and can furnish mono- and bis-sulfenylindoles just by varying the amount of iodine and oxidants. A plausible reaction mechanism for mono- and bis-sulfenylations of indoles is proposed in Scheme 26. Firstly, the electrophilic species RSI A obtained from reaction of RSH I2 reacts with indole to afford intermediate B, which releases a proton to give the monosulfenyl indole. Then the second sulfenylation of indole still occurs at the 3-position of indole leading to 3, 3-disubstituted indolenine intermediate C, which migrate one of the sulfide groups to the 2-position to furnish 2, 3-disulfenyl indoles. Finally, I2 is oxidized by TBHP to enter the next catalytic cycle (Scheme 26).
In 2018, Lenardao group[25] also found that bis-sulfen- ylindoles could be provided just varying the amount of SeO2 based on the reaction conditions of monosulfenylation. The available protocol features good substituent tolerance on both disulfide and indole, selectivity, mild conditions and so on (Scheme 27). The proposed mechanism for this transformation was depicted in Scheme 10. The formation of the bis-sulfenylated indole followed a similar pathway to that shown in cycle A, with the exception that the mono-sulfen- ylated indole is the starting material instead the indole, as previously described (Scheme 10, cycle C).
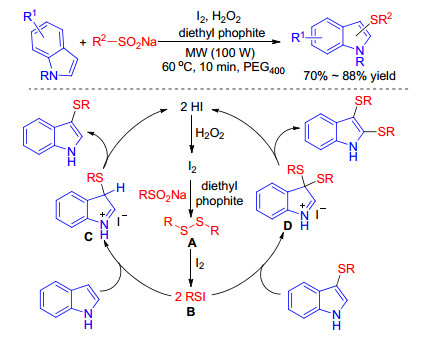
An iodine-catalyzed method using hydrogen peroxide as the oxidizing agent in PEG400 under microwave conditions for the synthesis of mono- and di-sulfenylindoles via sulfenylation of indole with sodium alkyl- and aryl-sulfinates was established by Barman group in 2017.[46] This presented protocol offered mono- or di-sulfenylindoles by varying the amount of iodine, H2O2 and diethyl phosphite in good to excellent yields with excellent functional group tolerance and can be easily performed within short reaction times under mild reaction conditions. The proposed plausible mechanism is similar to Wang’s worker except that the electrophilic RSI B was formed by the reaction of I2 and disulfide which is generated from sodium sulfinate in the presence of I2 or H2O2 with diethyl phosphite. Moreover, I2 is regenerated by the oxidation of H2O2 not TBHP with the formation of H2O in the end (Scheme 28).
In recent years, the construction of C—S bonds through direct C—H sulfenylation reactions has rapidly advanced and become an eco-friendly synthetic tool for pharmacists and organic chemists. Herein we present an overview of the recent progress in C—H bond sulfenylation of indoles under transition metal-free conditions. Because its rich-elec- tron property, many methods have been established for the sulfenylation of indoles at C(2) or/and C(3) position with various sulfenylating reagents such as thiols, disulfides, Bunte salts, sodium sulfinates, etc. Although much attention has been attracted and great advances have been achieved, there are still some puzzles that need to be addressed. At present, most of sulfenylation of indoles occurred at C(3) position, so C(2)—H sulfenylation and C(2)/C(3)—H disulfenylation need further to be probed. The regioselectivity of the reaction is also an issue worth to be explored. More- over, the synthesis of sulfenylindoles having reactive hydrogen functional groups (peculiarly NH2 and CO2H) has not been well documented to date. Finally, the mechanism of the reaction is worth in-depth study, which helps us to design and achieve more efficient and eco-friendly sulfenylations of indoles. Direct C—H bond sulfenylation of indoles is a vigorous research field filled with opportunities and challenges. We believe that more and more attention will be attracted to this area, and more and more innovative achievements will emerge in the future.
(a) Poulsen, T. B.; Jørgensen, K. A. Chem. Rev. 2008, 108, 2903.
(b) Joucla, L.; Djakovitch, L. Adv. Synth. Catal. 2009, 351, 673.
(c) Cacchi, S.; Fabrizi, G. Chem. Rev. 2011, 111, 215.
(a) Garbe, T. R.; Kobayashi, M.; Shimizu, N.; Takesue, N.; Ozawa, M.; Yukawa, H. J. Nat. Prod. 2000, 63, 596.
(b) Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C. O.; Sim, C. J.; Im, K. S.; Jung, J. H. J. Nat. Prod. 2005, 68, 711.
(c) Cacchi, S.; Fabrizi, G. Chem. Rev. 2005, 105, 2873.
(d) Humphrey, G. R.; Kuethe, J. T. Chem. Rev. 2006, 106, 2875.
Ragno, R.; Coluccia, A.; Regina, G. L.; Martino, G. D.; Piscitelli, F.; Lavecchia, A.; Novellino, E.; Bergamini, A.; Ciaprini, C.; Sinistro, A.; Maga, G.; Crespan, E.; Artico, M.; Silvestri, R. J. Med. Chem. 2006, 49, 3172. doi: 10.1021/jm0512490
Regina, G. L.; Edler, M. C.; Brancale, A.; Kandil, S.; Coluccia, A.; Piscitelli, F.; Hamel, E.; Martino, G. D.; Matesanz, R.; Díaz, J. F.; Scovassi, A. I.; Prosperi, E.; Lavecchia, A.; Novellino, E.; Artico, M.; Silvestri, R. J. Med. Chem. 2007, 50, 2865. doi: 10.1021/jm061479u
(a) Berger, J. P.; Doebber, T. W.; Leibowitz, M.; Moller, D. E.; Mosley, R. T.; Tolman, R. L.; Ventre, J.; Zhang, B. B.; Zhou, G. WO 0130343, 2001.
(b) Ramakrishna, V. S. N.; Shirsath, V. S.; Kambhampati, R. S.; Vishwakarma, S.; Kandikere, N. V.; Kota, S.; Jasti, V. WO 2007020653, 2007.
Unangst, P. C.; Connor, D. T.; Stabler, S. R.; Weikert, R. J.; Carethers, M. E.; Kennedy, J. A.; Thueson, D. O.; Chestnut, J. C.; Adolphson, R. L.; Conroy, M. C. J. Med. Chem. 1989, 32, 1360. doi: 10.1021/jm00126a036
(a) Lee, C. F.; Liu, Y. C.; Badsara, S. S. Chem. Asian J. 2014, 9, 706.
(b) Eichman, C. C.; Stambuli, J. P. Molecules 2011, 16, 590.
(c) Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596.
(d) Liu, H.; Jiang, X. Chem. Asian J. 2013, 8, 2546.
(e) Li, G.; Dong, D.; Yu, X.; Wang Z. New J. Chem. 2019, 43, 1667.
(f) Dong, D.; Chen, W.; Yang, Y.; Gao, X.; Wang, Z. ChemistrySelect 2019, 4, 2480.
(a) Xu, X.-M.; Chen, D.-M.; Wang, Z.-L. Chin. Chem. Lett. 2020, 31, 49.
(b) Dong, D.-Q.; Hao, S.-H.; Yang, D.-S.; Li, L.-X.; Wang, Z.-L. Eur. J. Org. Chem. 2017, 6576.
(c) Liu, Y.-Y.; Xiong, J.; Wei, L. Chin. J. Org. Chem. 2017, 37, 1667(in Chinese). (刘云云, 熊进, 韦丽, 有机化学, 2017, 37, 1667.)
(d) Xu, X.-M.; Chen, D.-M.; Wang, Z.-L. Chin. J. Org. Chem. 2019, 39, 3338(in Chinese). (徐鑫明, 陈德茂, 王祖利, 有机化学, 2019, 39, 3338.)
(e) Wang, Y.; Liu, Y. Acta Chim. Sinica 2019, 77, 418.
(f) Shamsabadi, A.; Chudasama, V. Org. Biomol. Chem. 2019, 17, 2865.
(g) Guo, Y.; Xiang, Y.; Wei, L.; Wan, J.-P. Org. Lett. 2018, 20, 3971.
(h) Gao, Y.; Liu, Y.; Wan, J.-P. J. Org. Chem. 2019, 84, 2243.
(i) Reddy Kandimalla, S.; Prathima Parvathaneni, S.; Sabitha, G.; Subba Reddy, B. V. Eur. J. Org. Chem. 2019, 1687.
Nalbandian, C.; Miller, E.; Toenjes, S.; Gustafson, J. L. Chem. Commun. 2017, 53, 1494. doi: 10.1039/C6CC09998J
Zou, J.-F.; Huang, W.-S.; Li, L.; Xu, Z.; Zheng, Z.-J.; Yang, K.-F.; Xu, L.-W. RSC Adv. 2015, 5, 30389. doi: 10.1039/C5RA03606B
Hostier, T.; Ferey, V.; Ricci, G.; Gomez-Pardo, D.; Cossy, J. Chem. Commun. 2015, 51, 13898. doi: 10.1039/C5CC05421D
(a) Freckleton, M.; Baeza, A.; Benavent, L.; Chinchilla, R. Asian J. Org. Chem. 2018, 7, 1006.
(b) Dalpozzo, R. Org. Chem. Front. 2017, 4, 2063.
(c) Liu, Y.-Y.; Zhang, Y.; Hu, C.; Wan, J.; Wen, C. RSC Adv. 2014, 4, 35528.
(d) Zhou, X.; Li, X. RSC Adv. 2014, 4, 1241.
(e) Yang, F.; Tian, S. Angew. Chem., Int. Ed. 2013, 52, 4929.
(f) Katrun, P.; Hongthong, S.; Hlekhlai, S.; Pohmakotr, M.; Reutrakul, V.; Soorukram, D.; Jaipetchb, T.; Kuhakarn, C. RSC Adv. 2014, 4, 18933.
(g) Rao, H.; Wang, P.; Wang, J.; Li, Z.; Sun, X.; Cao, S. RSC Adv. 2014, 4, 49165.
(h) Viglianisi, C.; Marcantoni, E.; Carapacchi, V.; Menichetti, S.; Marsil, L. Eur. J. Org. Chem. 2014, 2014, 6405.
Zhang, H.; Bao, X.; Song, Y.; Qu, J.; Wang, B. Tetrahenron 2015, 71, 8885. doi: 10.1016/j.tet.2015.09.070
He, Y.-Q.; Liu, S.; Wen, P.; Tian, W.; Ren, X.-Y.; Zhou, Q.; Ma, H.-J.; Huang, G.-S. ChemistrySelect 2016, 1, 1567. doi: 10.1002/slct.201600257
Yi, S.; Li, M.; Mo, W.; Hu, X.; Sun, N.; Jin, L.; Shen, Z. Tetrahedron Lett. 2016, 57, 1912. doi: 10.1016/j.tetlet.2016.03.073
Malik, S.; Equbal, D.; Lavekar, A. G.; Sinha, A. K. Org. Biomol. Chem. 2016, 14, 6111. doi: 10.1039/C6OB00930A
Choudhury, P.; Roy, B.; Basu, B. Asian J. Org. Chem. 2017, 6, 1569. doi: 10.1002/ajoc.201700275
Ohkado, R.; Ishikawa, T.; Iida, H. Green Chem. 2018, 20, 984. doi: 10.1039/C8GC00117K
Fan, W.; Yang, Z.; Jiang, B.; Li, G. Org. Chem. Front. 2017, 4, 1091. doi: 10.1039/C6QO00851H
Bai, F.-C.; Zhang, S.; Wei, L.; Liu, Y.-Y. Asian J. Org. Chem. 2018, 7, 371. doi: 10.1002/ajoc.201700677
Kong, D.; Huang, T.; Liang, M.; Wu, M.; Lin, Q. Org. Biomol. Chem. 2019, 17, 830. doi: 10.1039/C8OB02800A
Guo, W.; Tan, W.; Zhao, M.; Tao, K.; Zheng, L.-Y.; Wu, Y.; Chen, D.; X.-L. RSC Adv. 2017, 7, 37739. doi: 10.1039/C7RA08086G
Chen, M.; Luo, Y.; Zhang, C.; Guo, L.; Wang, Q.; Wu, Y. Org. Chem. Front. 2019, 6, 116. doi: 10.1039/C8QO00726H
(a) Chen, S.-Q.; Wang, Q.-M.; Xu, P.-C.; Ge, S.-P.; Zhong, P.; Zhang, X.-H. Phosphorus, Sulfur Silicon Relat. Elem. 2016, 191, 100.
(b) Rahaman, R.; Devi, N.; Barman, P. Tetrahedron Lett. 2015, 56, 4224.
(c) Devi, N.; Rahaman, R.; Sarma, K.; Khan, T.; Barman, P. Eur. J. Org. Chem. 2017, 2017, 1520.
(d) Rafique, J.; Saba, S.; Rosrio, A. R.; Braga, A. L. Chem.-Eur. J. 2016, 22, 11854.
(e) Bettanin, L.; Saba, S.; Doerner, C. V.; Franco, M. S.; Godoi, M.; Rafique, J.; Braga, A. L. Tetrahedron 2018, 74, 3971.
Thurow, S.; Penteado, F.; Perin, G.; Alves, D.; Santi, C.; Monti, B.; Schiesser, C. H.; Lenardao, E. J. Org. Chem. Front. 2018, 5, 1983. doi: 10.1039/C8QO00360B
Ye, L.-M.; Chen, J.; Mao, P.; Zhang, X.-J.; Yan, M. Tetrahedron Lett. 2017, 58, 2743. doi: 10.1016/j.tetlet.2017.05.090
Yu, Y.; Zhou, Y.; Song, Z.-Q.; Liang, G. Org. Biomol. Chem. 2018, 16, 4958. doi: 10.1039/C8OB00948A
Liu, C.; Ding, L. Org. Biomol. Chem. 2015, 13, 2251. doi: 10.1039/C4OB02575J
Rahaman, R.; Devi, N.; Bhagawati, J. R.; Barman, P. RSC Adv. 2016, 6, 18929. doi: 10.1039/C5RA26425A
Al-Saedy, M. A. E.; Joseph, A.-C. M. A. N.; Harrity, P. A. Synlett 2017, 28, 349.
Liu, C.; Fan, J.; Wu, M.; Chen, J.-H.; Zhao, Y.; Xie, M.-H. Chin. J. Chem. 2018, 36, 819. doi: 10.1002/cjoc.201800164
Zhao, X.; Wei, A.; Yang, B.; Li, T.-J.; Li, Q.; Qiu, D.; Lu, K. J. Org. Chem. 2017, 82, 9175. doi: 10.1021/acs.joc.7b01226
Zhao, X.; Zheng, X.; Tian, M.-M.; Sheng, J.; Tong, Y.-F.; Lu, K. Tetrahedron 2017, 73, 7233. doi: 10.1016/j.tet.2017.11.019
Yang, X.; Bao, Y.; Dai, Z.; Zhou, Q.; Yang, F. Green Chem. 2018, 20, 3727. doi: 10.1039/C8GC01764F
Siauciulis, M.; Sapmaz, S.; Pulis, A. P.; Procter, D. J. Chem. Sci. 2018, 9, 754. doi: 10.1039/C7SC04723A
(a) Gullapalli, K.; Ragam, R.; Narayanarao, V. RSC Adv. 2015, 5, 22718.
(b) Lu, K.; Deng, Z.; Li, M.; Li, T.; Zhao, X. Org. Biomol. Chem. 2017, 15, 1254.
(c) Zhao, X.; Wei, A.; Li, T.; Su, Z.; Chen, J.; Lu, K. Org. Chem. Front. 2017, 4, 232.
He, Y.; Jiang, J.; Bao, W.; Deng, W.; Xiang, J. Tetrahedron Lett. 2017, 58, 4583. doi: 10.1016/j.tetlet.2017.10.040
Equbal, D.; Saima, R. S.; Lavekar, A. G.; Sinha, A. K. J. Org. Chem. 2019, 84, 2660. doi: 10.1021/acs.joc.8b03097
Ghosh, A.; Lecomte, M.; Kim-Lee, S.-H.; Radosevich, A. T. Angew. Chem., Int. Ed. 2019, 58, 2864. doi: 10.1002/anie.201813919
Denmark, S. E.; Beutner, G. L. Angew. Chem., Int. Ed. 2008, 47, 1560. doi: 10.1002/anie.200604943
Yang, Y.; Zhang, S.; Tang, L.; Hu, Y.; Zha, Z.; Wang, Z. Green Chem. 2016, 18, 2609. doi: 10.1039/C6GC00313C
Rahaman, R.; Devi, N.; Sarma, K.; Barman, P. RSC Adv. 2016, 6, 10873. doi: 10.1039/C5RA24851E
(a) Ge, W.; Wei, Y. Green Chem. 2012, 14, 2066.
(b) Azeredo, J. B.; Godoi, M.; Martins, G. M.; Silveira, C. C.; Braga, A. L. J. Org. Chem. 2014, 79, 4125.
(c) Xiao, F. H.; Xie, H.; Liu, S. W.; Deng, G. J. Adv. Synth. Catal. 2014, 356, 364.
Qi, H.; Zhang, T.; Wan, K.-F.; Luo, M.-M. J. Org. Chem. 2016, 81, 4262. doi: 10.1021/acs.joc.6b00636
Li, J.; Cai, Z.-J.; Wang, S.-Y.; Ji, S.-J. Org. Biomol. Chem. 2016, 14, 9384. doi: 10.1039/C6OB01528J
Rahaman, R.; Barman, P. Eur. J. Org. Chem. 2017, 2017, 6327. doi: 10.1002/ejoc.201701293

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:



























