图式 1.
有机小分子催化不对称酰基化动力学拆分
Scheme 1.
Organocatalyzed acylation KR of amines

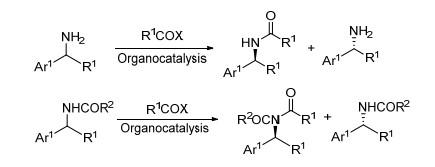
手性胺类化合物广泛存在于自然界, 是蛋白质、核酸、激素、抗生素和生物碱等生理活性物质的重要组成部分.同时, 因为高密度的结构信息和生成氢键的能力, 手性胺也是重要的药物, 如用于镇痛、镇咳、催眠的Codeine[1], 治疗抑郁症的Sertraline[2], 减肥药物Sibutramine[3]以及HIV蛋白酶抑制剂Lopinavir[4]等.另外, 手性胺作为重要的中间体和砌块, 被广泛应用于有机合成中.在有机不对称催化中, 手性胺作为金属配体或小分子催化剂也起着重要的作用[5].因为手性胺的广泛应用价值, 它的合成一直吸引着化学家们的巨大兴趣.目前, 合成手性胺的方法已经有许多报道, 而动力学拆分(Kinetic Resolution, 简称KR)[6]则是其中一类重要方法.
在手性化合物的众多不对称合成方法中, KR是最古老的一种. KR在现有方法中是独一无二的, 因为很多情况下它可以制备ee值非常高的对映异构体, 远远超出现代分析技术的检测极限.这一特点引起了学术界和产业界的广泛兴趣, 并被认为是自然界中手性起源之谜的可能解释之一.另一个重要特点是KR几乎可以与所有的对映选择性合成方法结合使用, 并且几乎可以用于合成所有的手性底物.因此, KR已被广泛应用于外消旋化合物的手性分离, 其中最负盛名的一种应用是通过生物酶或小分子催化的酰基化反应来分离拆分手性产物与手性原料.
值得注意的是, 无论生物酶法还是化学法都能实现一级或二级醇通过酰基化的高效拆分[7], 并且已在工业界实现了大规模应用.相对于醇类化合物, 胺类化合物(一级或二级胺)通常具有较强的亲核性, 因此用任何常规酰基化试剂进行酰基化反应时, 都会导致快速的酰基化反应, 从而降低反应的的选择性; 同时, 胺类化合物也容易与金属催化剂配位, 从而降低反应活性和选择性.因此, 胺类化合物的动力学拆分更具有挑战性, 发展也较为缓慢. 2001年, 自Fu课题组[8a]报道了首例有机小分子催化不对称酰基化动力学拆分的研究工作以来(Scheme 1), Seidel等[8b~8d]课题组也相继报道了基于不对称酰基化的拆分成果.这些不对称酰基化的拆分都局限于一级胺、二级环状胺以及氮上具有较低亲核性的酰胺[9]等底物.
近年来随着不对称催化反应的不断发展, 采用非酰基化的不对称催化反应来实现胺类化合物的动力学拆分越来越受到化学家们的关注, 并且获得了越来越多的研究成果.根据氮原子是否参与反应, 对该类非酰基化化学动力学拆分进行了梳理分类, 并根据反应类型的不同对动力学拆分研究现状进行了综述.
近年来, 采用非酰基化的化学动力学拆分制备光学纯手性胺的方法得到化学家们越来越多的关注与研究.其中, 氮原子参与催化反应的例子已经得到了许多报道.根据氮原子参与反应的类型可将其分为非酶催化的氧化还原动力学拆分、氮杂环丙烷不对称开环动力学拆分及2H-氮杂丙烯啶的不对称加成动力学拆分、C—X活化动力学拆分、取代动力学拆分及其他类型反应的动力学拆分等.
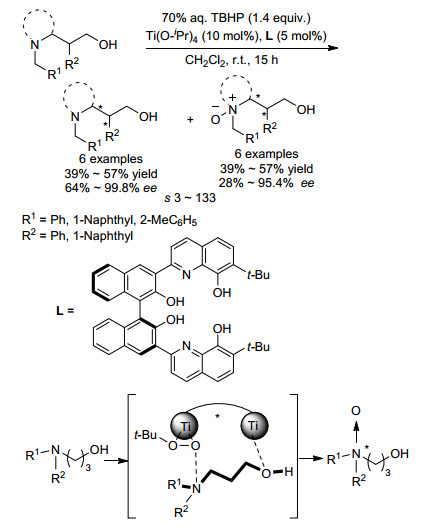
通过二级醇类化合物不对称脱氢生成潜手性酮, 从而合成手性二级醇的方法被称为氧化动力学拆分(OKR), 目前许多使用过渡金属催化的醇类化合物的OKR法已经被开发出来[10].相比之下, 胺类化合物的OKR法存在许多挑战, 主要是因为氮原子容易被氧化, 胺类化合物作为路易斯碱通常会和金属配位, 从而使催化剂失活.因此, 胺类化合物的OKR研究最先从叔胺类底物开始.早在20世纪八、九十年代, Sharpless课题组[11]就通过Ti催化的选择性N氧化实现了叔胺类化合物的OKR.得到了手性β-氨基醇类化合物.而在2016年, Yamamoto课题组[12]发展了Ti催化的γ-羟基叔胺化合物的高效OKR体系, 获得了光学活性的γ-氨基醇类产物(Scheme 2).该课题组利用Ti(OiPr)4与手性N, O配体配位形成双中心催化剂, 同时与底物中的羟基和氨基配位, 通过催化剂的调控让羟基官能团远离氨基, 从而使得不对称氧化选择性发生在氨基位置.该反应以TBHP作为氧化剂, 拆分系数s最高可达133, 回收的γ-氨基醇对映选择性最高可达99.4 %.
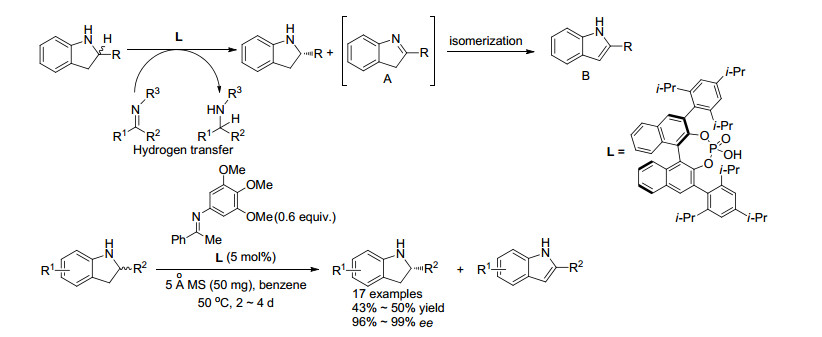
最近几年, 通过仲胺不对称脱氢形成亚胺从而实现二级胺的OKR也有报道. 2013年, Akiyama课题组[13]报道了首例伯胺的高效OKR, 他们利用不对称转移氢化反应实现了二氢吲哚类化合物的氧化动力学拆分(Scheme 3).该课题组以手性磷酸为催化剂, 避免了过渡金属催化剂因底物配位而失活的缺陷; 同时以酮亚胺为氢受体, 回收得到大于99% ee的手性二氢吲哚原料, 而这些氮上无保护的2位芳基取代二氢吲哚类化合物使用之前报道的不对称氢转移反应是无法合成的.通过控制实验, 该课题组发现在标准反应条件下, R构型的底物能被选择性氧化, 而S构型的底物基本没有反应.
2015年, Akiyama课题组[13]利用相同的体系, 实现了四氢喹啉及其衍生物的高效OKR (Scheme 4).该反应体系对2位苯基类和直链烷基类底物都有很好的效果, 特别是当取代基为正戊基时, 可回收98% ee的原料, 拆分系数s达到458.当底物拓展到含有杂原子的四氢喹啉类化合物时, 回收原料的对映选择性有了明显的下降, 例如原料含有S原子时, 对映选择性只有73%;而底物环状结构的扩大也导致拆分效果明显下降, 对于苯并氮杂䓬底物, 因为结构中含有七元环, 只能回收62% ee的原料.
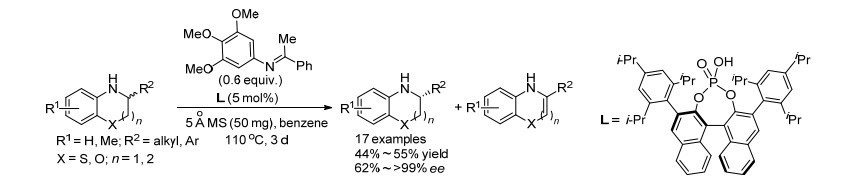
2016年, Akiyama课题组[15]利用自氧化还原反应同样实现了二氢吲哚类化合物的动力学拆分(Scheme 5).在该课题组[13]之前报道的二氢吲哚的OKR反应中, 需要加入酮亚胺作为氢受体, 但酮亚胺需要首先制备并保存.于是, 该课题组考虑利用二氢吲哚与醛原位缩合生成的亚胺作为氢受体, 通过分子内氢转移, 发生自氧化还原反应来实现二氢吲哚的OKR.该反应依然以手性磷酸作为催化剂, 可回收33%~99% ee的手性二氢吲哚原料.该反应体系对底物具有较好的官能团容忍性, 并且回收得到R构型的二氢吲哚原料.而之前该课题组使用的OKR体系则得到S构型的原料.
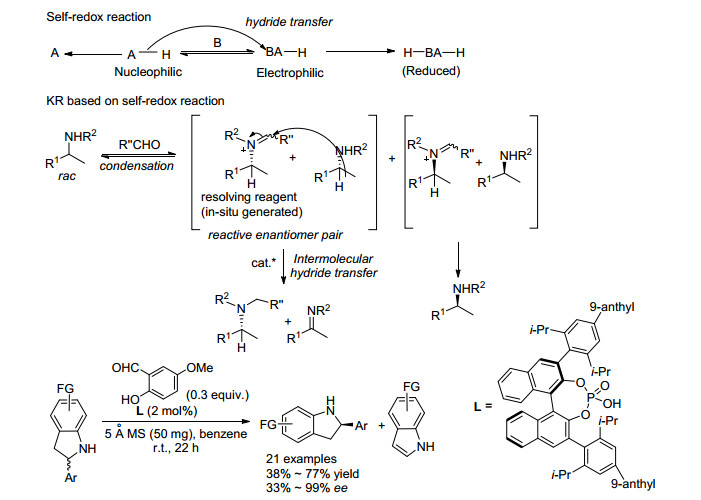
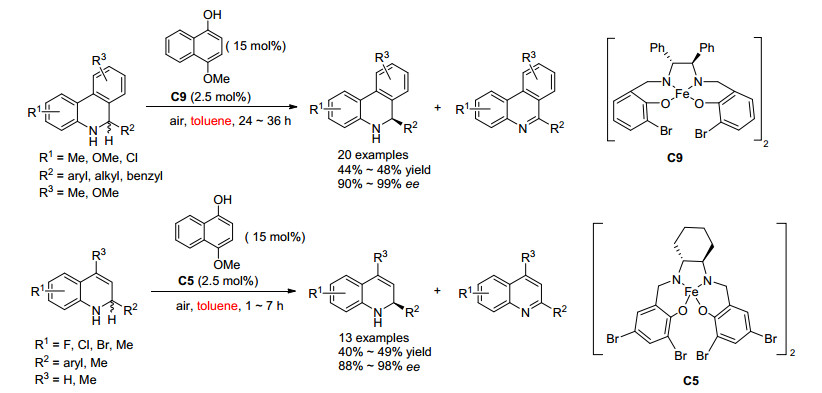
2019年, 刘磊课题组[16]首次报道了利用O2为氧化剂, Fe催化的环状仲胺的OKR (Scheme 6), 将二级胺的OKR体系由小分子催化拓展到金属催化.该反应以4-甲氧基-1-萘酚为添加剂, 在空气条件下即可实现环状仲胺的高效OKR, 对映选择性最高可达99%.对于5, 6-二氢菲咯烷类底物, 苯环上取代基为供电子基时取得了很好的效果; 对于1, 2-二氢喹啉类底物, 苯环上取代基为供电子基或吸电子基都能得到很高的对映选择性.通过机理研究, 该课题组发现添加剂4-甲氧基-1-萘酚在反应中起了重要的作用, 若没有添加剂, 则反应回收的原料只有35% ee; 单晶结构显示, 添加剂会与催化剂配位, 形成新的催化剂结构, 从而提高反应的对映选择性.
氮杂环丙烷是一类具有生物活性的三元杂环化合物, 许多天然产物中含有氮杂环丙烷结构, 它们具有抗病毒、抗肿瘤的生物活性[17].当氮杂环丙烷中氮原子上连有磺酰基、酰基或膦酰基等吸电子取代基时, 会降低三元环上的电子云密度, 更容易受到亲核试剂的进攻, 在开环反应中表现出较高的活性, 从而合成含有不同官能团的胺、氨基酸和氨基硫醇等化合物.因此, 利用氮杂环丙烷的不对称开环反应实现胺的动力学拆分越来越受到化学家的关注.
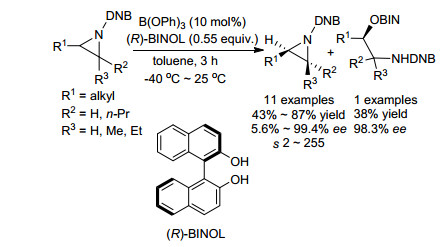
2012年, Morgan课题组[18]报道了利用手性BINOL和氮杂环丙烷的不对称开环反应实现氮杂环丙烷动力学拆分的例子(Scheme 7).该反应以B(OPh)3作为Lewis酸催化剂, 避免了N-DNB氮杂环丙烷重排生成噁唑啉的副反应, 并可回收最高99.4% ee的R构型原料, 拆分系数s最高可达255.空间位阻增大导致该反应拆分效果明显下降, 例如, R1基团为叔丁基时, 只得到对映选择性为5.6%的产物, 拆分系数s则只有2.
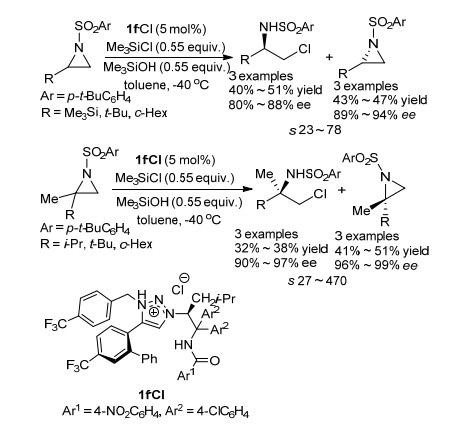
2012年, Ooi课题组[19]报道了有机小分子催化的氮杂环丙烷的不对称开环动力学拆分(Scheme 8).该课题组使用手性氯化1, 2, 3-三唑鎓盐为催化剂, Me3SiOH为添加剂, 利用Me3SiCl为亲核试剂对2-取代氮杂环丙烷底物进行不对称开环反应, 实现了2-取代氮杂环丙烷类化合物的动力学拆分.该课题组研究发现, 使用2-取代氮杂环丙烷为反应底物, 回收得到大于89% ee手性氮杂环丙烷原料的同时得到产物β-氯α仲胺, 此时的产物没有区域选择性; 然而当底物为2, 2-取代氮杂环丙烷时, 回收大于96% ee手性氮杂环丙烷原料的同时开环产物会出现区域选择性, 得到两种化合物, 其中主要产物为β-氯-α-叔胺, 对映选择性最高达97%.
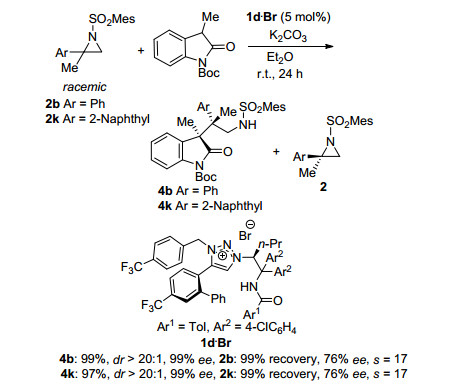
2013年, Ooi课题组[20]继续对2, 2-二取代氮杂环丙烷类化合物的动力学拆分进行研究, 发现当使用3-取代氧化吲哚作为亲核试剂, 可以同时获得高对映选择性和高区域选择性的产物(Scheme 9).该反应使用手性溴化1, 2, 3-Triazolium盐为催化剂, 当取代基为2-Naphthyl时, 可获得99% ee, dr大于20:1的开环产物, 并回收76% ee的2, 2-二取代氮杂环丙烷原料, 拆分系数s为17.
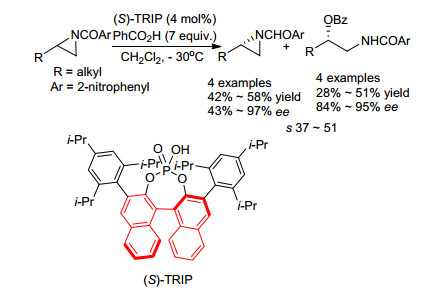
2014年, List课题组[21]报道了手性磷酸催化的端位氮杂环丙烷的不对称开环动力学拆分(Scheme 10), 该课题组使用苯甲酸作为亲核试剂, 可实现2-取代氮杂环丙烷的高效拆分, 拆分系数s最高可达51.该反应体系中底物的取代基为长链烷基和环烷基.
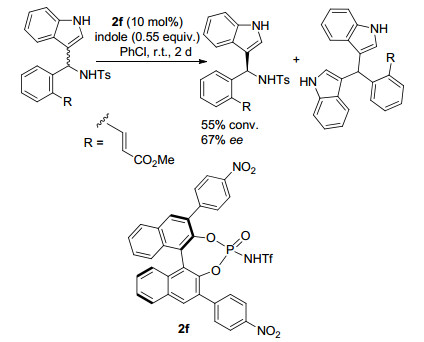
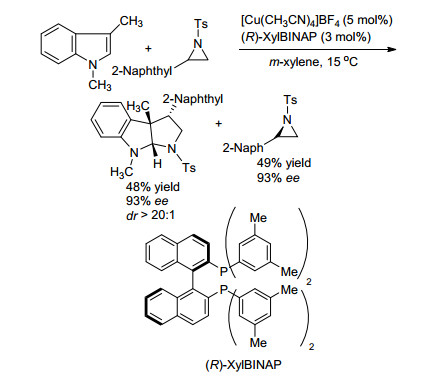
2015年, 柴卓课题组[22]报道了利用不对称[3+2]环加成反应实现氮杂环丙烷动力学拆分的例子(Scheme 11).该课题组以Cu(CH3CN)4BF4为金属, R-xylBINAP为手性配体, 通过氮杂环丙烷与吲哚类化合物的不对称[3+2]环加成反应实现了N-Ts氮杂环丙烷类底物的动力学拆分.对于2位取代基为2-萘基的底物.得到对映选择性为93%的环加成产物, 并回收对映选择性为93%的原料, 拆分系数s为60.
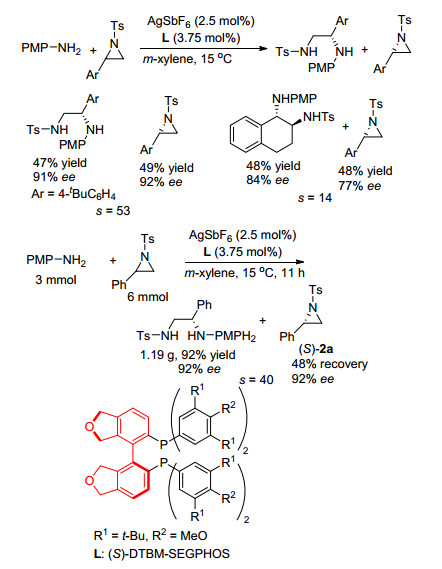
2017年, 柴卓课题组[23]继续报道了以胺作为亲核试剂实现氮杂环丙烷不对称开环动力学拆分的例子(Scheme 12).该课题组发现, 当使用Ag为金属, PMPNH2作为亲核试剂时, 可实现两2位取代的N-Ts氮杂环丙烷的动力学拆分, 拆分系数s最高可达53.同时, 对于2位取代基为Ph的底物, 可实现克级规模的动力学拆分.
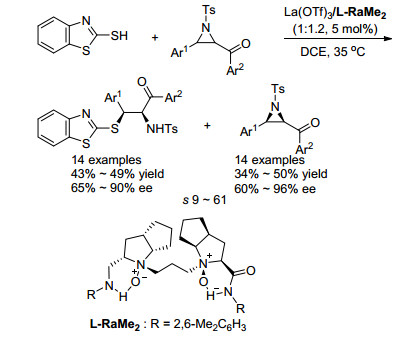
2019年, 冯小明课题组[24]报道了以巯基苯并噻唑作为亲核试剂, La(OTf)3/L-RaMe2配合物为手性催化剂, 实现1, 2-二取代氮杂环丙烷动力学拆分的例子.该反应拆分系数s最高达61 (Scheme 13).该课题组通过改变溶剂的用量, 成功实现了克级规模原料的动力学拆分, 并且得到了良好的效果.
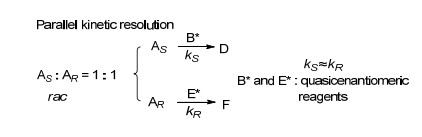
平行动力学拆分(Parallel Kinetic Resolution)是指两个具有互补立体选择性的手性试剂B*和E*, 与消旋底物的S构型(AS)和R构型(AR)分别反应得到两个非对映产物D和F, 从而实现底物构型分离的方法.在同样的反应条件下, S构型(AS)和R构型(AR)具有相近的反应活性(kS≈kR), 但立体选择性相反; 而在整个反应过程中, AS和AR的浓度基本相同, 从而能同时得到两种高产率与高对映选择性的产物(Scheme 14)[25].
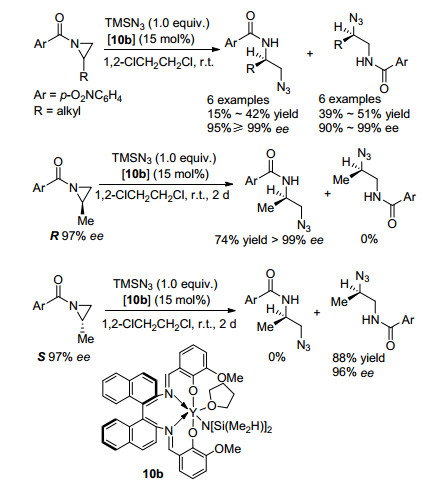
2009年RajanBabu课题组[26]报道了Y催化的氮杂环丙烷的开环平行动力学拆分的例子(Scheme 15).该课题组以TMSN3为亲核试剂, 10b为手性催化剂, 在1, 2-二氯乙烷条件下室温反应2~6 h, 即可实现2-烷基取代氮杂环丙烷的高效平行动力学拆分, 得到两种R构型的酰胺产物, 对映选择性最高可达99%. 2014年, 该课题组的机理研究表明, R构型的底物在催化条件下, TMSN3进攻3位; S构型的底物在催化条件下, TMSN3进攻2位[27].
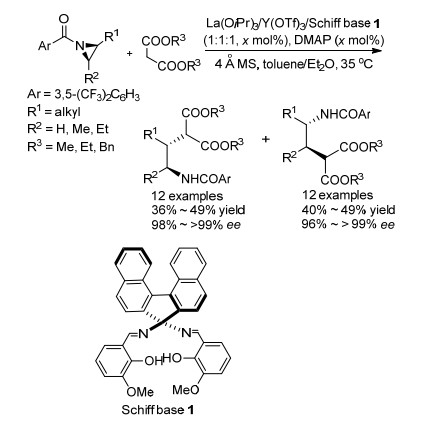
2014年Shibasaki课题组[28]报道了以丙二酸酯类化合物为亲核试剂实现氮杂环丙烷平行动力学拆分的例子(Scheme 16).该课题组以双核手性席夫碱为手性配体, 以La和Y作为双核金属, 通过丙二酸酯对氮杂环丙烷的区域选择性不对称开环实现了N-酰基氮杂环丙烷的平行动力学拆分, 得到大于98% ee的两个产物, 并且当底物由2取代拓展为2, 3-二取代时, 拆分效果依然很好.
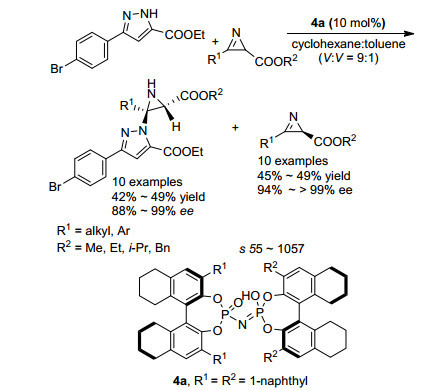
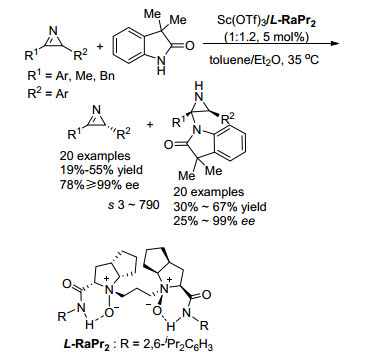
2H-氮杂丙烯啶是合成氮杂环丙烷的重要前体, 通过亲核试剂对2H-氮杂丙烯啶的不对称加成可以合成各种类型的手性氮杂环丙烷化合物. 2016年, Feng课题组[29]报道了利用Sc催化的2H-氮杂丙烯啶不对称加成动力学拆分获得手性氮杂环丙烷和手性2H-氮杂丙烯啶的例子(Scheme 17).该课题组以吲哚酮作为亲核试剂.以Sc(OTf)3/L-RaPr2配合物为手性催化剂, 可以取得优异的拆分结果, 拆分系数s最高达790.与之前文献报道不同的是, 亲核反应发生在吲哚酮亲核性较弱的1位氮原子而不是3位碳原子, 该反应体系对底物官能团容忍性较高, 不管R1是芳基还是烷基, 都能给出优秀的结果; 同时, R2基团芳环上的吸电子基和供电基都不影响效率.
2016年.张锁秦课题组[30]报道了利用吡唑类化合物对2H-氮杂丙烯啶的不对称加成, 从而实现2H-氮杂丙烯啶动力学拆分的例子(Scheme 18).该课题组利用手性磷酸作为催化剂.以3位取代基为4-溴苯基的吡唑酸乙酯为亲核试剂, 可选择性对(S)-2H-氮杂丙烯啶羧酸酯加成, 从而实现2H-氮杂丙烯啶的动力学拆分, 得到高对映选择性的产物和原料.拆分系数s最高可达到1051.
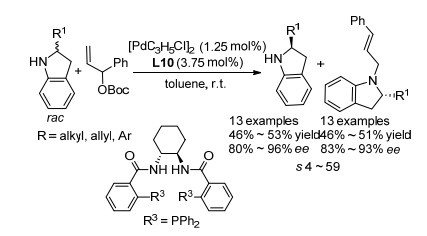
不对称烯丙基取代反应是通过碳-碳和碳-杂原子成键从而构建手性中心的强有力方法.目前, 将不对称烯丙基取代应用于胺类化合物的拆分也有了报道. 2009年, 侯雪龙课题组[31]报道了利用钯催化的不对称烯丙基取代反应实现二氢吲哚类化合物动力学拆分的例子(Scheme 19).该反应利用手性P, N类配体, 以烯丙基Boc酯为亲电试剂, 在室温条件下即可实现二氢吲哚类化合物的动力学拆分.该反应体系对2-芳基二氢吲哚底物有较好的拆分效果, 特别是取代基为对甲氧基苯基时, 可得到92% ee的产物并回收80% ee的原料, 拆分系数s可达59.然而, 当2位取代基为烷基时, 拆分效果很差, 拆分系数s小于10;同时, 对于四氢喹啉类化合物, 拆分结果同样不好.
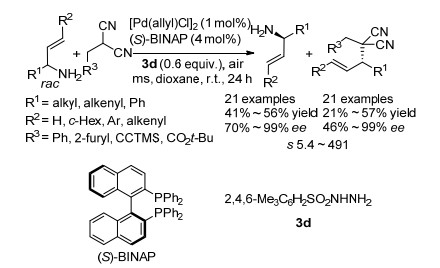
2015年, 田仕凯题组[32]报道了利用钯催化不对称烯丙基烷基化反应实现了一级烯丙基胺的动力学拆分的例子(Scheme 20).该课题组[33]之前研究了磺酰肼与一级烯丙基胺的氧化偶联反应, 并提出了磺酰肼加速一级烯丙基胺烯丙基烷基化的可能机理.于是, 该课题组将磺酰肼应用于一级烯丙基胺的动力学拆分中, 以提高拆分反应的活性, 并取得了良好的效果.该课题组利用手性BINAP作为配体, 在空气下反应, 即实现了一级烯丙基胺的高效动力学拆分, 拆分系数s最高可到491, 但是该反应体系的底物受限于芳基取代的烯丙基胺.
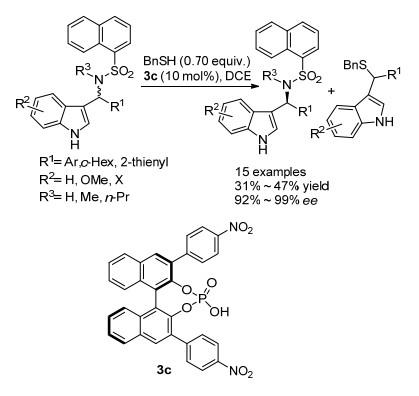
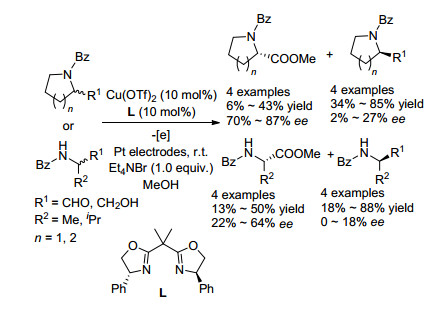
2009年, 田仕凯课题组[34]报道了硫醇作为亲核试剂与N-苄基磺胺类化合物的偶联反应(Scheme 21). 2012年, 该课题组[35]利用手性磷酸实现了硫醇对N-(3-吲哚基)(苯基)甲基磺胺类化合物的不对称亲核取代动力学拆分[35].该反应体系在温和条件下进行, 并且拆分效果很好, 回收的N-(3-吲哚基)(苯基)甲基磺胺类原料对映选择性大于90%.当底物拓展到叔胺类化合物时, 依然能得到很高的对映选择性, 但是当3-吲哚基的NH基团被NCH3基团取代时, 反应效果很差, 回收原料的对映选择性只有15%, 表明NH基团是实现该类化合物高效动力学拆分的基本骨架.
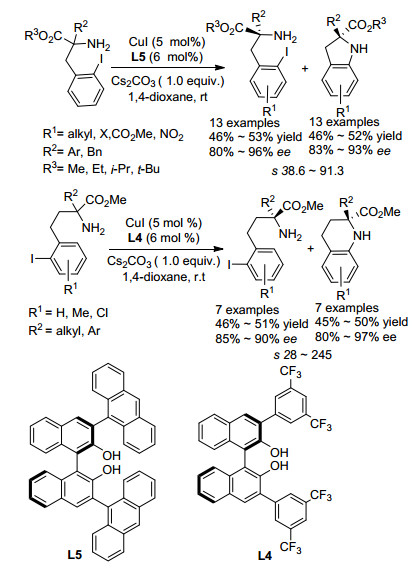
碳卤活化是重要的有机合成方法, 并且在有机合成中得到了广泛的应用.近年来, 将不对称碳氢活化及碳卤活化应用于胺的动力学拆分也有了一些研究. 2013年, Cai课题组[36]报道利用C—I活化, 通过分子内的不对称N芳基化, 实现了α叔胺类化合物的动力学拆分(Scheme 22).该课题组利用CuI作为催化剂, 手性BINOL衍生物作为配体, 在温和条件下快速地实现了2-氨基-3-(2-碘代芳基)丙酸酯类叔胺化合物的动力学拆分, 以高对映选择性回收原料的同时也得到了高对映选择性的具有季碳手性中心的二氢吲哚类产物, 拆分选择系数s最高可达91.3.该课题组将底物扩展到2-氨基- 4-(2-碘代芳基)丁酸酯类化合物时, 同样得到了很好的拆分结果.四氢喹啉类产物对映选择性最高可达97%, 拆分系数s最高可达245.
2008年, Enders课题组[37]报道了手性磷酰胺催化的苄胺的立体化学消融动力学拆分(Scheme 23).该反应只有当R基团为3-丙烯酸甲酯基时动力学拆分才能实现, 并可获得67% ee的手性苄胺原料.
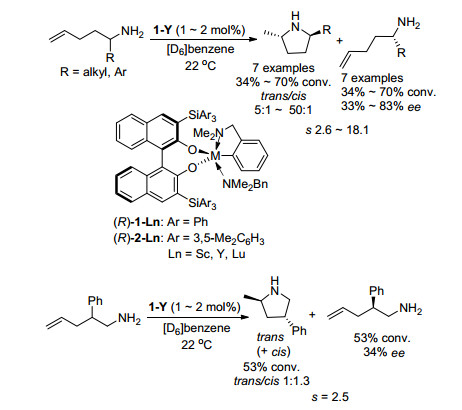
2009年, Hultzsch课题组[38]报道了稀土金属催化的γ-氨基-γ-取代烯烃化合物的氢胺化动力学拆分(Scheme 24).该反应使用手性双酚和联萘酚作为配体, 通过γ-氨基-γ-取代烯烃的分子类不对称氢胺化反应实现了动力学拆分, 得到了非对映选择性(反式/顺式高达50:1)的环状胺类化合物, 并回收了中等到良好对映选择性的原料, 拆分系数s最高达19.该研究发现, 随着烷基取代基的空间位阻增加, 含有脂肪族取代基的底物拆分效果不断下降, 产物的反/顺-非对映选择性增加; 当底物由γ-氨基-γ-取代烯烃拓展到γ-氨基-β-取代烯烃时, 其拆分效果同样下降, 产物的反/顺-非对映选择性小于2:1, 回收的原料对映择性也很差.
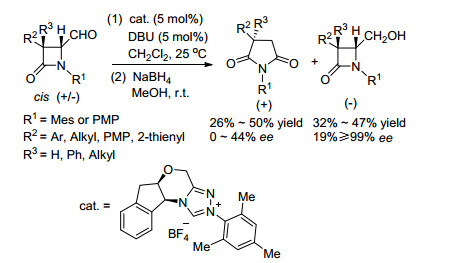
2008年游书力课题组[39]报道了手性NHC催化的β内酰胺的扩环动力学拆分的例子(Scheme 25).该小组利用β内酰胺的扩环反应实现了cis-4-甲酰基-β内酰胺的高效动力学拆分, 回收的cis-4-甲酰基-β-内酰胺对映选择性最高可达到99%, 但是该反应的产物对映选择性很低.
2013年冯小明课题组[40]报道了通过有机小分子催化的不对称氧化胺化实现氧杂吖丙啶类化合物的动力学拆分的例子(Scheme 26).该课题组利用吖内酯底物的多位点反应性, 通过吖内酯的不对称氧化胺化成功实现了氧杂吖丙啶类化合物物的动力学拆分, 回收高对映选择性的光学氧杂吖丙啶原料的同时, 得到中等到良好对映选择性的4-恶唑烷酮产物.
2014年池永贵课题组[41]报道了通过NHC催化的不对称[3+4]环化反应, 实现了甲亚胺亚胺动力学拆分(Scheme 27).该课题组利用甲亚胺亚胺和共轭烯醛的不对称环化, 以最高93%的对映选择性回收了原料, 并获得了ee大于93%的产物, 拆分系数s最高达339.
胺类化合物(一级或二级胺)氮原子的强亲核性和强配位性通常导致动力学拆分反应效果不佳.于是.通过氮原子不参与不对称催化反应的方法来实现高效动力学拆分成为化学家们研究的另一种策略.氮原子不参与不对称催化反应, 不仅能避免胺类化合物自身固有缺点, 而且还可以在底物结构中引入另一个手性中心, 使得拆分产物有更多潜在应用前景.目前, 氮原子不参与的化学动力学拆分也得到了一些研究和发展.
手性氨基酸是生物体的组成部分, 同时也是重要的有机合成原料及中间体, 具有广泛的药理和生理活性.因此, 合成光学活性的天然和非天然氨基酸一直吸引着化学家的兴趣. 2008年, Onomura课题组[42]报道了氨基醛类化合物及氨基醇类化合物的氧化动力学拆分(Scheme 28).该课题组以(R, R)-Ph-BOX作为手性配体, 实现了Cu催化的N-保护的1.2-氨基醛和1.2-氨基醇的不对称电化学氧化反应, 以较低的产率得到高对映选择性的氨基酸甲酯, 表明了氨基醛类化合物及氨基醇类化合物的动力学拆分的可行性.
之后, Onomura课题组[43]继续研究, 发现当使用N-碘代琥珀酰亚胺(NIS)作为氧化剂时, 可代替电化学氧化, 实现铜催化1.2-氨基醛的选择性OKR (Scheme 29), 得到光学活性氨基酸甲酯化合物, 同时以氨基醛二甲基缩醛的形式回收原料.该催化体系对环状和非环状底物都有良好的催化活性, 能以较高的产率获得对映选择性最高99%的氨基酸甲酯化合物, 选择系数s最高可达368;但是, 该体系回收的氨基醛二甲基缩醛类原料只有中等的对映选择性.
2009年, Sibi课题组[44]报道了利用铜催化的选择性Diels-Alder环加成反应, 实现吡唑烷酮类化合物的动力学拆分(Scheme 30).该课题组使用手性BOX配体, 利用环戊二烯和不饱和吡唑烷酮酰亚胺的不对称环加成反应, 得到了高对映选择性的手性吡唑烷酮类化合物, 对映选择性最高可达到99%.该课题组研究表明, 当底物R2基团为芳基类时, 能得到很好的拆分效果.
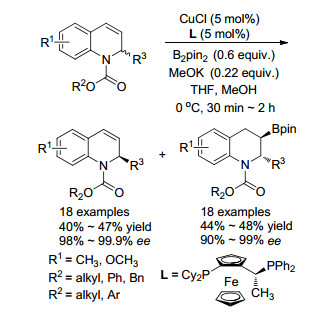
2017年, 侯国华课题组[45]报道了利用铜催化的不对称硼化反应, 实现1, 2-二氢喹啉化合物的动力学拆分(Scheme 31).该课题组使用CuCl作为金属盐, JosiPhos-1作为手性配体, 高效、高选择性地实现了2-取代-1, 2-二氢喹啉的动力学拆分.该反应催化体系能够在30 min内完成底物的动力学拆分, 以优异的选择性(dr>99:1, 99.7% ee)获得了手性硼氢化产物, 同时以99.4% ee的对映选择性回收了未反应的原料.该方法具有很好的底物普适性, 2位芳基或烷基取代的底物均能得到优秀的拆分效果, 拆分选择系数s最高可达569.
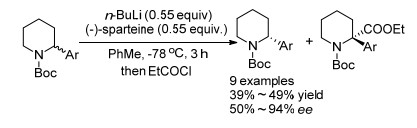
2014年Coldham课题组[46]报道了利用n-BuLi/(-)- sparteine或n-BuLi/(+)-sparteine作为手性碱, 通过不对称去质子化实现了N-Boc-2-芳基哌啶的动力学拆分, 并取得了不错的效果(Scheme 32).该反应对取代芳香环上有强吸电子基的底物效果很差, 这可能是由于强酸性底物锂化困难.
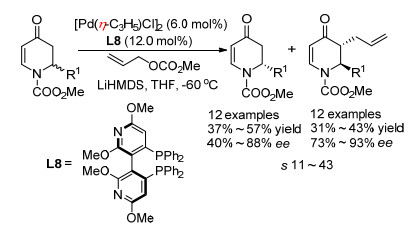
2014年侯雪龙课题组[47]利用钯催化不对称烯丙基烷基化反应, 实现了2-取代2, 3-二氢-4-吡啶酮类化合物的动力学拆分(Scheme 33).该反应以(S)-P-PHOS作为手性配体, 以烯丙基碳酸甲酯作为亲电试剂, 在低温下反应, 得到较高对映选择性的反式烯丙基化产物的同时, 能够以中等到良好的对映选择性回收2-取代2, 3-二氢-4-吡啶酮类原料, 拆分系数s在11到43之间, 2位取代基对产物的对映选择性影响较小, 但是对对回收原料的对映选择性影响很大.
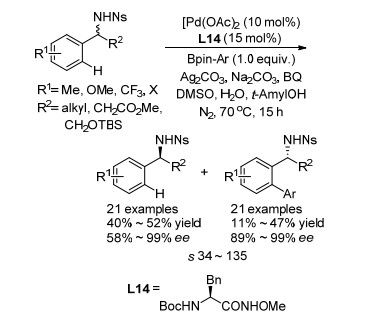
目前, 钯催化的不对称C—H活化已经得到了许多研究和发展, 将不对称C—H交叉偶联应用于外消旋苄胺动力学拆分反应, 显得十分有吸引力, 因为这种方法将得到手性苄胺和手性邻芳基苄胺两种具有广泛应用价值的骨架. 2016年, Yu课题组[48]报道了利用不对称C—H官能化实现苄胺类化合物的动力学拆分(Scheme 34).该反应利用氨基酸衍生物Boc-L-Bn-NHOMe为手性配体, 与芳基硼酸酯在N2条件下发生偶联反应, 得到了高对映选择性邻芳基苄胺产物的同时回收了高对映选择性的苄胺原料.该反应具有良好的拆分效果, 拆分系数s最高可达135, 并且对各种取代苄胺和取代芳基硼酸酯都有良好的官能团耐受性.
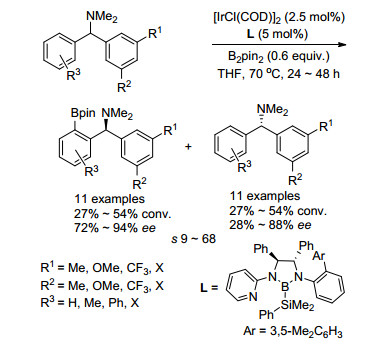
2019年, 徐森苗课题组[49]利用Ir催化的不对称C—H硼化, 实现了外消旋二芳甲基胺的C—H活化动力学拆分(Scheme 35).当外消旋二芳甲基胺的两个芳基取代基不同, 在进行C—H活化时, 就会出现区域选择性.如果催化剂能够区分两个光学异构体, 即可实现二芳甲基胺类化合物的动力学拆分.于是该课题组利用手性双齿硼酸配体, 成功实现了芳基取代基不同的二芳甲基胺类的动力学拆分, 得到对映选择性最高93%的硼化产物.然而只能回收对映选择性中等到良好的二芳甲基胺原料, 根据苯环取代基的电子性质和位置, 这些反应给出的选择系数s值在9~68之间.
虽然近年来胺的动力学拆分的研究有了新的发展, 但相较于醇的动力学拆分, 就以下几方面仍然有待进一步发展. (1)拓展更多的氮参与反应的胺的不对称催化动力学拆分方法, 从而实现各类胺的手性拆分. (2)开发氮不参与反应的不对称反应的动力学拆分方法, 不但能实现更多类型胺的动力学拆分, 同时也能为高效构建多手性中心的含氮化合物提供新方法. (3)研究开发胺的动态动力学拆分, 突破动力学拆分的最高只有50%收率的限制.
Yoshimura, H.; Oguri, K.; Tsukamoto, H. Tetrahedron Lett. 1968, 9, 483. doi: 10.1016/S0040-4039(01)98789-1
Lautens, M.; Rovis, T. J. Org. Chem. 1997, 62, 5246. doi: 10.1021/jo971115x
Jeffery, J. E.; Kerrigan, F.; Miller, T. K.; Smith, G. J.; Tometzki, G. B. J. Chem. Soc., Perkin Trans. 1996, 2583.
Stoner, E. J.; Cooper, A. J.; Diclcman, D. A.; Kolaczkowslti, L.; Lallaman, J. E.; Liu, J. H.; Oliver-Shaffer, P A.; Patel, K. M.; Paterson, J. B.; Plata, D. J.; Riley, D. A.; Sham, H. L.; Stengel, P J.; Tien, J. H. Org. Proc. Res. Dev. 2000, 4, 264. doi: 10.1021/op990202j
France, S.; Guerin, D. J.; Miller, S. J.; Lectka, T. Chem. Rev. 2003, 103, 2985. doi: 10.1021/cr020061a
(a) Vedejs, E.; Jure, M. Angew. Chem., Int. Ed. 2005, 44, 3974.
(b) Kagan, H. B.; Fiaud, J.-C. Top. Stereochem. 1988, 18, 249.
Keith, J. M.; Larrow, J. F.; Jacobsen, E. N. Adv. Synth. Catal. 2001, 343, 5. doi: 10.1002/1615-4169(20010129)343:1<5::AID-ADSC5>3.0.CO;2-I
(a) Arai, S.; Bellemin-Laponnaz, S.; Fu, G. C. Angew. Chem., Int. Ed. 2001, 40, 234.
(b) De, C. K.; Klauber, E. G.; Seidel, D. J. Am. Chem. Soc. 2009, 131, 17060.
(c) Klauber, E. G.; De, C. K.; Shah, T. K.; Seidel, D. J. Am. Chem. Soc. 2010, 132, 13624.
(d) Mittal, N.; Lippert, K. M.; De, C. K.; Klauber, E. G.; Emge, T. J.; Schreiner, P. R.; Seidel, D. J. Am. Chem. Soc. 2015, 137, 5748.
(e) Birman, V. B.; Jiang, H.; Li, X.; Guo, L.; Uffman, E. W. J. Am. Chem. Soc. 2006, 128, 6536.
(f) Yang, X.; Bumbu, V. D.; Liu, P.; Li, X.; Jiang, H.; Uffman, E. W.; Guo, L.; Zhang, W.; Jiang, X.; Houk, K. N.; Birman, V. B. J. Am. Chem. Soc. 2012, 134, 17605.
(g) Arseniyadis, S.; Valleix, A.; Wagner, A.; Mioskowski, C. Angew. Chem., Int. Ed. 2004, 43, 3314.
(h) Arseniyadis, S.; Subhash, P. V.; Valleix, A.; Mathew, S. P.; Blackmond, D. G.; Wagner, A.; Mioskowski, C. J. Am. Chem. Soc. 2005, 127, 6138.
(a) Birman, V. B.; Jiang, H.; Li, X.; Guo, L.; Uffman, E. W. J. Am. Chem. Soc. 2006, 128, 6536.
(b) Yang, X.; Bumbu, V. D.; Liu, P.; Li, X.; Jiang, H.; Uffman, E. W.; Guo, L.; Zhang, W.; Jiang, X.; Houk, K. N.; Birman, V. B. J. Am. Chem. Soc. 2012, 134, 17605.
(c) Fowler, B. S.; Mikochik, P. J.; Miller, S. J. J. Am. Chem. Soc. 2010, 132, 2870.
(d) Bumbu, V. D.; Yang, X.; Birman, V. B. J. Am. Chem. Soc. 2011, 133, 13902.
(e) Bumbu, V. D.; Yang, X.; Birman, V. B. Org. Lett., 2013, 15, 279.
For representative reports on OKR, see: (a) Ferreira, E. M.; Stoltz, B. M. J. Am. Chem. Soc. 2001, 123, 7725.
(b) Muller, J. A.; Sigman, M. S. J. Am. Chem. Soc. 2003, 125, 7005.
(c) Nishibayashi, Y.; Yamauchi, A.; Onodera, G.; Uemura, S. J. Org. Chem. 2003, 68, 5875.
(d) Radosevich, A. T.; Musich, C.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 1090.
(e) Pawar, V. D.; Bettigeri, S.; Weng, S.-S.; Kao, J.-Q.; Chen, C.-T. J. Am. Chem. Soc. 2006, 128, 6308.
(f) Chen, T.; Jiang, J.-J.; Xu, Q.; Shi, M. Org. Lett. 2007, 9, 865.
(g) Arita, S.; Koike, T.; Kayaki, Y.; Ikariya, T. Angew. Chem., Int. Ed. 2008, 49, 2447.
(h) Tomizawa, M.; Shibuya, M.; Iwabuchi, Y. Org. Lett. 2009, 11, 1829.
(i) Kunisu, T.; Oguma, T.; Katsuki, T. J. Am. Chem. Soc. 2011, 133, 12937.
(a) Miyano, S.; Lu, L. D.-L.; Viti, S. M.; Sharpless, K. B. J. Org. Chem. 1983, 48, 3608.
(b) Miyano, S.; Lu, L. D.-L.; Viti, S. M.; Sharpless, K. B. J. Org. Chem. 1985, 50, 4350.
(c) Hayashi, M.; Okamura, M.; Toba, T.; Oguni, N.; Sharpless, K. B. Chem. Lett. 1990, 547.
Bhadra, S.; Yamamoto, H. Angew. Chem., Int. Ed. 2016, 55, 13043. doi: 10.1002/anie.201606354
Saito, K.; Shibata, Y.; Yamanaka, M.; Akiyama, T. J. Am. Chem. Soc. 2013, 135, 11740. doi: 10.1021/ja406004q
(a) Tsutsui, H.; Narasaka, K. Chem. Lett. 1999, 28, 45.
(b) Tsutsui, H.; Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2002, 75, 1451.
Saito, K.; Akiyama, T. Angew. Chem., Int. Ed. 2016, 55, 3148. doi: 10.1002/anie.201510692
Lu, R.; Cao, L.; Guan, H.; Liu, L. J. Am. Chem. Soc. 2019, 141, 6318. doi: 10.1021/jacs.9b00615
(a) Tanner, D. Angew. Chem., Int. Ed. Engl. 1994, 33, 599.
(b) Fruit, C.; Müller, P. Chem. Rev. 2003, 103, 2905.
Cockrell, J.; Wilhelmsen, C.; Rubin, H.; Martin, A.; Morgan, J. B. Angew. Chem., Int. Ed. 2012, 51, 9842. doi: 10.1002/anie.201204224
Ohmatsu, K.; Hamajima, Y.; Ooi, T. J. Am. Chem. Soc. 2012, 134, 8794. doi: 10.1021/ja3028668
Ohmatsu, K.; Ando, Y.; Ooi, T. J. Am. Chem. Soc. 2013, 135, 18706. doi: 10.1021/ja411647x
Monaco, M.R.; Poladura, B.; De Los Bernardos, M. D.; Leutzsch, M.; Goddard, R.; List, B. Angew. Chem., Int. Ed. 2014, 53, 7063. doi: 10.1002/anie.201400169
Zhu, Y. M.; Yang, P. J.; Wang, S.; Liu, Z.; Yang, G.; Chai, Z. J. Am. Chem. Soc. 2015, 137, 10088. doi: 10.1021/jacs.5b05820
Yang, P. J., Zhang, H.; Wang, S.; Yang, G.; Chai, Z. Angew. Chem., Int. Ed. 2017, 56, 650. doi: 10.1002/anie.201610693
Zhang, F.; Zhang, Y.; Tan, Q.; Lin, L.; Liu, X.; Feng, X. Org. Lett. 2019, 21, 5928. doi: 10.1021/acs.orglett.9b02058
Chen, X.; Vedejs, W. J. Am. Chem. Soc. 1997, 119, 2584. doi: 10.1021/ja963666v
Wu, B.; Parquette, J. R.; RajanBabu, T. V. Science 2009, 326, 1662. doi: 10.1126/science.1180739
Wu, B.; Gallucci, J. C.; Parquette, J. R.; RajanBabu, T. V. Chem. Sci. 2014, 5, 1102. doi: 10.1039/C3SC52929K
Xu, Y.; Kaneko, K.; Kanai, M.; Shibasaki, M.; Matsunaga, S. J. Am. Chem. Soc. 2014, 136, 9190. doi: 10.1021/ja5039165
Hu, H.; Liu, Y.; Lin, L.; Zhang, Y.; Liu, X.; Feng, X. Angew. Chem., Int. Ed. 2016, 55, 10098.
An, D.; Guan, X.; Guan, R.; Jin, L.; Zhang, G.; Zhang, S. Chem. Commun. 2016, 52, 11211. doi: 10.1039/C6CC06388H
Zheng, B. H.; Hou, X.-L. Org. Lett. 2009, 11, 1789. doi: 10.1021/ol9002543
Wang, Y., Xu, Y.-N.; Fang, G.-S.; Kang, H.-J.; Gu, Y.; Tian, S.-K. Org. Biomol. Chem. 2015, 13, 5367. doi: 10.1039/C5OB00671F
Li, M.-B.; Li, H.; Wang, J.; Liu, C.-R.; Tian, S.-K. Chem. Commun. 2013, 49, 8190. doi: 10.1039/c3cc44914a
Liu, C.-R.; Li, M.-B.; Yang, C.-F.; Tian, S.-K. Chem.-Eur. J. 2009, 15, 793. doi: 10.1002/chem.200801665
Wu, X.-S.; Tian, S.-K. Chem. Commun. 2012, 48, 898. doi: 10.1039/C1CC16630A
Yang, W.; Long, Y.; Zhang, S.; Zeng, Y.; Cai, Q. Org. Lett. 2013, 15, 3598. doi: 10.1021/ol401449b
Narine, A. A.; Toulgoat, F.; Bisschops, T.; Enders, D. Angew. Chem., Int. Ed. 2008, 47, 5661. doi: 10.1002/anie.200801354
Reznichenko, A. L.; Hampel, F.; Hultzsch, K. C. Chem.-Eur. J. 2009, 15, 12819. doi: 10.1002/chem.200902229
Li, G.-Q.; Li, Y.; Dai, L.-X.; You, S.-L. Adv. Synth. Catal. 2008, 350, 1258. doi: 10.1002/adsc.200800071
Dong, S.; Liu, X.; Zhu, Y.; He, P.; Lin, L.; Feng, X. J. Am. Chem. Soc. 2013, 135, 10026. doi: 10.1021/ja404379n
Wang, M.; Huang, Z.; Xu, J.; Chi, Y. R. J. Am. Chem. Soc. 2014, 136, 1214. doi: 10.1021/ja411110f
Minato, M.; Arimoto, H.; Nagasue, Y.; Demizu, Y.; Onomura, O. Tetrahedron 2008, 64, 6675. doi: 10.1016/j.tet.2008.05.015
Minato, D.; Nagasue, Y.; Demizu, Y.; Onomura, O. Angew. Chem., Int. Ed. 2008, 47, 9458. doi: 10.1002/anie.200804188
Sibi, M. P.; Kawashima, K.; Stanley, L. M. Org. Lett. 2009, 11, 3894. doi: 10.1021/ol901504p
Kong, D.; Han, S.; Wang, R.; Li, M.; Zi, G.; Hou, G. Chem. Sci. 2017, 8, 4558. doi: 10.1039/C7SC01556A
Cochrane, E. J.; Leonori, D.; Hassallb, L. A.; Coldham, I. Chem. Commun. 2014, 50, 9910. doi: 10.1039/C4CC04576A
Lei, B.-L.; Zhang, Q.-S.; Yu, W.-H.; Ding, Q.-P.; Ding, C.-H.; Hou, X.-L. Org. Lett. 2014, 16, 1944. doi: 10.1021/ol500498m
Xiao, K.-J.; Chu, L.; Chen, G.; Yu, G.-Q. J. Am. Chem. Soc. 2016, 138, 7796. doi: 10.1021/jacs.6b04660
Zou, X.; Zhao, H.; Li, Y.; Gao, Q.; Ke, Z.; Xu, S. J. Am. Chem. Soc. 2019, 141, 5334. doi: 10.1021/jacs.8b13756

图式 4 手性磷酸催化的四氢喹啉及环状二级胺衍生物氧化动力学拆分
Scheme 4 Chiral phosphoric acid-catalyzed OKR of cyclic secondary amine derivatives including tetrahydroquinolines

图式 5 手性磷酸催化的二氢吲哚自氧化还原动力学拆分
Scheme 5 Chiral phosphoric acid-catalyzed self-redox KR of indolines

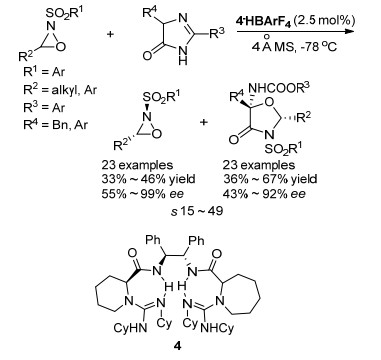
图式 9 小分子催化的2, 2-二取代氮杂环丙烷的动力学拆分
Scheme 9 Organocatalyzed KR of 2, 2-disubstituted aziridines

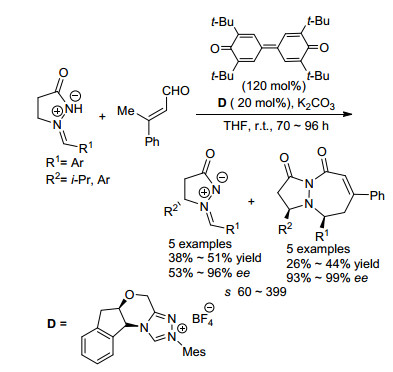
图式 18 手性磷酸催化的2H-azirine动力学拆分
Scheme 18 Chiral phosphoric acid-catalyzed KR of 2H-azirines

图式 19 钯催化的二氢吲哚烯丙基取代动力学拆分
Scheme 19 Pd-catalyzed asymmetric allylic amination KR of indolines

图式 20 钯催化烯丙基胺的烯丙基烷基化动力学拆分
Scheme 20 Pd-catalyzed asymmetric allylic alkylation KR of primary allylic amines

图式 21 手性磷酸催化的N-苄基磺胺取代动力学拆分
Scheme 21 Chiral phosphoric acid-catalyzed KR of N-benzylic sulfonamides

图式 24 稀土金属催化的烯胺化合物不对称氢胺化动力学拆分
Scheme 24 Ln-catalyzed asymmetric hydroamination KR of aminoalkenes

图式 25 手性NHC催化的顺式4-甲酰-β内酰胺动力学拆分
Scheme 25 Chiral NHC-catalyzed KR of cis-4-formyl-β-lac- tams

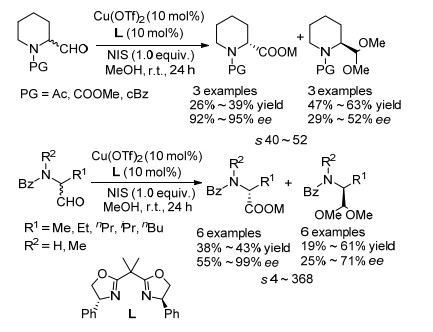
图式 28 铜催化的氨基醇和氨基醛的电化学氧化动力学拆分
Scheme 28 Cu-catalyzed electrochemical OKR of aminoalcohols and aminoaldehydes

图式 31 铜催化1, 2-二氢喹啉化合物不对称硼化动力学拆分
Scheme 31 Cu-catalyzed borylation KR of 1, 2-dihydroquino- lines

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:






















