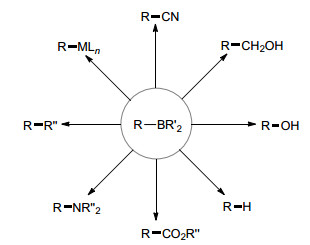
图 1.
有机硼化合物的转化
Figure 1.
Transformations of organoboranes

有机硼化合物是一类重要的有机砌块, 碳-硼键可以转化为碳-碳、碳-氢、碳-杂等多种化学键, 其合成和应用在有机合成化学中占据极其重要的地位, 如图 1所示[1~5].例如Suzuki[6]因为发现有机硼试剂参与的碳-碳交叉偶联反应(Suzuki交叉偶联反应), 获得了2010年的诺贝尔化学奖; Brown[7]发展了第一例硼-氢键向不饱和分子的加成反应(硼氢化反应), 获得了1979的诺贝尔化学奖.
烷基硼酸酯类化合物, 特别是烷基频哪醇硼酸酯, 对空气和水分不敏感, 能够通过常规的有机合成手段分离纯化, 且存储、运输、使用便捷, 因此其合成和应用受到了广泛关注.传统上, 烷基硼酸酯类化合物是利用有机金属试剂(如格氏试剂、有机锂试剂、有机铝试剂等)与三溴硼烷或三烷基硼酸酯在低温下反应[8~11], 用醇淬灭反应后制备的.然而, 上述合成有机硼酸酯的经典方法仅适用于合成简单硼酸酯类化合物, 并且反应条件苛刻、操作复杂、官能团兼容性差, 限制了其在合成上的广泛应用.
自从1956年Brown等[7]第一次发现烯烃的硼氢化反应以来, 烯烃的硼氢化反应被认为是制备官能化有机硼烷的最有效的方法之一[12]. 1985年, Männig和Nöth课题组[13]首次实现了室温下铑催化儿茶酚硼烷与烯烃的硼氢化反应.过渡金属催化的烯烃硼氢化反应, 具有简单高效、原子经济且选择性好的优点, 是构建烷基硼酸酯类化合物的最有效方法之一.过渡金属催化的烯烃硼氢化反应机理与反应底物、催化剂、配体以及反应条件密切相关.在过去的几十年里, 铑、钌、钯、铱等贵金属催化剂在烯烃的硼氢化加成反应中占据主导地位, 尤其是铑催化剂, 具有非常高的催化效率和区域选择性, 在有机合成中得到了广泛的应用[14~18].但是这些贵金属在地壳中储量稀少、价格昂贵、毒性大、后处理繁琐, 在一定程度上限制了其广泛应用.
近年来, 无过渡金属催化的、简单碱促进的羰基化合物的硼氢化反应得到了广泛深入地研究, 但是应用于不饱和烯烃的硼氢化反应却鲜有报道[19, 20], 并且反应通常需要较高的温度, 官能团容忍性差, 只能得到anti- Markovnikov加成产物.随着对绿色化学和可持续发展的追求, 人们试图找到替代贵金属的催化剂[21].在地壳中储量丰富的廉价过渡金属被认为是贵金属的理想替代品, 其中第四周期第VIII族元素铁、钴、镍得到了化学工作者的广泛关注.尽管铁、钴、镍用于烯烃的硼氢化反应研究较晚, 但是发展迅速, 并且以其价格低、毒性小等优势具有广阔的应用前景.近年来不少课题组[22~26]对廉价金属催化的硼氢化反应的研究进展进行了总结.例如2013年, 黄正等[22]总结了铁催化烯烃硼氢化反应; 2018年, Obligacion和Chirik[23]总结了地球上含量丰富金属催化的烯烃硼氢化和硅氢化反应; 同年, 陆展等[24]总结了烯烃的不对称官能化反应; 2019年, Zhang等[26]对支链选择性的烯烃硼氢化反应进行了系统的评述.值得注意的是, 2018年, 陆展等在文献[25]中, 系统总结了第一排过渡金属(铜、镍、钴、铁等)催化的烯烃氢金属化反应的研究进展和现状, 展望了未来的发展趋势.虽然上述文献从不同角度、不同程度阐述了铁、钴、镍等廉价金属催化烯烃的硼氢化反应, 但是目前, 尚未有聚焦于第四周期第VIII族元素铁、钴、镍催化简单烯烃的硼氢化反应的综述. 2018~2019年出现了不少新的铁、钴、镍催化烯烃的硼氢化反应报道.基于此, 本文总结了自1994年以来, 地球含量相对丰富的铁、钴、镍三种廉价金属催化的简单烯烃的硼氢化反应研究进展, 分析了相关反应的特点和面临的挑战, 为进一步解决这些问题提供策略和思路.
铁作为地壳中储量最丰富的过渡金属, 由于其低价格、低毒性等优点, 获得化学工作者的重视.我们小组[27~31]在研究铁催化的杂环化反应的过程中, 注意到铁作为催化剂在偶联、氧化、加氢等反应中也展现了良好的催化活性[32~39]. 2009年, Ritter课题组报道了基于铁催化的烯烃硼氢化反应[40], 打破了贵金属催化剂在烯烃的硼氢化反应的垄断地位[41].在此之后, 不断有新型铁催化体系用于该反应类型中, 反应的化学选择性和区域选择性不断提高, 反应范围也得以扩展.
2013年, 黄正课题组[42]在铁催化下实现了单取代烯烃高化学选择性和高区域选择性的硼氢化反应, 如Scheme 1(a)所示.通过对配体构型及种类的探索, 他们首次使用富电子的氮膦配体与铁离子形成的络合物4与三乙基硼氢化钠作为烯烃硼氢化反应的催化体系, 显示出远高于已知贵金属催化体系的催化效率.该反应使用廉价、环境友好的催化剂, 反应条件温和, 产物分离简单, 官能团相容性良好, 对于芳基和烷基取代的端烯都能以较好的收率得到目标产物, 能够合成其它方法难以获得的烷基硼酸酯, 使其成为制备烷基硼酸酯及衍生物的实用方法.美中不足的是, 该方法并不适用于空间位阻较大的内烯烃. 2015年, Rauchfuss课题组[43]制备了以膦-亚胺吡啶作为配体的铁络合物7, 并用于烯烃的硼官能化反应中.其中FeBr2(PNPhPy)催化下1-辛烯与HBPin顺利进行反应, 以95%产率生成anti-Markov- nikov加成产物, 如Scheme 1(b)所示. 2016年, Webster课题组[44]报道了β-双烯酮亚胺络合Fe(II)化合物10催化下烯烃的硼氢化反应, 如Scheme 1(c)所示.该催化体系不仅适用于活化和未活化端烯烃, 还适用于空间位阻大的内烯烃, 而且反应条件温和, 不需要使用还原剂或其他添加剂, 也不需要使用过量的硼化试剂.
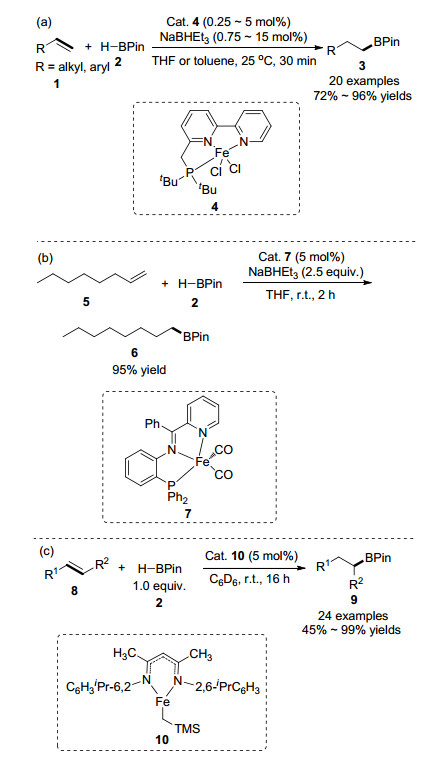
2013年, Chirik课题组[45]利用三齿鳌合的双(亚氨基)吡啶配体与铁的络合物16为催化剂, 实现了环烯烃和顺式-1, 2-二取代烯烃的氢硼化反应, 如Scheme 2(a)所示.该反应具有高反应活性和区域选择性, 并且能够在无有机溶剂条件下反应, 从而最大限度地减少浪费, 方便产品的分离提纯. 2014年, 陆展课题组[46]发展了一种新型手性三齿配体亚氨基吡啶噁唑啉17, 实现了铁催化1, 1-二取代芳基烯烃的高度区域选择性和对映选择性的anti-Markovnikov硼氢化反应, 如Scheme 2(b)所示.作者提出亚氨基吡啶基团能够稳定铁离子和手性噁唑啉基团, 从而控制反应的对映选择性. 2015年, Szymczak等[47]报道了在室温下, 酰胺衍生的N, N, N- Fe(II)络合物催化烯烃的anti-Markovnikov硼氢化反应.他们发现, 通过对配体的远端位置进行后期修饰, 相比于18, 远端N原子烷基化催化剂19更具有亲电性, 能够显著增加烯烃氢硼化反应速率, 调控反应的区域选择性.例如, Fe(bMepi)(THF)OTf 18和甲基化的Fe(bMepiMe)-OTf2 19分别作为催化剂用于cis-4-辛烯(15)和HBPin的加成反应.在18催化下, 完全生成的anti-Markovnikov硼氢化产物.在相同的条件下使用19作为催化剂, 则得到直链型和支链型的混合物, 如Scheme 2(c)所示.
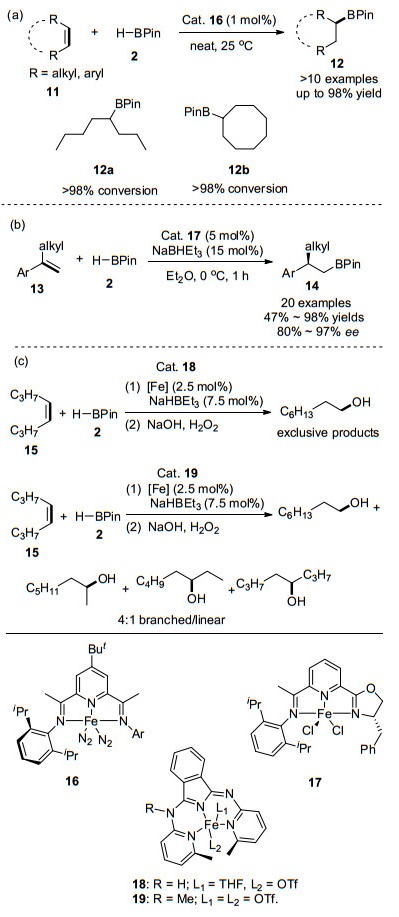
2014年, Darcel课题组[48]报道了一例非常有趣的紫外光辅助的烯烃硼氢化反应.在UV (λ=350 nm)照射下, 配合物[(IMes)Fe(CO)4] 20能高效地催化在烯烃与HBPin的硼氢化反应, 且该反应仅能得到anti-Markov- nikov加成产物, 如Eq. 1所示.该反应对空间位阻较大的内烯烃也有较强的反应活性.
|
|
(1) |
2019年, Gomes研究小组[49]合成了一系列5-取代-2-亚氨基吡咯配位的Co(II)化合物[Fe-{κ2N, N'-5-R-NC4H2-2-C(H)=N(2, 6-iPr2C6H3)}(Py)Cl] (24~26, Py=吡啶基), 并在K(HBEt3)活化作用下, 成功催化端位烯烃与HBPin的反应, 生成anti-Markovnikov为主的直链型加成产物, 如Scheme 3(a)所示.当使用苯乙烯参与该反应时, 反应选择性大幅度下降, 并且作者发现Co(II)化合物的2-亚胺基吡咯上的空间位阻大的取代基团会利于生成Markovnikov支链型加成产物, 如Scheme 3(b)所示.
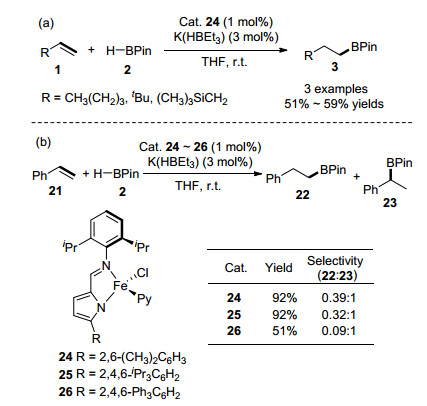
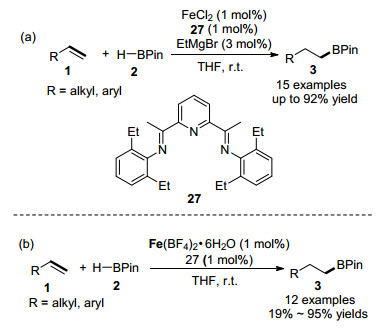
除了直接选用制备好的以铁离子为中心的络合物作为催化剂以外, 还能够利用简单的铁盐和配体组成催化体系用于烯烃的硼氢化反应中. 2013年, Thomas课题组[50]报道了一种官能团兼容性高、底物范围广、操作简单且具有区域和立体选择性的铁催化的烯烃的硼氢化加成反应, 如Scheme 4(a)所示.其中, 反应中使用的铁催化体系是由铁盐FeCl2、双(亚氨基)吡啶配体27以及乙基溴化镁原位产生的, 大大简化了实际操作步骤.该铁催化体系也适合于苯乙烯与三乙基氢化锗的加成反应, 能够生成直链型的锗氢化产物. 2019年, 该课题组[51]又报道了在六水合四氟硼酸铁(II)、双(亚氨基)吡啶配体27组成的预催化体系作用下, 烯烃与HBPin能够顺利地进行anti-Markovnikov加成反应, 如Scheme 4(b)所示.
与频哪醇硼烷(HBPin)相比, 联硼酸频那醇酯(B2Pin2, 29)具有稳定性高, 对空气和湿气不敏感的优点. 2015年, 周宇涵课题组[52]报道了氯化亚铁催化的芳基乙烯与联硼酸频那醇酯(B2Pin2)的anti-Markovnikov硼氢化反应, 如Eq. 2所示.该反应具有催化剂装载量小、官能团兼容性好、区域选择性高等优点.值得一提的是, 这是首例不需要添加配体的铁催化的烯烃硼氢化反应.
|
|
(2) |
2016年, Thomas课题组[53]研究了一系列新颖的氮杂环卡宾配体与Fe(II)的配合物催化的乙烯基芳烃的硼氢化反应.与之前的报道的倾向于生成anti-Markov- nikov加成产物不同, 该反应受到催化剂的影响, 具有可控和可切换的区域选择性:当使用带有芳氧基氮杂环卡宾配体的Fe(II)配合物34作为催化剂时, 烯烃与儿茶酚硼烷31反应更倾向于生成anti-Markovnikov加成产物32, 而当采用带有烷氧基氮杂环卡宾配体的Fe(II)配合物35作为催化剂时, 苯乙烯衍生物与HBPin反应生成Markovnikov加成产物33 (Scheme 5).研究表明, 该类型配体有利于提高催化剂的催化活性, 使得硼氢化反应在室温下快速进行. 2016年, Webster课题组[54, 55]通过改变β-双烯酮亚胺配体的结构, 也能实现对烯烃硼氢化反应区域选择性的调控.
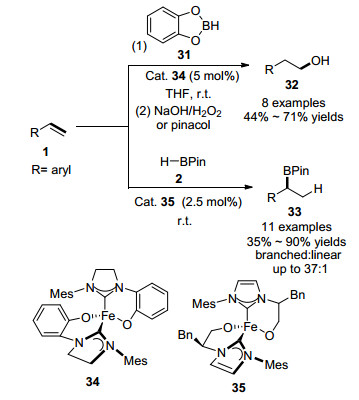
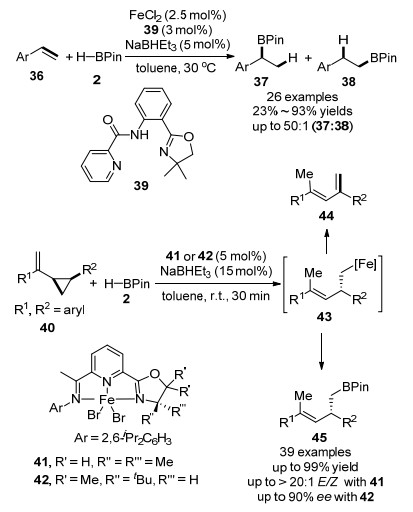
2017年, 陆展课题组[54]报道了氯化亚铁与噁唑啉基苯基吡啶酰胺39组成的催化体系作用下的乙烯基芳烃的硼氢化反应, 高度选择性地生成了Markovnikov加成产物37, 如Scheme 6所示.该反应区域选择性高, 对绝大部分芳基烯烃, 支链产物和直链产物比例高达50:1.此外, 该反应操作简单, 很容易在实验室中实现克级规模的合成.随后, 该课题组报道了第一例铁催化的烯基环丙烷的Markovnikov硼氢化反应.铁催化剂41能够区域选择性地断裂环丙烷的碳碳键, 立体专一地生成了高烯丙基硼酸酯化合物45, 烯烃E:Z比值高达20:1.使用手性配体42, 反应还能够以较好的ee值得到手性的烯丙基硼烷.作者通过氘代实验, 中间体捕捉等控制实验, 证实反应经历了Fe-H复合物对碳碳双键的插入过程, 生成关键中间体43(作者分离得到了43经过β-H消除生成共轭二烯烃44副产物).同时作者还证实了铁催化剂在实现反应立体专一性上起了重要作用.该反应为铁催化烯烃硼氢化反应的机理和Markovnikov选择性提供了依据[55].
自从Zaidlewicz课题组[56]于1997年报道了首例二氯化钴催化下共轭二烯烃与邻苯二氧硼烷的1, 2-硼氢化反应以来, 化学工作者陆续发展了一系列新的钴催化硼氢化体系.与铁催化体系相比, 钴催化剂同样具有良好的催化效果, 不仅拓宽了催化剂的范围, 还为发展快速、高效的烯烃硼氢化反应提供了新的方法和研究思路.
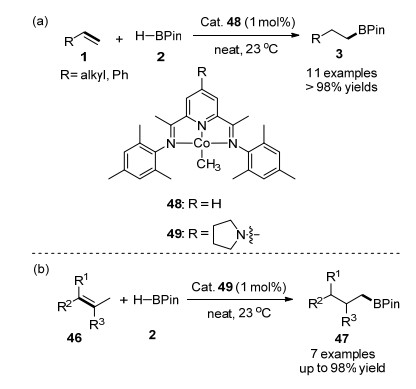
2013年, Chirik课题组[57]利用双(亚氨基)吡啶钴甲基络合物48作为催化剂, 首次实现了钴催化的HBPin与末端烯烃的区域选择性的硼氢化加成反应, 以高收率和高选择性地生成anti-Markovnikov硼酸酯产物3, 如图 7(a)所示.在该催化剂吡啶的4-位引入N-吡咯基合成催化剂49, 可以显著提升催化剂的催化活性, 实现空间位阻大的二取代、三取代以及四取代的脂肪族或芳香族烯烃的硼氢化.对于内烯烃而言, 能够通过异构化反应生成烷基链末端位置的硼酸酯加成产物, 该方法为远程碳-氢键的官能化提供了简便的方法, 如图Scheme 7(b)所示.
几乎同时, 黄正课题组在前期研究[(tBuPNN)FeCl2]催化烯烃硼氢化基础上[42], 进一步合成N, N, P-三齿配体与氯化钴的络合物50, 也实现了芳基烯烃和脂肪族末端烯烃与HBPin的硼氢化加成反应[58], 得到anti- Markovnikov产物(Eq. 3).该反应不仅具有优异的区域和化学选择性和广泛的官能团耐受性, 而且效率也很高, TON值能高达19800.随后, Stradiotto和Turculet等[59]也报道了类似的N, N, P-三配位钴催化剂用于烯烃的硼氢化反应.
|
|
(3) |
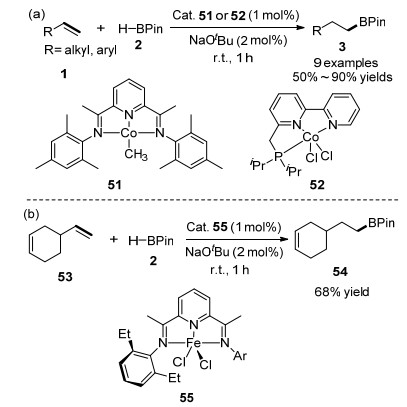
2017年, Thomas课题组[60]首次利用叔丁醇钠活化的钴(II)络合物51、52等催化烯烃的硼氢化反应, 生成anti-Markovnikov产物, 如Scheme 8(a)所示.他们发现叔丁醇钠对于铁预催化剂也有良好的活化作用, 并且利用EtBIPFeCl2 55作为催化剂也实现了烯烃的anti- Markovnikov硼氢化反应, 如Scheme 8(b)所示.随后, 该课题组[51]报道了在六水合四氟硼酸钴(II)、双(亚氨基)吡啶配体组成的预催化体系作用下, 烯烃与HBPin顺利地进行反应, 生成anti-Markovnikov加成产物(Eq. 4).
|
|
(4) |
2017年, Fout课题组[61]利用一种富电子低价态的钴催化剂(DIPPCCC)CoN2 56, 实现了端烯烃的anti-Markov- nikov硼氢化反应(Eq. 5).该反应具有很好的官能团兼容性, 例如携有芳基、氨基、酯基、酮基、环氧基等官能团的烯烃与HBPin进行反应时, 都能够以较高产率获得anti-Markovnikov加成产物.
|
|
(5) |
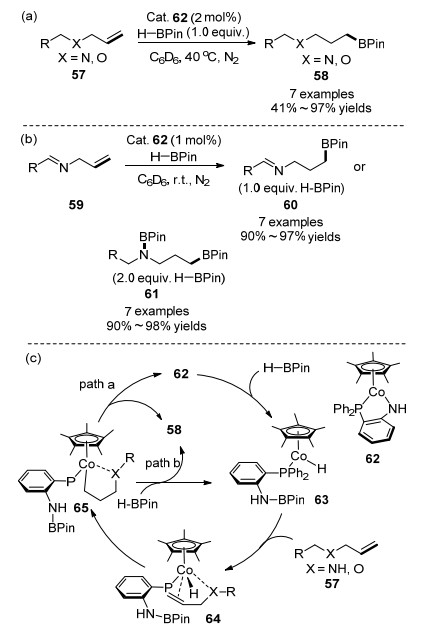
2018年, 佟庆笑和王文光课题组[62]报道了携有膦胺配体的半三明治型钴(II)配合物Cp*Co(1, 2-Ph2P- C6H4NH) (62)催化的烯烃的anti-Markovnikov硼氢化反应, 如Scheme 9(a)所示.值得注意的是, 该反应不仅适用于C=C双键, 而且也适用于C=N双键.例如对于N-烯丙基亚胺化合物59, 可以通过控制HBPin的量, 选择性生成60或61, 如Scheme 9(b)所示.作者还通过相关的控制实验, 证实反应真正活性物种是CoII—H 63, 并提出了相关的机理如Scheme 9(c)所示. HBPin对催化剂62加成, 生成活性的CoII—H物种63, 后者与烯烃双键配位生成中间体64.当烯烃β-位是杂原子(O原子或N原子)时, 杂原子能够协助配位, 因此能够促进该反应顺利进行.中间体64通过Co-H迁移插入, 产生中间体65.中间体65经过路径a和b均可以生成目标产物.
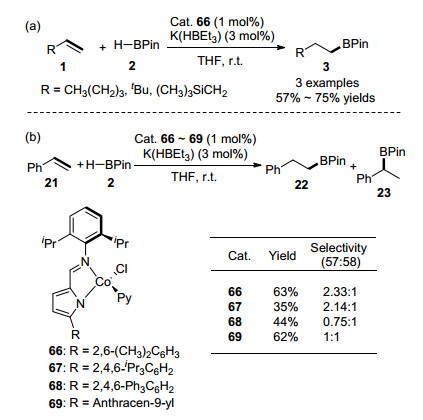
2018年, Gomes研究小组[63]合成了一系列新型的顺磁性15-电子Co(II)化合物[Co-{κ2N, N'-5-R-NC4H2-2-C(H)=N(2, 6-iPr2-C6H3)}(Py)Cl] (66~69, Py=吡啶基), 并发现该钴化合物能够有效催化烯烃与HBPin的anti- Markovnikov硼氢化加成反应(选择性高达>99:1), 如Scheme 10(a)所示.当使用苯乙烯参与该反应时, 反应选择性大幅度下降, 并且作者发现Co(II)化合物的2-亚胺基吡咯上的空间位阻大的取代基团会利于生成Markovnikov支链型加成产物, 如Scheme 10(b)所示.
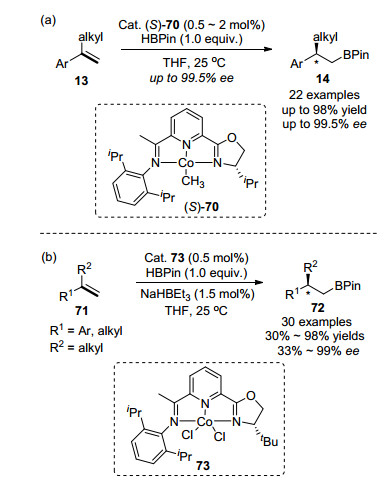
此外, 钴和手性配体相结合, 在催化烯烃的不对称硼氢化反应中也取得了突破性进展. 2014年, 黄正课题组[64]报道了新型手性亚胺吡啶-噁唑啉的钴配合物70的合成方法及其在1, 1-二取代芳基烯烃不对称硼氢化反应中的应用, 得到具有手性的anti-Markovnikov加成产物, ee高达99.5%, 如Scheme 11(a)所示.同年, 陆展课题组[65]也报道了结构相似的不对称钴催化剂IPO-CoCl2 73催化下的1, 1-二取代芳基烯烃anti-Markovnikov硼氢化反应, 并且手性产物72的绝对构型与黄正课题组报道的相同, 如Scheme 11(b)所示.
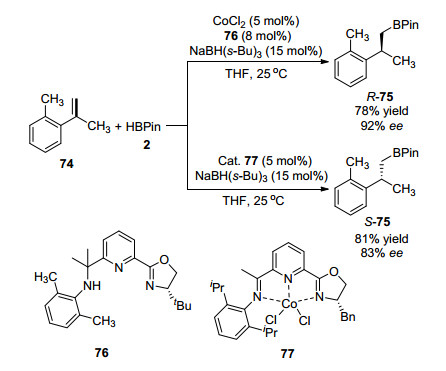
2016年, 陆展课题组[66]研究发现受配体结构控制立体结构的钴催化苯乙烯衍生物的不对称硼氢化加成反应.以2-甲基(2'-甲基)苯乙烯(74)和HBPin的反应为例, 柔性噁唑啉氨基异丙基吡啶配体76和氯化钴组成的催化体系能以产率78%、ee值92%生成产物R-构型产物R-75; 而在OIP·CoCl2(刚性噁唑啉亚胺吡啶合氯化钴)77催化下, 以产率81%, ee值83%生成S-构型产物S-75, 如Scheme 12所示.该反应能够顺利放大制备量至克级, 生成的手性烷基硼酸酯可以通过交叉偶联反应形成新的不同的C—C、C—N、C—O键等.
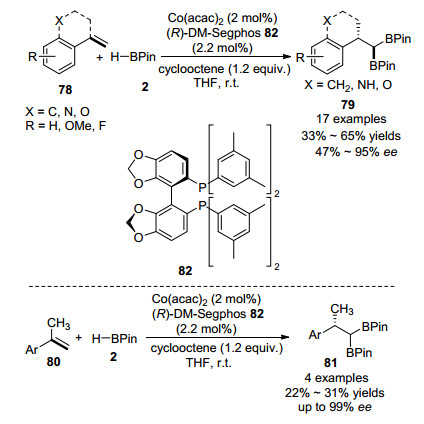
2018年, Ge课题组[67]报道了在Co(acac)2和手性配体(R)-DM-Segphos 82组成的催化体系作用下, 1, 1-二取代烯烃78、80与HBPin发生的不对称双硼化反应, 以高对映选择性生成一系列偕-二硼烷烃化合物79、81 (Scheme 13).该化合物可以很容易地转化为多种手性分子和天然产物.
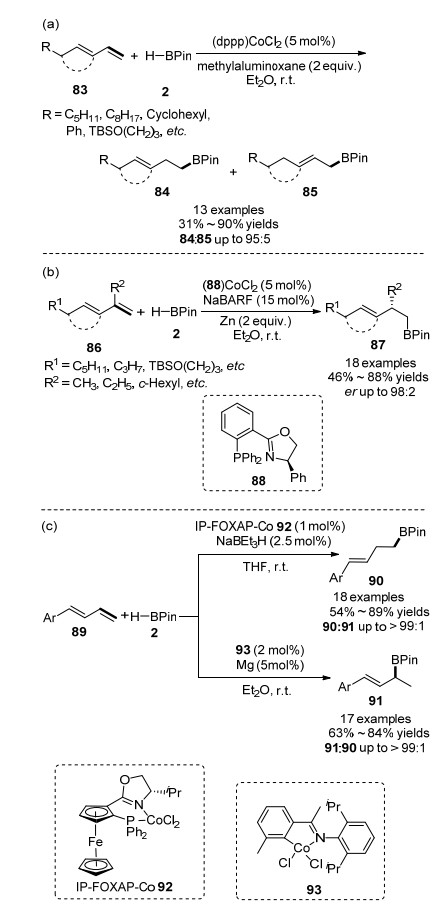
2019年, RajanBabu课题组[68]报道了一种新型钴催化1, 3-共轭二烯烃与HBPin的反应.与之前报道的此类烯烃发生1, 4-加成方式不同, 他们通过对反应条件的筛选, 发现在NaBARF (四[(3-5-三氟甲基)苯基]硼酸钠)作用下钴(II)盐与三甲基铝、甲基铝氧烷或活化的金属锌构成的催化体系能够实现1, 3-共轭二烯烃的1, 2-硼氢化加成反应, 并且以anti-Markovnikov型产物为主.其中(dppp)CoCl2催化线型的1, 3-二烯烃和2-取代1, 3-二烯烃效果最好, 1, 2-加成产物84与1, 4-加成产物85的比例>95:5, 如Scheme 14(a)所示.该课题组[68]进一步研究了在手性膦噁唑啉-Co(II)化合物、金属锌和NaBARF组成的催化体系作用下, 2-取代1, 3-二烯86的不对称硼氢化反应以较高产率、高区域选择性、高对映选择性地生成对应1, 2-加成产物87, 如Scheme 14(b)所示.随后, 刘桂霞和黄正等[69]报道了配体控制区域选择性的钴催化1-芳基-1, 3-共轭二烯烃与HBPin的1, 2-硼氢化加成反应.其中, 在钴配合物IP-FOXAP-Co 92催化作用下, 该反应进行anti-Markonikv 1, 2-硼氢化加成反应, 生成高烯丙基硼酸酯90; 在联有亚胺-吡啶配体的二氯化钴配合物93的催化作用下, 该反应进行Markonikv 1, 2-硼氢化加成反应, 生成烯丙基硼酸酯91, 如Scheme 14(c)所示.
虽然钴催化的烯烃硼氢化反应发展迅速, 得到了广泛研究, 但是绝大部分反应产物都是以anti-Markov- nikov规则为主, 或者生成两者的混合物. 2015年, Chirik课题组[70]研究三联吡啶合钴(II) 94催化的烯烃异构化-硼氢化反应过程中, 才发现首例钴催化的高度遵守Markovnikov规则的苯乙烯硼氢化反应, 支链产物与直链产物比例为25:1. 2016年, Hollis等[71]发现钴催化剂95在催化苯乙烯与HBPin的硼氢化反应时, 产物也以支链产物23为主(Eq. 6).
|
|
(6) |
直到2017年才有系统研究钴催化的烯烃Markov- nikov硼氢化反应的报道. Thomas课题组[72]以双吡啶-噁唑啉钴(II) (96)为催化剂, 实现了乙烯基芳烃的Markovnikov硼氢化反应, 如Eq. 7所示.在反应过程中, NaOtBu激活了原位生成的预催化剂, 高产率(高达92%)高选择性(高达>98:2)地制备了仲硼酸酯化合物37. 2017年, Zhang课题组[73]基于柔性NNN配体合成了双核钴催化剂Co(II)-NNN络合物97, 在催化芳基烯烃硼氢化反应时, 主要得到了Markovnikov硼氢化产物37.对于富电子芳基乙烯, 例如4-甲基氧基苯乙烯, 支链型产物37和直链型产物38比例高达97:3, 但是对于贫电子的芳基乙烯, 例如4-氟苯乙烯, 该比例迅速下降到55:45.随后, 该课题组[74]设计合成了一种空气中稳定的多聚物催化剂98, 该催化剂对富电子和贫电子的芳基乙烯, 都能以较好的区域选择性得到支链型产物37. 2018年, Geetharani等[75]将钴催化剂固载到氮杂卡宾上, 合成了Co(I)催化剂99, 在温和的反应条件下, 选择性地对乙烯基芳烃进行Markovnikov硼氢化反应, 高产率地合成了一系列仲、叔烷基硼酸酯.其中, 烯烃的适用范围广泛, 包括携有供电子基团或吸电子基团的单取代芳基乙烯、1, 1-双取代烯烃、1, 1, 2-三取代烯烃以及环烯烃, 并且反应不需要额外加入碱.但是, 上述催化剂催化脂肪族的端烯烃的硼氢化反应时, 都无一例外地得到了anti-Markovnikov加成的产物.
|
|
(7) |
2018年, Findlater等[76]以Co(acac)3为催化剂, 商业可购买的三苯基膦为配体, 在NaOtBu促进下条件下, 高产率、高区域选择性地生成Markovnikov产物, 如Eq. 8所示.需要指出的是, 烷基取代的烯烃如1-己烯, 以大于99:1的选择性得到了anti-Markovnikov加成产物3.
|
|
(8) |
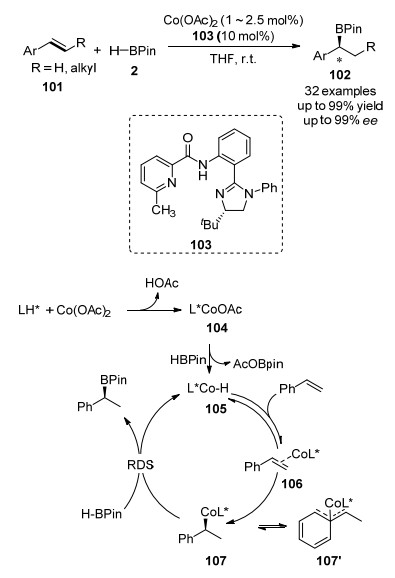
最近, 钴催化的不对称Markovnikov-硼氢化反应也取得了突破. 2019年, 陆展课题组[77]采用N-(2-咪唑啉取代苯基)-6-甲基吡啶酰胺(103)为配体, 以醋酸钴为催化剂, 可以有效地将HBPin以Markovnikov方式加成到乙烯基芳烃101的C=C双键上, 高对映选择性的生成手性的仲-硼酸酯产物102 (ee高达99%).该反应具有操作简单, 官能团耐受性好, 化学、区域和立体选择选择性高的优点.作者还通过X-射线单晶衍射, 证实了Co金属中心与配体中吡啶和酰胺配位, 同时酰胺上的质子转移到咪唑上面, 从而形成一个三齿配位的手性环境, 从而诱导手性的产生.最后, 作者通过控制实验、同位素标记、动力学研究等手段, 初步揭示了反应的机理(Scheme 15).首先, 乙酸钴通过配体交换, 生成Co(II)催化剂104.接着, 催化剂104和HBpin通过σ-复分解反应生成活性钴氢105.烯烃经过配位、插入过程生成中间体107, 后者再次与HBpin通过σ-复分解反应, 生成手性的产物硼酸酯, 并再生活性催化剂105.
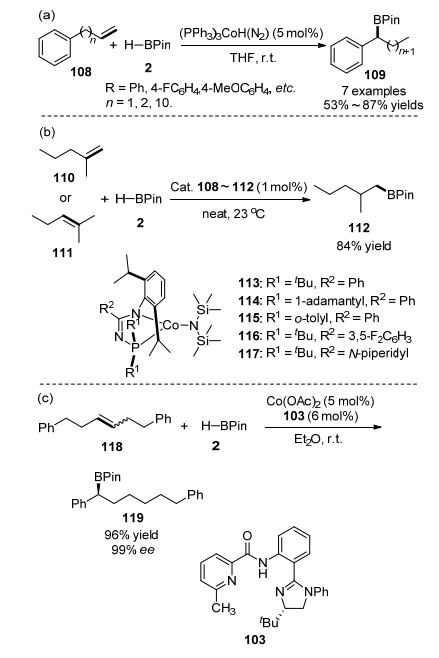
综上所述, 钴催化烯烃硼氢化反应具有催化活性高, 催化剂装载量小, 区域选择性高, 催化剂对空气稳定, 操作简单等优点.但是, 我们也注意到钴催化的反应, 主要是anti-Markovnikov加成为主, Markovnikov硼氢化反应底物仍然局限在芳基烯烃.到目前为止, 烷基取代烯烃的Markovnikov硼氢化反应仍然是一个巨大的挑战. 2015年, Chirik课题组[78]报道了在(PPh3)3CoH(N2)催化下, 烯烃首先发生异构化, 随后能够与加入的HBPin进行硼氢化反应, 如Scheme 16(a)所示.之后, 钴催化烯烃异构化-不对称硼氢化反应取得了新的进展, 2017年, Turculet等[79]制备了一系列N-膦脒基配位的钴化合物113~117, 并用于烯烃的异构化-硼氢化反应, 其中端烯烃、内烯烃都能够顺利进行.与此同时, 他们[79]也制备了类似的铁化合物, 同样可以催化烯烃的异构化-硼氢化反应, 如Scheme 16(b)所示; 2018年, 陆展课题组[80]报道了钴催化的内烯烃的异构化-硼氢化反应, 高区域、高对映选择性地实现了远程C—H键的硼官能化, 如Scheme 16(c)所示.
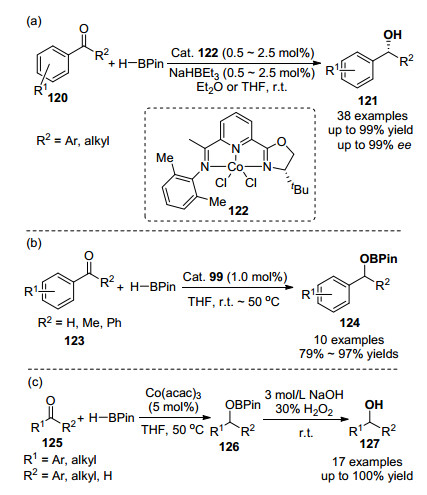
醛和酮的化学选择性催化氢硼化反应已在有机化学中得到广泛应用, 成为合成硼酸酯中间体的重要方法, 但是大多数反应主要使用的是贵金属金属、主族元素和稀土金属作为催化剂, 存在催化剂太过昂贵或催化剂本身难以合成等诸多弊端[81, 82].如何使用一些含量丰富、毒性小的廉价金属, 实现醛、酮的化学和立体选择性硼氢化反应, 是该领域面临的主要挑战之一.最近研究发现, 铁[83]、钴[75, 76, 84]、镍[85]、锰[86]等非贵金属催化剂, 在醛、酮催化硼氢化反应表现出了很高的催化活性和选择性.值得一提的是, 其中一些催化体系不仅能高效地催化烯烃的硼氢化反应, 还能够实现醛和酮的化学、立体选择性催化硼氢化反应. 2015年, 陆展课题组[84]在报道不对称钴催化剂IPO-CoCl2 73催化下的1, 1-二取代芳基烯烃anti-Markovnikov硼氢化反应后, 报道了该系列催化剂催化芳基酮的不对称硼氢化反应.甲基取代的催化剂101的对映选择性最高, 高效地实现了芳基酮120到光学纯的苄醇121的转化, 如Scheme 17(a)所示.该反应有优秀的官能团耐受性, 能兼容卤素、胺、醚、硫醚、酯和酰胺等官能团. 2018年, Geetharani等[75]在研究NHC-固载钴催化剂99催化的烯烃Markov- nikov-选择性硼氢化反应(Eq. 7)时, 也研究了该催化体系催化的醛、酮硼氢化反应, 其催化效率更高, 仅需要1 mol%的催化剂, 就能以优秀的产率(79%~97%)得到硼酸酯衍生物124, 如Scheme 17(b)所示.与此同时, Findlater等[76]发现, 与烯烃的Markovnikov硼化反应相比(Eq. 8), Co(acac)3在催化醛和酮的硼氢化反应时, 不需要额外加入叔丁醇钠和三苯基膦等添加物, 如Scheme 17(c)所示.
相比于铁和钴, 镍催化的烯烃硼氢化反应研究的较早, 但是研究却不是很多. 1994年, Kabalka等[87]发现廉价的无水碘化镍(II)可以在萘基锂存在的情况下, 作为烯烃与儿酚硼烷(HBcat)硼氢化反应的预催化剂. 1996年, Srebnik小组[88]在研究过渡金属催化烯烃与HBPin的反应过程中发现, 络合物NiCp(PPh3)Cl能够催化对称的内烯烃(trans-4-辛烯)和苯乙烯进行硼氢化反应, 其中4-辛烯和HBPin反应, 高度区域选择性生成支链型产物, 苯乙烯则高度区域选择性地生成anti-Markovnikov产物.尽管文中反应例子很少, 但是加深了人们对镍催化烯烃区域选择性硼氢化反应的认识, 促进了对镍催化烯烃(α, β-不饱和羰基化合物[89, 90]和1, 3-共轭二烯[91~93])硼氢化体系的研究.
2016年, Schomaker课题组[94]报道了NHC-膦配位的镍(II)预催化体系IMes(Cy3P)NiCl2 (128), 在KOtBu活化下可以实现苯乙烯衍生物和HBPin进行了高度区域选择性的硼氢化反应, 生成一系列Markovnikov加成产物苄基硼酸酯, 如Eq. 9所示.这是首例镍催化的Markovnikov硼氢化反应, 同时适用于富电子和贫电子的芳基乙烯化合物, 具有高产率、高区域选择性和高官能团耐受性等优点.
|
|
(9) |
过渡金属催化的烯烃硼氢化反应通常需要加入当量的无机或者有机碱以活化反应, 这不仅增加了反应成本, 而且产生大量固体废物, 不利于工业化大规模应用. 2017年, 叶萌春和王永秋课题组[95]发现Ni(cod)2和tBu3P组成的催化体系能够在无碱条件下催化烯烃与B2(Pin)2反应, 较高产率地生成anti-Markovnikov加成产物, 如Eq. 10所示.其中, 甲醇作为溶剂对无碱条件和高反应活性起着至关重要的作用.该反应适用于多种类型的烯烃, 包括芳香族和脂肪族烯烃, 其中芳香族烯烃几乎完全生成anti-Markovnikov加成产物, 而脂肪族烯烃的区域选择性则取决于取代基的空间位阻. 2018年, Kamei等[96]报道了类似的Ni(cod)2和P(Cy)3催化体系, 反应中添加水作为在中性条件下质子的来源.
|
|
(10) |
2019年, Mandal等[97]研究了常温下, 镍催化乙烯基芳烃和脂肪族烯烃的硼氢化反应.他们发现, 在镍-非那烯配合物Ni(PLY)2(THF)2 (131)作为催化剂, 金属钾作为还原剂的条件下, 乙烯基芳烃和HBPin进行Markovnikov硼氢化反应; 而脂肪族烯烃则生成anti- Markovnikov加成的硼酸酯3 (Eq. 11).作者[98]发现, TEMPO对反应有明显的抑制作用, 结合其前期烯烃的硅氢化反应的研究, 作者认为该反应经历自由基过程, 其中非那烯配体中心自由基的生成是反应的关键.
|
|
(11) |
烯烃的硼氢化反应是生成有机硼化合物最直接、最重要的方法之一.贵金属催化剂, 如Ru、Pd、Rh、Ir等, 尽管在烯烃的硼氢化反应中表现出了非常高的活性和选择性, 但是随着人们对贵金属的低丰度、高成本和环境问题的关注, 研究廉价过渡金属催化的烯烃硼氢化反应日益迫切. 2009年以后, 铁、钴、镍催化烯烃硼氢化反应研究已经取得了长足的研究进展, 新的催化体系不断的呈现, 在催化活性、反应的区域和立体选择性控制方面展现出无与伦比的优势.特别是国内陆展课题组、黄正课题组等围绕铁、钴催化烯烃的不对称硼氢化反应进行了大量的研究.目前铁、钴、镍催化体系已扩展到烯烃异构化-硼氢化[69, 78~80]、炔烃的硼氢化[50, 72, 99~106]、C=O双键(醛、酮等)的硼氢化[76, 83, 84, 86, 107~110]等反应, 极大地丰富了廉价金属催化体系.尽管铁、钴、镍催化烯烃硼氢化反应研究已经取得了长足的研究进展, 但仍存在一些亟待改进和探索的科学问题:烯烃的适用性需要进一步扩展, 底物大多局限于单取代或者末端烯烃等简单烯烃, 并且官能团耐受性差; 目前的催化体系对区域、立体选择性地控制有待进一步提升, 例如镍催化烷基烯烃的Markovnikov硼氢化反应仍然是巨大的挑战.相信在不久的将来, 廉价过渡金属催化烯烃的硼氢化反应会得到更大的突破和改善, 在有机硼化合物合成领域发挥更重要的作用.
Brown, H. C.; Singaram, B. Pure Appl. Chem. 1987, 59, 879. doi: 10.1351/pac198759070879
Stymiest, J. L.; Bagutski, V.; French R. M.; Aggarwal, V. K. Nature 2008, 456, 778. doi: 10.1038/nature07592
Bagutski, V.; French R. M.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2010, 49, 5142. doi: 10.1002/anie.201001371
Bull, J. A. Angew. Chem., Int. Ed. 2012, 51, 8930. doi: 10.1002/anie.201203876
Mlynarski, S. N.; Karns, A. S.; Morken, J. P. J. Am. Chem. Soc. 2012, 134, 16449. doi: 10.1021/ja305448w
Suzuki, A. Angew. Chem., Int. Ed. 2011, 50, 6723. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21618370
Brown, H. C.; Subba Rao, B. C. J. Am. Chem. Soc. 1956, 78, 2582. doi: 10.1021/ja01592a070
Mattraw, H. C.; Erickson, C. E.; Laubengayer, A. W. J. Am. Chem. Soc. 1956, 78, 4901. doi: 10.1021/ja01600a024
McCusker, P. A.; Ashby, E. C.; Makowski, H. S. J. Am. Chem. Soc. 1957, 79, 5179. doi: 10.1021/ja01576a026
Washburn, R. M.; Levens, E.; Albright, C. F.; Billig, F. A.; Cernak, E. S. Adv. Chem. Ser. 1959, 23, 102.
Matteson, D. S.; Liedtke, J. D. J. Am. Chem. Soc. 1965, 87, 1526. doi: 10.1021/ja01085a021
Dhillon, R. S. Hydroboration and Organic Synthesis, Springer Berlin Heidelberg, New York, 2007, pp. 59~99.
Mannig, D.; Noth, H. Angew. Chem., Int. Ed. 1985, 24, 878. doi: 10.1002/anie.198508781
Burgess K.; Ohlmeyer, M. J. Chem. Rev. 1991, 91, 1179. doi: 10.1021/cr00006a003
Beletskaya, I.; Pelter, A. Tetrahedron 1997, 53, 4957. doi: 10.1016/S0040-4020(97)00001-X
Crudden, C. M.; Edwards, D. Eur. J. Org. Chem. 2003, 4695.
Vogels, C. M.; Westcott, S. A. Curr. Org. Chem. 2005, 9, 687. doi: 10.2174/1385272053765060
Carroll, A.-M.; O'Sullivan, T. P.; Guiry, P. J. Adv. Synth. Catal. 2005, 347, 609. doi: 10.1002/adsc.200404232
Wu, Y.; Shan, C.; Ying, J.; Su, J.; Zhu, J.; Liu, L. L.; Zhao, Y. Green Chem. 2017, 19, 4169. doi: 10.1039/C7GC01632H
Ma, D. H.; Jaladi, A. K.; Lee, J. H.; Kim, T. S.; Shin, W. K.; Hwang, H.; An, D. ACS Omega 2019, 4, 15893. doi: 10.1021/acsomega.9b01877
Li, Y.; Cheng, Y.; Shan, C.; Zhang, J.; Xu, D.; Bai, R.; Qu, L.; Lan, Y. Chin. J. Org. Chem. 2018, 38, 1885(in Chinese). (李园园, 程玉华, 单春晖, 张敬, 徐冬冬, 白若鹏, 屈凌波, 蓝宇, 有机化学, 2018, 38, 1885.)
Zhang, L.; Huang, Z. Synlett 2013, 1745.
Obligacion, J. V.; Chirik, P. J. Nat. Rev. Chem. 2018, 2, 15. doi: 10.1038/s41570-018-0001-2
Chen, J.; Lu, Z. Org. Chem. Front. 2018, 5, 260. doi: 10.1039/C7QO00613F
Chen, J.; Guo, J.; Lu, Z. Chin. J. Chem. 2018, 36, 1075. doi: 10.1002/cjoc.201800314
Fan, W.; Li, L.; Zhang, G. J. Org. Chem. 2019, 84, 5987. doi: 10.1021/acs.joc.9b00550
Wang, Y.; Li, W.; Che, G.; Bi, X.; Liao, P.; Zhang, Q.; Liu, Q. Chem. Commun. 2010, 46, 6843. doi: 10.1039/c0cc01758b
Wang, Y.; Bi, X.; Li, D.; Liao, P.; Wang, Y. Yang, J. Zhang, Q. Liu, Q. Chem. Commun. 2011, 47, 809. doi: 10.1039/C0CC03802D
Wang, Y.; Bi, X.; Li, W.; Li, D.; Zhang, Q.; Liu, Q.; Ondon, B. S. Org. Lett. 2011, 13, 1722. doi: 10.1021/ol200246x
Li, Q.; Wang, Y.; Fang, Z.; Liao, P.; Barry, B.; Che, G.; Bi, X. Synthesis 2013, 609. doi: 10.1002/chin.201325051
Sun, B.; Ma, Q.; Wang, Y.; Zhao, Y.; Liao, P.; Bi, X. Eur. J. Org. Chem. 2014, 34, 7552.
Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Chem. Rev. 2004, 104, 6217. doi: 10.1021/cr040664h
Correa, A.; Mancheno, O.; Bolm, C. Chem. Soc. Rev. 2008, 37, 1108. doi: 10.1039/b801794h
Baucer, E. B. Curr. Org. Chem. 2008, 12, 1341. doi: 10.2174/138527208786241556
Greenhalgh, M. D.; Thomas, S. P. Chem. Cat. Chem. 2014, 6, 1520.
Bauer, I.; Knölker, H.-J. Chem. Rev. 2015, 115, 3170. doi: 10.1021/cr500425u
Mako, T. L.; Byers, J. A. Inorg. Chem. Front. 2016, 3, 766. doi: 10.1039/C5QI00295H
Shang, R.; Ilies, L.; Nakamu, E. Chem. Rev. 2017, 117, 9086. doi: 10.1021/acs.chemrev.6b00772
Li, Y.; Hu, Y.; Wu, X. Chem. Soc. Rev. 2018, 47, 172. doi: 10.1039/C7CS00529F
Wu, J. Y.; Moreau, B.; Ritter, T. J. Am. Chem. Soc. 2009, 131, 12915. doi: 10.1021/ja9048493
Cao, Y.; Zhang, Y.; Zhang, L.; Zhang, D.; Leng, X.; Huang, Z. Org. Chem. Front. 2014, 1, 1101. doi: 10.1039/C4QO00206G
Zhang, L.; Peng, D.; Leng, X.; Huang, Z. Angew. Chem., Int. Ed. 2013, 52, 3676. doi: 10.1002/anie.201210347
Gilbert-Wilson, R.; Chu, W.-Y.; Rauchfuss, T. B. Inorg. Chem. 2015, 54, 5596. doi: 10.1021/acs.inorgchem.5b00692
Espinal-Viguri, M.; Woof, C. R.; Webster, R. L. Chem.-Eur. J. 2016, 22, 11605. doi: 10.1002/chem.201602818
Obligacion, J. V.; Chirik, P. J. Org. Lett. 2013, 15, 2680. doi: 10.1021/ol400990u
Chen, J.; Xi, T.; Lu, Z. Org. Lett. 2014, 16, 6452. doi: 10.1021/ol503282r
Tseng, K. T.; Kampf, J. W.; Szymczak, N. K. ACS Catal. 2015, 5, 411. doi: 10.1021/cs501820w
Zheng, J.; Sortais, J.-B.; Darcel, C. Chem. Cat. Chem. 2014, 6, 763. https://www.researchgate.net/publication/256664970_ChemInform_Abstract_Selective_Reduction_of_Carboxylic_Acids_to_Aldehydes_Through_Manganese_Catalyzed_Hydrosilylation
Cruz, T. F. C.; Pereira, L. C. J.; Waerenborgh, J. C.; Veiros, L. F.; Gomes. P. T. Catal. Sci. Technol. 2019, 9, 3347. doi: 10.1039/C8CY02319K
Greenhalgh, M. D.; Thomas, S. P. Chem. Commun. 2013, 49, 11230. doi: 10.1039/c3cc46727a
Agahi, R.; Challinor, A. J.; Carter, N. B.; Thomas, S. P. Org. Lett. 2019, 21, 993. doi: 10.1021/acs.orglett.8b03986
Liu, Y.; Zhou, Y.; Wang, H.; Qu, J. RSC Adv. 2015, 5, 73705. doi: 10.1039/C5RA14869C
Macnair, A. J.; Millet, C. R. P.; Nichol, G. S.; Ironmonger, A.; Thomas, S. P. ACS Catal. 2016, 6, 7217. doi: 10.1021/acscatal.6b02281
Chen, X.; Cheng, Z.; Lu, Z. Org. Lett. 2017, 19, 969. doi: 10.1021/acs.orglett.7b00227
Chen, C.; Shen, X.; Chen, J.; Hong, X.; Lu, Z. Org. Lett. 2017, 19, 5422. doi: 10.1021/acs.orglett.7b02691
Zaidlewicz, M.; Meller, J. Tetrahedron Lett. 1997, 38, 7279. doi: 10.1016/S0040-4039(97)01691-2
Obligacion, J. V.; Chirik, P. J. J. Am. Chem. Soc. 2013, 135, 19107. doi: 10.1021/ja4108148
Zhang, L.; Zuo, Z.; Leng, X.; Huang, Z. Angew. Chem., Int. Ed. 2014, 53, 2696. doi: 10.1002/anie.201310096
Ruddy, A. J.; Sydora, O. L.; Small, B. L.; Stradiotto, M.; Turculet, L. Chem. Eur. J. 2014, 20, 13918. doi: 10.1002/chem.201403945
Docherty, J. H.; Peng, J.; Dominey, A. P.; Thomas, S. P. Nat. Chem. 2017, 9, 595. doi: 10.1038/nchem.2697
Ibrahim, A. D.; Entsminger, S. W.; Fout. A. R. ACS Catal. 2017, 7, 3730. doi: 10.1021/acscatal.7b00362
Pang, M.; Wu, C.; Zhuang, X.; Zhang, F.; Su, M.; Tong, Q.; Tung, C.-H.; Wang, W. Organometallics 2018, 37, 1462. doi: 10.1021/acs.organomet.8b00114
Cruz, T. F. C.; Lopes, P. S.; Pereira, L. C. J.; Veiros, L. F.; Gomes, P. T. Inorg. Chem. 2018, 57, 8146. doi: 10.1021/acs.inorgchem.8b00568
Zhang, L.; Zuo, Z.; Wan, X.; Huang, Z. J. Am. Chem. Soc. 2014, 136, 15501. doi: 10.1021/ja5093908
Chen, J.; Xi, T.; Ren, X.; Cheng, B.; Guo J.; Lu, Z. Org. Chem. Front. 2014, 1, 1306. doi: 10.1039/C4QO00295D
Zhang, H.; Lu, Z. ACS Catal. 2016, 6, 6596. doi: 10.1021/acscatal.6b02278
Teo, W. J.; Ge, S. Angew. Chem., Int. Ed. 2018, 57, 12935. doi: 10.1002/anie.201805705
Duvvuri, K.; Dewese, K. R.; Parsutkar, M. M.; Jing, S. M.; Mehta, M. M.; Gallucci, J. C.; RajanBabu, T. V. J. Am. Chem. Soc. 2019, 141, 7365. doi: 10.1021/jacs.8b13812
Peng, S.; Yang, J.; Liu, G.; Huang, Z. Sci. China Chem. 2019, 62, 336. doi: 10.1007/s11426-018-9418-7
Palmer, W. N.; Diao, T.; Pappas, I.; Chirik, P. J. ACS Catal. 2015, 5, 622. doi: 10.1021/cs501639r
Reilly, S. W.; Webster, C. E.; Hollis, T. K.; Valle, H. U. Dalton Trans. 2016, 45, 2823. doi: 10.1039/C5DT04752H
Peng, J.; Docherty, J. H.; Dominey, A. P.; Thomas, S. P. Chem. Commun. 2017, 53, 4726. doi: 10.1039/C7CC01085K
Zhang, G.; Wu, J.; Wang, M.; Zeng, H.; Cheng, J.; Neary, M. C.; Zheng, S. Eur. J. Org. Chem. 2017, 2017, 5814. doi: 10.1002/ejoc.201701047
Zhang, G.; Wu, J.; Li, S.; Cass, S.; Zheng, S. Org. Lett. 2018, 20, 7893. doi: 10.1021/acs.orglett.8b03431
Verma, P. K.; Sethulekshmi, A. S.; Geetharani, K. Org. Lett. 2018, 20, 7840. doi: 10.1021/acs.orglett.8b03356
Tamang, S. R.; Bedi, D.; Shafiei-Haghighi, S.; Smith, C. R.; Crawford, C.; Findlater, M. Org. Lett. 2018, 20, 6695. doi: 10.1021/acs.orglett.8b02775
Chen, X.; Cheng, Z.; Lu, Z. ACS Catal. 2019, 9, 4025. doi: 10.1021/acscatal.8b05135
Scheuermann, M. L.; Johnson, E. J.; Chirik, P. J. Org. Lett. 2015, 17, 2716. doi: 10.1021/acs.orglett.5b01135
Ogawa, T.; Ruddy, A. J.; Sydora, O. L.; Stradiotto, M.; Turculet, L. Organometallics 2017, 36, 417. doi: 10.1021/acs.organomet.6b00823
Chen, X.; Cheng, Z.; Guo, J.; Lu, Z. Nat. Comm. 2018, 9, 3939. doi: 10.1038/s41467-018-06240-y
Chong, C. C.; Kinjo, R. ACS Catal. 2015, 5, 3238. doi: 10.1021/acscatal.5b00428
Shegavi, M. L.; Bose, S. K. Catal. Sci. Technol. 2019, 9, 3307. doi: 10.1039/C9CY00807A
Tamang, S. R.; Findlater, M. J. Org. Chem. 2017, 82, 12857. doi: 10.1021/acs.joc.7b02020
Guo, J.; Chen, J.; Lu, Z. Chem. Commun. 2015, 51, 5725. doi: 10.1039/C5CC01084E
King, A. E.; Stieber, S. C. E.; Henson, N. J.; Kozimor, S. A.; Scott, B. L.; Smythe, N. C.; Sutton, A. D.; Gordon, J. C. Eur. J. Inorg. Chem. 2016, 2016, 1635. doi: 10.1002/ejic.201600143
Zhang, G.; Zeng, H.; Wu, J.; Yin, Z.; Zheng, S.; Fittinger, J. C. Angew. Chem., Int. Ed. 2016, 55, 14369. doi: 10.1002/anie.201607579
Kabalka, G. W.; Narayana, C.; Reddy, N. K. Synth. Commun. 1994, 24, 1019. doi: 10.1080/00397919408020777
Pereira, S.; Srebnik, M. Tetrahedron Lett. 1996, 37, 3283. doi: 10.1016/0040-4039(96)00576-X
Hirano, K.; Yorimitsu, H.; Oshima, K. Org. Lett. 2007, 9, 5031. doi: 10.1021/ol702254g
Lillo, V.; Geier, M. J.; Westcott, S. A.; Fernández, E. Org. Biomol. Chem. 2009, 7, 4674. doi: 10.1039/b909341a
Ely, R. J.; Morken, J. P. J. Am. Chem. Soc. 2010, 132, 2534. doi: 10.1021/ja910750b
Ely, R. J.; Morken, J. P. Org. Lett. 2010, 12, 4348. doi: 10.1021/ol101797f
Ely, R. J.; Yu, Z.; Morken, J. P. Tetrahedron Lett. 2015, 56, 3402. doi: 10.1016/j.tetlet.2015.01.123
Touney, E. E.; Hoveln, R. V.; Buttke, C. T.; Freidberg, M. D.; Guzei, I. A.; Schomaker, J. M. Organometallics 2016, 35, 3436. doi: 10.1021/acs.organomet.6b00652
Li, J.-F.; Wei, Z.-Z.; Wang, Y.-Q.; Ye, M. Green Chem. 2017, 19, 4498. doi: 10.1039/C7GC02282D
Kamei, T.; Nishino, S.; Shimada, T. Tetrahedron Lett. 2018, 59, 2896. doi: 10.1016/j.tetlet.2018.06.024
Vijaykumar, G.; Bhunia, M.; Mandal, S. K. Dalton Trans. 2019, 48, 5779. doi: 10.1039/C9DT00468H
Vijaykumar, G.; Pariyar, A.; Ahmed, J.; Shaw, B. K.; Adhikari, D.; Mandal, S. K. Chem. Sci. 2018, 9, 2817. doi: 10.1039/C7SC04687A
Haberberger, M.; Enthaler, S. Chem. Asian J. 2013, 8, 50. doi: 10.1002/asia.201200931
Obligacion, J. V.; Neely, J. M., Yazdani, A. N.; Pappas, I.; Chirik, P. J. J. Am. Chem. Soc. 2015, 137, 5855. doi: 10.1021/jacs.5b00936
Zuo, Z.; Huang, Z. Org. Chem. Front. 2016, 3, 434. doi: 10.1039/C5QO00426H
Nakajima, K.; Kato, T.; Nishibayashi, Y. Org. Lett. 2017, 19, 4323. doi: 10.1021/acs.orglett.7b01995
Gorgas, N.; Alves, L. G.; Stoeger, B.; Martins A. M.; Veiros, L. F.; Kirchner, K. J. Am. Chem. Soc. 2017, 139, 8130. doi: 10.1021/jacs.7b05051
Ferrand, L.; Lyu, Y.; Rivera-Hernández, A.; Fallon, B. J.; Amatore, M.; Aubert, C.; Petit, M. Synthesis 2017, 49, 3895. doi: 10.1055/s-0036-1588996
Ben-Daat, H.; Rock, C. L.; Flores, M.; Groy, T. L.; Bowmanb, A. C.; Trovitch, R. J. Chem. Commun. 2017, 53, 7333. doi: 10.1039/C7CC02281F
Zuo, Z.; Wen, H.; Liu, G.; Huang, Z. Synlett 2018, 29, 1421. doi: 10.1055/s-0037-1609682
Li, L.; Liu, E.; Cheng, J.; Zhang, G. Dalton Trans. 2018, 47, 9579. doi: 10.1039/C8DT02134A
Wu, J.; Zeng, H.; Cheng, J.; Zheng, S.; Golen, J. A.; Manke, D. R.; Zhang, G. J. Org. Chem. 2018, 83, 16, 9442.
Zhang, G.; Cheng, J.; Davis, K.; Bonifacioa, M. G.; Zajaczkowskia, C. Green Chem. 2019, 21, 1114. doi: 10.1039/C9GC00078J
Zhang, G.; Li, S.; Wu, J.; Zeng, H.; Mo, Z.; Davisa, K.; Zheng, S. Org. Chem. Front. 2019, 6, 3228. doi: 10.1039/C9QO00834A

图式 1 N, N-两齿配合铁催化anti-Markovnikov硼氢化反应
Scheme 1 N, N-Two-coordinate Fe-catalyzed anti-Markovnikov hydroboration

图式 2 N, N, N-三齿配合铁催化苯乙烯和HBPanti- Markovnikov硼氢化反应
Scheme 2 N, N, N-Three-coordinate Fe-catalyzed anti-Markov- nikov hydroboration of styrene with HBPin

图式 3 [Fe-{κ2N, N'-5-R-NC4H2-2-C(H)=N(2, 6-iPr2C6H3)}-(Py)Cl]催化烯烃的硼氢化反应
Scheme 3 [Fe-{κ2N, N'-5-R-NC4H2-2-C(H)=N(2, 6-iPr2C6H3)}-(Py)Cl]-catalyzed hydroboration of alkenes

图式 5 Fe(II)催化下烯烃的Markovnikov硼氢化反应
Scheme 5 Fe(II)-catalyzed Markovnikov hydroboration of alkene

图式 6 Fe催化下烯烃的Markovnikov硼氢化反应
Scheme 6 Fe-catalyzed Markovnikov hydroboration of alkenes

图式 7 N, N, N-三齿配合钴催化下烯烃和HBPin反应
Scheme 7 N, N, N-Three-coordinate cobalt-catalyzed reaction of alkenes with HBPin

图式 8 Co(II)、Fe(II)络合物催化烯烃的硼氢化反应
Scheme 8 Co(II), Fe(II)-catalyzed hydroboration of alkenes

图式 10 [Co-{κ2N, N'-5-R-NC4H2-2-C(H)=N(2, 6-iPr2-C6H3)}-(Py)Cl]催化烯烃的硼氢化反应
Scheme 10 [Co-{κ2N, N'-5-R-NC4H2-2-C(H)=N(2, 6-iPr2-C6H3)}(Py)Cl]-catalyzed hydroboration of alkenes

图式 11 钴催化1, 1-二取代芳基乙烯不对称硼氢化反应
Scheme 11 Cobalt-catalyzed enantioselective hydroboration of 1, 1-disubstituted aryl alkenes

图式 12 钴催化双重立体控制不对称硼氢化反应
Scheme 12 Dual stereocontrol asymmetric cobalt-catalyzed hydroboration

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:












 下载:
下载:






