


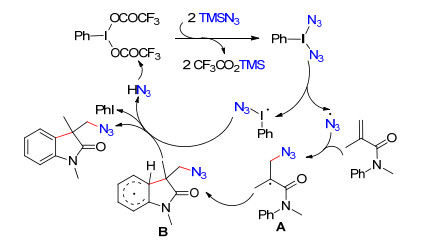
图式 1.
烯酰胺与三甲基硅叠氮的叠氮化/芳基化反应机理
Scheme 1.
Proposed mechanism for the azidoarylation of arylacrylamides with TMSN3

C—N键是诸多生物活性分子的重要结构单元, 广泛存在于化学药物、天然产物及功能材料中[1~6].因而发展简洁高效的方法构建C—N键一直是工业和学术界的关注焦点, 并已取得了很大进展, 涌现了许多经典构建策略.近几十年来, 过渡金属催化体系下Ullmann反应及Buchwald-Hartwig等传统偶联反应构建C—N键发展迅速, 在天然产物及药物合成中发挥着越来越重要的关键作用[7~11].近十年来, 过渡金属催化C—H键直接官能团化成为构建C—N键的强有力工具, 已逐渐成为有机合成化学的研究热点[12~17].然而, 传统偶联反应存在需要使用预先官能团化底物的缺陷, 不仅增长了反应步骤, 而且限制了底物类型多样性.此外, 过渡金属催化C—H键直接官能团化不可避免地需要使用过渡金属催化剂, 使得此类策略依然存在配体和/或催化剂价格昂贵、金属残留等亟待解决的问题.因此, 急需发展出绿色高效的C—N键构建新方法.
基于上述原因, 近年来无过渡金属体系下自由基反应构建C—N键引起了人们的广泛关注[18, 19].该策略通常在氧化剂作用下引发自由基, 具有反应条件温和、反应活性较高及官能团耐受性优良等特点, 为构建C—N键开辟了一条新途径.其中, C(sp2)—H键参与的自由基反应研究备受瞩目, 其通常在构建C—N键的同时能构建多种化学键, 大幅度地提高了化学反应的步骤经济性.本文拟结合课题组近年来在该方面的研究工作[36, 37, 43, 45], 依据氮源类型的不同(唑、叠氮、亚硝酸叔丁酯、腙、酰胺及其它氮源), 对近年来(2013年以来)无过渡金属体系下C(sp2)—H键的自由基反应构建C—N键研究进行综述.
唑衍生物具有独特的结构特点和生理活性, 在有机化学、药物化学及材料科学中扮演着极其重要的角色[20].借助氧化体系的调控, 唑可以产生含氮自由基以构建C—N键从而实现复杂唑衍生物的高效制备. 2013年, 朱成建课题组[21]实现了“四丁基碘化铵-过氧化叔丁醇”体系下烯烃、唑及水三组分的双官能团化反应(Eq. 1).该反应具有良好的区域选择性, 各种1-取代、1, 1-二取代及1, 2-二取代烯烃都能顺利地以75%~91%的收率实现双官能团化反应.
|
|
(1) |
2018年, 何延红、官智等[22]首次报道了光催化下贫电子烯烃与唑的氧化偶联反应(Eq. 2).该方法以玫瑰红作为有机光催化剂, 反应条件温和, 原料廉价易得.当在标准反应条件下添加2, 6-二叔丁基-4-甲基苯酚(BHT)或四甲基哌啶氮氧化物(TEMPO)作为自由基捕获剂时, 通过高分辨质谱检测到了唑自由基与上述两种捕获剂偶联的中间体, 从而证实了唑自由基的存在.
|
|
(2) |
叠氮化合物参与的有机化学反应引起了化学家的广泛关注, 不仅因为其是有机合成中重要的通用中间体和结构单元, 而且具有显著的生物活性[23~26].近年来, 借助自由基策略实现C—H键的直接叠氮化反应研究得到了迅猛发展, 为烯烃的叠氮化提供了一条新颖而简洁的途径[27]. 2013年, Antonchick课题组[27]发展了三氟醋酸碘苯促进的烯酰胺与三甲基硅叠氮的叠氮化/芳基化反应(Eq. 3).溶剂对反应影响较大, 最佳反应溶剂为二氯甲烷, 而在乙醚及甲醇中未检测到目标产物.该反应在室温下反应1 h就能以中等到良好的收率得到系列具有广泛生物活性的2-吲哚酮衍生物.可能的反应过程如下:首先, 三氟醋酸碘苯与三甲基硅叠氮经两次配体交换及热裂解产生叠氮自由基.随后, 叠氮自由基与烯酰胺的碳碳双键经自由基加成反应得到烷基自由基中间体A, 其进一步与芳烃发生环化反应得到中间体B.最后, 经过再次芳构化得到最终产物2-吲哚酮衍生物.
|
|
(3) |
随后, 张荣华课题组[28]通过叠氮源的优化进一步实现了烯酰胺与叠氮化钠的自由基串联环化反应(Eq. 4).该反应机理与Antonchick等[27]的报道类似, 但使用过硫酸钾作为氧化剂, 丙酮/水作为混合溶剂.
|
|
(4) |
2014年, Nevado课题组[29]发现N-磺酰基烯酰胺在菲咯啉的促进下能发生芳基化/叠氮化反应(Eq. 5), 该反应经过自由基加成、1, 4-芳基迁移及去磺酰化的过程, 以中等收率高区域选择性得到系列α-芳基-β-叠氮基酰胺.该特殊反应路径实现的关键在于烯酰胺的特定结构:氮原子上同时被磺酰基和芳基取代.
|
|
(5) |
2016年, 施敏课题组[30]报道了1, 7-二烯的卤代/环化和卤代/叠氮化串联反应合成3, 1-苯并噁嗪衍生物(Eq. 6), 该方法以三甲基硅叠氮作为叠氮源、N-溴代琥珀酰亚胺作为溴源, 同时在一个分子中构建一个C—N键、一个C—O键和两个C—Br键.
|
|
(6) |
2017年, 柳忠全课题组[31]通过调控自由基极性, 实现了简单烯烃的分子间叠氮化/芳杂化串联反应(Eq. 7).该串联反应是由叠氮自由基对烯烃的加成反应引发的, 而氧化剂对于叠氮自由基的顺利产生至关重要:当使用五氧化二碘、2-碘酰基苯甲酸或乙酸基高碘酸盐作为氧化剂时, 该反应几乎未能引发; 而当使用醋酸碘苯或三氟乙酸碘苯作为氧化剂时, 反应则能顺利发生.
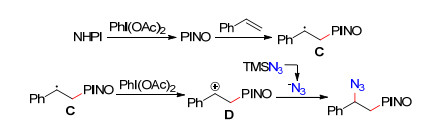
三甲基硅叠氮不仅能作为自由基源参与到烯烃的双官能团化反应, 而且能以亲核试剂的形式参与. 2015年, 夏晓峰等[32]实现了烯烃、三甲基硅叠氮及N-羟基邻苯二甲酰亚胺三组分的双官能团化反应(Eq. 8).反应中N-羟基邻苯二甲酰亚胺作为氧自由基源, 三甲基硅叠氮作为亲核试剂以叠氮负离子形式实现烯烃的叠氮化.可能反应机理如Scheme 2:在氧化剂醋酸碘苯作用下, 由N-羟基邻苯二甲酰亚胺产生氧中心自由基, 该自由基与烯烃通过自由基加成反应得到烷基自由基中间体C, 随后进一步被醋酸碘苯氧化成碳正离子中间体D, 最后叠氮负离子与碳正离子中间体D反应得到目标产物.
|
|
(7) |
|
|
(8) |
随着社会对能源优化的不断追求, 可见光催化逐渐成为有机合成化学家的研究工具. 2017年, 陆展课题组[33]报道了可见光促进下烯烃的叠氮化/羟基化反应(Eq. 9).该反应经两步实现, 首先得到的是β-叠氮过氧化物, 随后经还原得到β-叠氮醇衍生物.整个反应过程反应条件温和, 操作简便, 初始原料商业化可购买, 底物普适性优良.
|
|
(9) |
随后, 在陆展等[33]的研究基础上, 魏伟、岳会兰、杨道山等[34]进一步开发了绿色高效的可见光促进下烯烃的叠氮化/氧化策略制备α-叠氮酮衍生物(Eq. 10).在廉价易得的玫瑰红和二苯基二硒醚的共同催化下, 该反应在常温下空气中就能进行.条件优化表明溶剂对反应影响较大, 在乙腈中能以92%的收率得到目标产物, 而在四氢呋喃、1, 4-二氧六环、1, 2-二氯乙烷、甲苯、N, N-二甲基甲酰胺、乙醇中目标产物收率仅为0~30%, 作者尚未给出可能的原因.
|
|
(10) |
2018年, 朱晨课题组[35]发展了远端杂芳基迁移导向的烯烃叠氮化/杂芳基化方法(Eq. 11).反应以醋酸碘苯作为氧化剂, 在室温下能以中等至良好的收率得到系列杂芳基取代的烷基叠氮衍生物.此外, 制备的杂芳基取代的烷基叠氮衍生物能高效转化为哌啶、β-哌啶酸和三唑衍生物, 证明了该策略的实用性.
|
|
(11) |
2018年, 同样借助醋酸碘苯作为氧化剂, 魏文廷课题组[36]报道了一种1, 6-烯炔类化合物温和条件下卤化/叠氮化策略(Eq. 12).该方法具有较高的区域选择性, 叠氮自由基在该体系下高选择性与1, 6-烯炔的碳碳双键发生自由基加成反应, 随后经分子内自由基环化得到关键的烯基自由基中间体, 该烯基自由基中间体随即与卤素自由基经偶联反应得到最终产物.
|
|
(12) |
2019年, 魏文廷课题组[37]进一步发展了1, 6-烯炔类化合物与叠氮化合物的叠氮化反应(Eq. 13).与之前的工作相比[36], 该反应在发生叠氮化/环化反应之后, 烯基自由基中间体通过氢原子攫取从溶剂中获得一个氢原子从而终止了进一步的自由基偶联反应.
|
|
(13) |
近年来亚硝酸叔丁酯作为新型自由基硝化试剂得到了广泛的应用与发展, 其能避免传统亲核及亲电取代中NO2-和NO2+的使用, 使得制备过程更加简洁和通用[38, 39]. 2013年, Maiti等[40]实现了烯烃与亚硝酸叔丁酯的直接硝化反应(Eq. 14).该反应无需金属催化剂, 在TEMPO的促进下高选择性(E构型)得到硝化烯烃衍生物.控制实验表明, 空气在该反应中具有重要作用, 当反应在氮气氛围下进行时, 只能以5%的收率得到目标产物.作者进一步指出, 空气的作用在于将亚硝酸叔丁酯产生的亚硝基自由基氧化为硝基自由基.
|
|
(14) |
随后, Maiti等[41]进一步报道了α, β-不饱和羧酸与亚硝酸叔丁酯的脱羧/硝化反应(Eq. 15).同样使用TEMPO作为促进剂, 高选择性得到系列硝化烯烃衍生物, 该方法作为传统Henry反应制备该类化合物的改良方案, 反应条件温和, 底物适应性优良.
|
|
(15) |
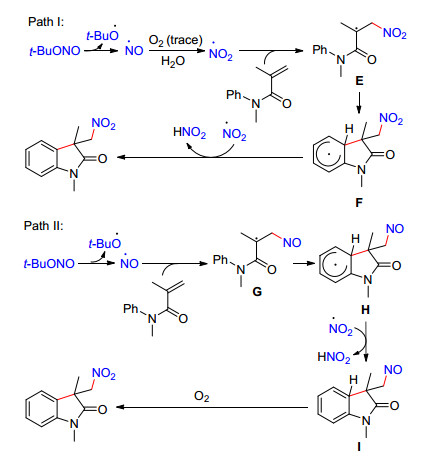
2014年, 焦宁课题组[42]首次报道了烯酰胺与亚硝酸叔丁酯的硝化/环化反应(Eq. 16).无需添加任何助剂, 多种官能团取代的烯酰胺均能通过C—N键和C—C键的构建制备系列2-吲哚酮衍生物.值得指出的是, 当用硝酸铁、硝酸银及亚硝酸银作为硝化试剂时, 该硝化/环化反应亦能发生, 只是目标产物收率较低(12%~38%).在最优反应条件下, 在反应完成后的体系中充入18O2继续在室温下搅拌反应12 h, 通过质谱检测到16O-产物与18O-产物比例为5:1, 表明一条可能的反应路径是亚硝基自由基先与烯烃发生加成反应, 随后被氧化为硝基取代的产物.此外, 当在最优反应条件下加入2 equiv. 18H2O, 16O-产物与18O-产物比例为1:1, 这表明水能促进硝基自由基的产生, 并且硝基自由基与烯烃的加成反应速率要快于亚硝基自由基.基于此, 作者提出了两种可能的反应机理.一种是硝基自由基先与碳碳双键发生自由基加成反应, 随后经分子内环化和氧化得到目标产物.另一种是亚硝基自由基先与碳碳双键发生加成反应, 接着经环化、氧化反应得到亚硝基取代的2-吲哚酮中间体, 最后被空气氧化为目标产物.
|
|
(16) |
2019年, 魏文廷课题组[43]巧妙地通过底物设计, 发展了烯酰胺与亚硝酸叔丁酯的硝化/羟基化反应(Eq. 17).当烯酰胺氮原子上保留一个氢原子时, 反应并未按Jiao等[42]报道的硝化/环化过程进行, 取而代之的是硝化/羟基化过程.作者提出可能的原因是Jiao等报道的底物均为N, N-二取代的烯酰胺, 由于受空间位阻的影响, 使得硝基自由基与碳碳双键加成之后得到的烷基自由基中间体更靠近芳环, 从而优先与芳环发生环化反应.
|
|
(17) |
2014年, 李金恒课题组[44]率先报道了1, 7-烯炔与亚硝酸叔丁酯的硝化/环化反应(Eq. 18).该反应无需借助任何助剂, 在温和条件下经烯烃硝化、6-exo-trig环化、C—H硝化及再次环化, 以良好的底物适应性及较高的收率得到系列吡咯并[4, 3, 2-de]喹啉酮衍生物.
随后, 魏文廷课题组[45]进一步发展了1, 6-烯炔与亚硝酸叔丁酯的硝化/环化反应(Eq. 19).该反应以1, 4-二氧六环作为反应溶剂, 在70 ℃空气氛围下反应12 h, 以高区域选择性得到系列2-吡咯烷酮衍生物.
|
|
(18) |
|
|
(19) |
腙自由基作为一种迷人而重要的氮中心自由基, 可以很容易的从最常见的腙中引发, 但尚未得到有机合成化学家们应有重视, 具有极大地开发潜力[46, 47]. 2013年, 韩丙课题组[48]发展了腙自由基促进的烯烃双官能团化反应合成吡唑啉的新方法(Eq. 20).反应中TEMPO一方面作为自由基引发剂诱导腙自由基对碳碳双键的加成反应, 另一方面作为碳自由基捕获剂而发生自由基偶联反应.
|
|
(20) |
2017年, Cai等[49]实现了碘(III)介导的β, γ-不饱和甲苯磺酰腙的分子内亚磺酰和硒化反应(Eq. 21).该方法反应条件温和, 以醋酸碘苯作为唯一的氧化剂, 以1, 8-二氮杂二环十一碳-7-烯作为碱, 构建了多种不同官能化的杂原子取代吡唑啉衍生物.
|
|
(21) |
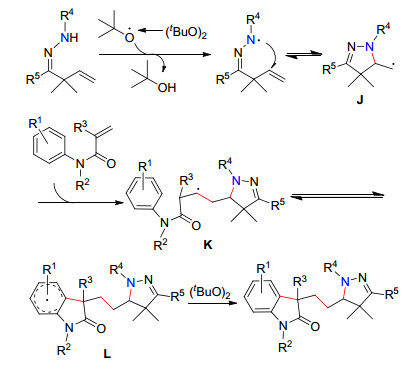
2015年, 韩丙课题组[50]进一步发展了腙基自由基引发的串联反应合成吡唑啉取代的2-吲哚酮的新策略(Eq. 22).在无金属和无溶剂条件下, 仅需使用二叔丁基过氧化物作为氧化剂引发β, γ-不饱和腙产生腙自由基, 随后经过串联环化/加成/环化过程, 一步反应中同时构建一个C—N键和两个C—C键.稍显遗憾的是, 该反应需要使用过量(6.5 equiv.)的氧化剂二叔丁基过氧化物, 具有潜在的危险性.该方法的可能反应机理如Scheme 4:首先, 在氧化剂作用下产生腙自由基, 其经5-exo- trig的分子内环化得到烷基自由基中间体J.随后, J与烯酰胺的自由基加成得到自由基中间体K, 其进一步与相邻的芳环经环化反应得到中间体L.最后, 中间体L在二叔丁基过氧化物作用下经进一步氧化芳构化得到最终产物.
|
|
(22) |
酰胺被广泛应用于药物化学、材料科学等领域, 对其结构的构建修饰一直是有机化学的研究热点[51].酰胺参与自由基反应一般有两种方式, 一种是作为亲核试剂, 另一种是作为氮自由基. 2014年, 杜云飞等[52]开发了“四丁基碘化铵-过氧化叔丁醇”促进的烯胺与酰胺衍生物的直接氧化偶联反应(Eq. 23).该方法反应时间短(0.2~3 h), 为多取代烯烃的快速制备提供了更好的选择.
|
|
(23) |
2017年, 孙凯等[53]发展了高价碘介导的杂芳烃氧化自由基胺化策略(Eq. 24).该反应无需任何金属催化剂, 高区域选择性的实现喹啉、吡啶及吡咯衍生物的C(2)位胺化反应, 具有广泛的官能团兼容性和较高的收率.当在反应体系中加入常用的自由基捕获剂TEMPO或BHT时, 目标产物收率急剧下降, 表明该反应涉及氮自由基过程.
|
|
(24) |
无过渡金属体系反应符合绿色化学发展的方向, 故而有机合成化学家期望开发出更多的氮源, 高效地实现C(sp2)—H键的自由基反应构建C—N键. 2013年, Minakata等[54]实现了碘/碘化铵催化下苯乙烯衍生物与亚氨基碘的氮杂环丙烷化反应(Eq. 25).该反应在室温下进行, 只需反应3 h.
|
|
(25) |
2017年, 王毅课题组[55]发展了N-氨基氰衍生物的脱羰基化/环化反应制备喹啉酮和二氢异喹啉酮类化合物的方法(Eq. 26).氧化剂对反应的顺利进行至关重要, 当使用无机氧化剂(醋酸碘苯或过硫酸钾)时反应收率极低(小于5%), 而当使用有机氧化剂过氧化二叔丁基时则收率大幅度提升.
|
|
(26) |
2019年, 王瑶课题组[56]报道了琥珀酰亚胺促进的烯烃、胺及碘叶立德三组分的丙二酰化/胺化反应(Eq. 27).采用琥珀酰亚胺作为质子传递物, 不仅可以加快烯烃捕获瞬态产生的高度不稳定自由基阳离子对的过程, 而且还可以显著提高目标产物收率.
2014年, 姜波、屠树江等[57]高选择性地实现了吲哚与芳基磺酰肼的磺化/重氮化反应(Eq. 28).反应在“四丁基碘化铵-过氧化叔丁醇”的体系下进行, 无需使用金属催化剂, 在吲哚C(2)位实现重氮化的同时在C(3)位实现磺化反应.当在反应体系中添加TEMPO时, 未能检测到目标产物, 证明该反应涉及自由基反应机理.此外, 在缺少过氧化叔丁醇时反应未能发生, 进一步证明了该方法经历自由基过程.
|
|
(27) |
|
|
(28) |
综上所述, 本文阐述了2013年以来无过渡金属体系下C(sp2)—H键的自由基反应构建C—N键反应, 根据氮源的不同, 从唑参与、叠氮参与、亚硝酸叔丁酯参与、腙参与、酰胺参与及其它氮源参与六个方面进行了归纳概述.同时, 注重不同反应方法之间优劣的比较和反应机理的分析, 可望为该类反应的进一步研究提供理论基础.此类反应的特点是利用氧化剂引发产生自由基, 从而为C—N键构建提供了一条简洁高效的途径.因此, 这类反应具有潜在的发展应用空间.但是, 由于自由基物种本身的高活性, 发展高对映选择性的C—N键构建方法仍然面临着挑战.此外, 部分自由基偶联反应涉及的机理还没被充分揭示.可以预期, 在不久的将来, 绿色反应条件下C(sp2)—H键参与高效构建C—N键的方法一定能够实现.
Humphrey, J.-M.; Chamberlin, A.-R. Chem. Rev. 1997, 97, 2243. doi: 10.1021/cr950005s
Collet, F.; Lescot, C.; Dauban, P. Chem. Soc. Rev. 2011, 40, 1926. doi: 10.1039/c0cs00095g
Cho, H. S.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2011, 40, 5068. doi: 10.1039/c1cs15082k
刘薇, 郑昕宇, 曾建国, 程辟, 有机化学, 2017, 37, 1. http://sioc-journal.cn/Jwk_yjhx/CN/Y2017/V37/I1/1Liu, W.; Zheng, X. Y.; Zeng, J. G.; Chen, P. Chin. J. Org. Chem. 2017, 37, 1 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2017/V37/I1/1
袁斯甜, 王艳华, 邱观音生, 刘晋彪, 有机化学, 2017, 37, 566. http://sioc-journal.cn/Jwk_yjhx/CN/Y2017/V37/I3/566Yuan, S.; Wang, Y.; Qiu, G.; Liu, J. Chin. J. Org. Chem. 2017, 37, 566 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2017/V37/I3/566
魏文廷, 朱文明, 吴益, 黄依铃, 梁洪泽, 有机化学, 2017, 37, 1916. doi: 10.6023/cjoc201703039Wei, W.; Zhu, W.; Wu, Y.; Huang, Y.; Liang, H. Chin. J. Org. Chem. 2017, 37, 1916 (in Chinese). doi: 10.6023/cjoc201703039
Davies, H. M. L.; Long, M.-S. Angew. Chem., Int. Ed. 2005, 44, 3518. doi: 10.1002/anie.200500554
Ullmann, F. Ber. Dtsch. Chem. Ges. 1903, 36, 2382. doi: 10.1002/cber.190303602174
Surry, D. S.; Buchwald, S. L. Angew. Chem., Int. Ed. 2008, 47, 6338. doi: 10.1002/anie.200800497
Hartwig, J. F. Acc. Chem. Res. 2008, 41, 1534. doi: 10.1021/ar800098p
刘学民, 陈雯, 倪邦庆, 陈新志, 钱超, 葛新, 有机化学, 2018, 38, 1703. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346529.shtmlLiu, X.; Chen, W.; Ni, B.; Chen. X.; Qian, C.; Ge, X. Chin. J. Org. Chem. 2018, 38, 1703 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract346529.shtml
Gunanathan, C.; Ben-David, Y.; Milstein, D. Science 2007, 317, 790. doi: 10.1126/science.1145295
Muthaiah, S.; Ghosh, S.-C.; Jee, J.-E.; Chen, C.; Zhang, J.; Hong, S.-H. J. Org. Chem. 2010, 75, 3002. doi: 10.1021/jo100254g
Ruiz-Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564. doi: 10.1021/acs.chemrev.6b00512
Kim, H.; Chang, S. ACS Catal. 2016, 6, 2341. doi: 10.1021/acscatal.6b00293
张文曼, 戴建军, 许华建, 有机化学, 2015, 35, 1820. http://sioc-journal.cn/Jwk_yjhx//CN/abstract/abstract345062.shtmlZhang, W.-M.; Dai, J.-J.; Xu, H.-J. Chin. J. Org. Chem. 2015, 35, 1820 (in Chinese). http://sioc-journal.cn/Jwk_yjhx//CN/abstract/abstract345062.shtml
Gui, J.; Pan, C.-M.; Jin, Y.; Qin, T.; Lo, J. C.; Lee, B. J.; Spergel, S. H.; Mertzman, M. E.; Pitts, W. J.; La Cruz, T. E.; Schmidt, M. A.; Darvatkar, N.; Natarajan, S. R.; Baran, P. S. Science 2015, 348, 886.
Liu, D.; Lei, A. Chem. Asian J. 2015, 10, 806. doi: 10.1002/asia.201403248
Luo, J.; Wei, W.-T. Adv. Synth. Catal. 2018, 360, 2076. doi: 10.1002/adsc.201800205
Senger, J.; Melesina, J.; Marek, M.; Romier, C.; Oehme, I.; Witt, O.; Sippl, W.; Jung, M. J. Med. Chem. 2016, 59, 1545. doi: 10.1021/acs.jmedchem.5b01493
Xue, Q.; Xie, J.; Xu, P.; Hu, K.; Cheng, Y.; Zhu, C. ACS Catal. 2013, 3, 1365. doi: 10.1021/cs400250m
Xin, J.-R.; He, Y.-L.; Guan, Z. Org. Chem. Front. 2018, 5, 1684. doi: 10.1039/C8QO00161H
Huryn, D. M.; Okabe, M. Chem. Rev. 1992, 92, 1745. doi: 10.1021/cr00016a004
Scriven, E. F. V.; Turnbull, K. Chem. Rev. 1988, 88, 297.
Megiatto, J. D.; Schuster, D. I. J. Am. Chem. Soc. 2008, 130, 12872. doi: 10.1021/ja8050519
Park, C.; Kim, H.; Kim, S.; Kim, C. J. Am. Chem. Soc. 2009, 131, 16614. doi: 10.1021/ja9061085
Matcha, K.; Narayan, R.; Antonchick, A. P. Angew. Chem., Int. Ed. 2013, 52, 7985. doi: 10.1002/anie.201303550
Qiu, J.; Zhang. R. Org. Biomol. Chem. 2014, 12, 4329. doi: 10.1039/C4OB00720D
Kong, W.; Merino, E.; Nevado, C. Angew. Chem., Int. Ed. 2014, 53, 5078.
Sun, Y.-M.; Yu, L.-Z.; Zhu, Z.-Z.; Hu, X.-B.; Gao, Y.-N.; Shi, M. Org. Biomol. Chem. 2017, 15, 634. doi: 10.1039/C6OB02566H
Liu, Z.; Liu, Z.-Q. Org. Lett. 2017, 19, 5649. doi: 10.1021/acs.orglett.7b02788
Xia, X.-F.; Gu, Z.; Liu, W.; Wang, H.; Xia, Y.; Gao, H.; Liu, X.; Liang, Y.-M. J. Org. Chem. 2015, 80, 290. doi: 10.1021/jo502327r
Yang, B.; Lu, Z. ACS Catal. 2017, 7, 8362. doi: 10.1021/acscatal.7b02892
Wei, W.; Cui, H.; Yue, H.; Yang, D. Green Chem. 2018, 20, 3197. doi: 10.1039/C8GC01245H
Wang, M.; Wu, Z.; Zhang, B.; Zhu, C. Org. Chem. Front. 2018, 5, 1896. doi: 10.1039/C8QO00301G
Ying, W.-W.; Chen, W.-T.; Bao, W.-H.; Gao, L.-H.; Wang, X.-Y.; Chen, G.-P.; Ge, G.-P.; Wei, W.-T. Synlett 2018, 29, 1664. doi: 10.1055/s-0037-1609752
Wang, Y.-N.; Li, Q.; Fang, Y.-L.; Gao, L.-H.; Song, S.-Z.; Dong, Y.; Wei, W.-T. Asian J. Org. Chem. 2019, 8, 832. doi: 10.1002/ajoc.201900210
Song, L. R.; Fan, Z.; Zhang, A. Org. Biomol. Chem. 2019, 17, 1351. doi: 10.1039/C8OB02750A
Ballini, R.; Araújo, N.; Gil, M. V.; Román, E.; Serrano, J. A. Chem. Rev. 2013, 113, 3493. doi: 10.1021/cr2002195
Maity, S.; Naveen, T.; Sharma, U.; Maiti, D. Org. Lett. 2013, 15, 3384. doi: 10.1021/ol401426p
Manna, S.; Jana, S.; Saboo, T.; Maji, A.; Maiti, D. Chem. Commun. 2013, 49, 5286.
Jiao, N.; Shen, T.; Yuan, Y. Chem. Commun. 2014, 50, 554. doi: 10.1039/C3CC47336H
Gao, L.-H.; Meng, X.-X.; Wang, Y.-N.; Song, S.-Z.; Ge, G.-P.; Dong, Y.; Wei, W.-T.; Liu, Y.-Y.; Li, Q. Asian J. Org. Chem. 2019, 8, 348.
Liu, Y.; Zhang, J.-L.; Song, R.-J.; Qian, P.-C.; Li, J.-H. Angew. Chem., Int. Ed. 2014, 53, 9017. doi: 10.1002/anie.201404192
Wei, W.-T.; Ying, W.-W.; Bao, W.-H.; Gao, L.-H.; Xu, X.-D.; Wang, Y.-N.; Meng, X.-X.; Chen, G.-P.; Li, Q. ACS Sustainable Chem. Eng. 2018, 6, 15301. doi: 10.1021/acssuschemeng.8b03752
Li, Z.; Zhang, C.; Zhu, L.; Liu, C.; Li, C. Org. Chem. Front. 2014, 1, 10.
Harej, M.; Dolenc, D. J. Org. Chem. 2007, 72, 7214. doi: 10.1021/jo071091m
Duan, X.-Y.; Yang, X.-L.; Fang, R.; Peng, X.-X.; Yu, W.; Han, B. J. Org. Chem. 2013, 78, 10692. doi: 10.1021/jo4016908
Yu, J.; Cai, C. Org. Biomol. Chem. 2018, 16, 490. doi: 10.1039/C7OB02892J
Duan, X.-Y.; Yang, X.-L.; Jia, P.-P.; Zhang, M.; Han, B. Org. Lett. 2015, 17, 6022. doi: 10.1021/acs.orglett.5b03003
Guo, J.; Li, W. M.; Xue, W. W.; Ye, X.-S. J. Med. Chem. 2017, 60, 2135. doi: 10.1021/acs.jmedchem.6b01644
Yuan, Y.; Hou, W.; Zhang-Negrerie, D.; Zhao, K.; Du, Y. Org. Lett. 2014, 16, 5410. doi: 10.1021/ol5026525
Zhao, F.; Sun, T.; Sun, H.; Xi, G.; Sun, K. Tetrahedron Lett. 2017, 58, 3132. doi: 10.1016/j.tetlet.2017.06.081
Kiyokawa, K.; Kosaka, T.; Minakata, S. Org. Lett. 2013, 15, 4858. doi: 10.1021/ol402276f
Liu, X.; Qian, P.; Wang, Y.; Pan, Y. Org. Chem. Front. 2017, 4, 2370. doi: 10.1039/C7QO00677B
Zhang, L.; Kong, X.; Liu, S.; Zhao, Z.; Yu, Q.; Wang, W.; Wang, Y. Org. Lett. 2019, 21, 2923. doi: 10.1021/acs.orglett.9b00983
Qiu, J.; Tu, S.; Hao, W.; Wang, D.; Wei, P.; Sun, J.; Jiang, B. Chem. Commun. 2014, 50, 14782. doi: 10.1039/C4CC06795A

图式 1 烯酰胺与三甲基硅叠氮的叠氮化/芳基化反应机理
Scheme 1 Proposed mechanism for the azidoarylation of arylacrylamides with TMSN3

图式 2 烯烃、三甲基硅叠氮及N-羟基邻苯二甲酰亚胺三组分的双官能团化反应可能机理
Scheme 2 Proposed mechanism for the three-component bifunctionalization of alkenes with TMSN3 and N-hydroxyphtha- limide

图式 3 烯酰胺与亚硝酸叔丁酯的硝化/环化反应可能机理
Scheme 3 Proposed mechanism for the nitro-cyclization of arylacrylamides with t-BuONO

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

























 下载:
下载:
