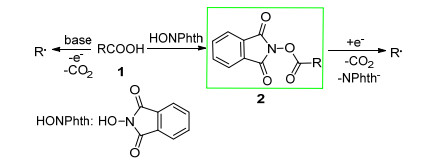
图式1.
烷基羧酸及其衍生物脱羧过程
Scheme 1.
Decarboxylative of alkyl carboxylic acids and their derivatives

羧酸及其衍生物广泛地存在于自然界之中, 具有来源丰富和、稳定性好等特点, 利用羧酸作为起始原料构筑各类有机分子已经成为有机合成化学中的热门研究领域之一[1~3].自由基反应是有机化学中非常重要的反应, 其中可见光诱导的自由基反应已经成为构筑各类化学键的有效合成策略[4~6].烷基羧酸及其衍生物在可见光作用下脱去一分子二氧化碳形成烷基自由基, 而生成的烷基自由基可以进行一系列后续转化, 提供了一条经由脱羧自由基官能团化制备有机分子的合成路线[7~9].可见光反应体系一般具有反应活性高、条件温和、官能团容忍性好及底物适用范围广等优点.本文按照可见光作用下烷基羧酸及其衍生物形成烷基自由基的后续反应类型进行分类, 对烯烃/炔烃加成、加成环化、芳杂环加成、自由基偶联、有机金属(M-R)交叉偶联及其它反应进行了归纳和总结.
在可见光氧化还原催化体系下, 烷基羧酸在碱的作用下经历单电子氧化过程失去一个电子, 脱除一分子二氧化碳形成烷基自由基.与此同时, 烷基羧酸通过简单的酯化反应, 能够制备具有良好氧化活性的烷基N-羟基邻苯二甲酰亚胺酯, 其在可见光催化体系经历单电子还原过程得到一个电子, 随后脱除二氧化碳形成烷基自由基(Scheme 1).在可见光氧化还原体系烷基羧酸及其衍生物可以分别经历氧化和还原过程得到烷基自由基, 这吸引了广大科研工作者的兴趣, 极大地促进了可见光诱导脱羧自由基反应的发展, 丰富了烷基羧酸及其衍生物在可见光催化领域的应用.
烷基羧酸及其衍生物在可见光条件下能够高效地形成烷基自由基, 通过自由基对不饱和键的加成反应构筑各类化学键是自由基化学经典的反应过程.烷基自由基对烯烃/炔烃的加成反应可能形成烷基化、烯基化、炔基化与双官能团化产物四种类型产物.
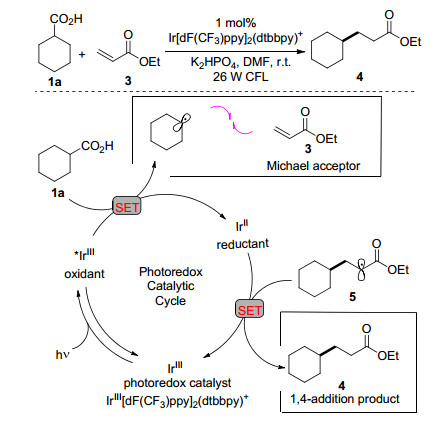
可见光促进的烷基羧酸及α-氨基酸的脱羧自由基加成反应近年来受到了广泛关注, MacMillan课题组[10]报道了可见光促进的烷基羧酸与缺电子烯烃的Michael加成反应(Scheme 2).在可见光的照射下铱催化剂进入到激发态, 而激发态的铱催化剂具有强氧化能力, 其单电子氧化环己羧酸并脱除二氧化碳得到环己基自由基, 环己基自由基与缺电子烯烃3加成构建新的碳-碳键形成自由基物种5, 随后经过单电子还原得到1, 4-加成产物4.值得注意的是, 利用这种新型的脱羧加成策略可以简单地通过三步法合成药物分子pregabalin. Nishibayashi课题组[11]在可见光条件下以对位氨基取代的苯乙酸作为苄基化试剂, 实现了苄基自由基对缺电子烯烃的加成反应. Ruepling课题组[12]研究了氨基酸在可见光作用下形成的一级及二级氨甲基自由基与不饱和羰基化合物的加成反应. Gonzalez-Gomez及Koike等[13, 14]研究了可见光非金属吖啶盐类催化剂的脱羧反应体系.近来, MacMillan课题组[15]将此可见光促进的脱羧自由基反应策略运用于蛋白质的选择性位点修饰, 成功地实现了胰岛素蛋白质的选择性修饰, 提供了一种全新的生物共轭加成方法, 将促进蛋白质修饰领域的快速发展. Flanagan等[16]开发了DNA标记的自由基受体与α-氨基酸的自由基加成反应体系.而α-酮酸经历脱羧基及脱羰基过程, 亦能高效形成烷基自由基, 并与烯烃发生加成反应[17].
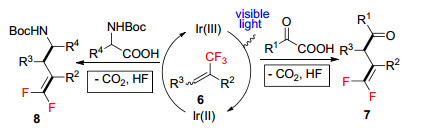
烷基羧酸作为自由基前驱体与含有杂原子烯烃化合物加成, 能够制备各类具有高附加值的有机化合物. α-三氟甲基炔烃常用来合成各类有机氟化物, 周磊等[18]报道了可见光诱导的α-氨基酸与α-三氟甲基炔烃6的脱羧/脱氟反应(Scheme 3), 在温和反应条件下高效地合成了一系列偕二氟类化合物7和8, 并且这些化合物能够进一步转化为具有高附加值的二氟甲基化合物或单氟杂环化合物, 该反应具有良好的底物普适性, 多种α-酮酸和α-氨基酸均能以中等到良好的收率获得偕二氟类化合物.硼酸及其衍生物在有机合成、高分子材料及医药行业都有十分重要的作用, Aggarwal课题组[19]报道了可见光条件下α-氨基羧酸与烯基硼酸酯9的脱羧自由基加成反应, 高效地制备一系列γ-氨基硼酸酯化合物10 (Eq. 1).他们通过氘代标记实验和密度泛函(DFT)理论计算认为, 该反应涉及α-硼自由基的单电子还原过程, 由于烷基硼酸酯可以发生一系列后续官能团化, 这种高效合成烷基硼酸酯方法对药物的开发具有重要的价值. α-芳基烯基膦酸酯表现出良好的生物学活性, 李燕等[20]研究了可见光条件下烷基羧酸与α-芳基烯基膦酸酯11的Giese反应, 简单高效地制备了α-芳基烷基膦酸酯化合物12 (Eq. 2), 反应具有良好的官能团容忍性和底物普适性, 还可以用于制备α-芳基取代的膦胺霉素类似物.
|
|
(1) |
|
|
(2) |
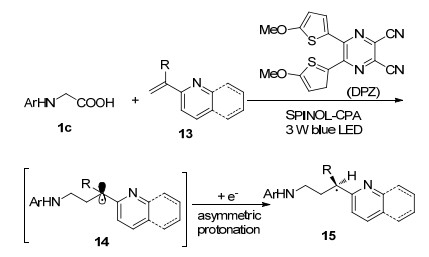
可见光促进的自由基手性合成一直是有机光化学中最具有挑战性的课题之一, 江智勇等[21]开发了可见光催化剂二腈基哌嗪化合物(DPZ), 结合手性磷酸的协同催化体系, 实现了N-芳基甘氨酸与α-支链-2-烯基吡啶/喹啉类化合物13的非对映选择性自由基加成(Scheme 4).在温和反应条件下以良好的收率及优异的对映选择性制备了一系列具有高附加值的手性α-叔碳芳杂烃化合物15.他们认为手性来源于氢键作用下的立体选择性氢质子化, 此外该反应被成功地应用于(R)-非尼拉敏药物分子制备.
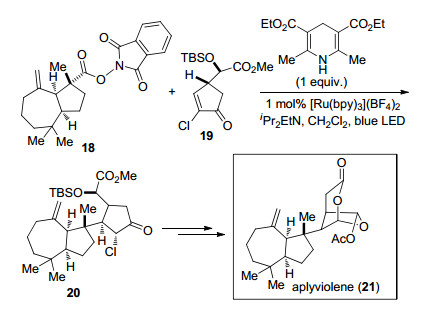
烷基羧酸的酯化产物烷基N-羟基邻苯二甲酰亚胺酯的脱羧烷基化反应近来受到了人们的关注, 早在1991年, Okada等[22]报道了在可见光作用下烷基N-羟基邻苯二甲酰亚胺酯2与缺电子烯烃16的Michael加成反应(Eq. 3).烷基N-羟基邻苯二甲酰亚胺酯在可见光催化剂[Ru(bpy)3]Cl2及氢给体1-苄基-1, 4-二氢烟酰胺(BNAH)的作用下, 通过单电子转移过程脱去二氧化碳形成烷基自由基, 随后与缺电子烯烃加成得到主产物17.由此可见, 烷基N-羟基邻苯二甲酰亚胺酯可以作为良好的烷基自由基前驱体.然而直到近年来可见光促进的脱羧自由基体系才得以快速发展, Overman与合作者[23]报道了在可见光作用条件下烷基N-羟基邻苯二甲酰亚胺酯高效形成三级烷基自由基, 随后与不饱和烯烃发生自由基加成反应, 从而高效构建四级碳中心分子, 此反应体系被成功地应用于天然产物二萜化合物aplyviolene (21)的全合成(Scheme 5).随后, 他们[24]将烯烃偶联试剂拓展到烯丙基和乙烯基卤代化合物, 这些化合物都能很好地兼容该反应体系, 但是反应速率稍微变慢.陈以昀[25]以烯基砜化合物为烯基化试剂, 完成了类似的反应, 反应具有非常好的底物普适性, 适用于一级、二级、三级、苄基与α-杂原子取代的活性酯, 而且反应不仅可以在有机溶剂中发生, 在中性水溶液中也能很好地进行.
|
|
(3) |
2016年, König课题组[26]报道了由来源丰富的氨基酸及脂肪酸通过简单的酯化反应制备一系列烷基N-羟基邻苯二甲酰亚胺酯, 其在有机染料光催化剂曙红的作用下高效地形成烷基自由基, 随后与缺电子烯烃22发生加成反应(Eq. 4).该反应体系展现出良好的底物普适性, 适用于α-氨基酸和α-酮酸及(一级、二级及三级)脂肪酸, 但是对于自然界中广泛存在的长链脂肪酸衍生物, 产率都相对偏低.付华课题组[27]使用廉价易得的苯硫酚类化合物作为光催化剂, 实现了烷基N-羟基邻苯二甲酰亚胺酯与缺电子烯烃24的脱羧自由基加成反应(Eq. 5), 其中4-三氟甲基苯硫酚表现出最佳的光催化性能.利用丰富的苯硫酚类化合物作可见光催化剂能够有效地降低反应成本, 值得注意的是, 该反应体系同样适用于烷基N-羟基邻苯二甲酰亚胺酯自身脱羧胺化, 及其与异氰联苯的环化反应.
|
|
(4) |
|
|
(5) |
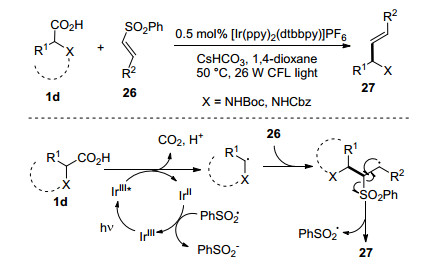
与简单烯烃的烯基化反应是有机化学重要的反应之一, 其中Heck反应已经成为有机合成中的基本反应.近来自由基与烯烃的直接加成制备烯基化产物受到了关注, 相比传统Heck烯基化反应, 该反应是通过自由基对烯烃加成后继续氧化得到烯基化产物. MacMillan课题组[28]报道了可见光条件下α-氨基酸与烯基砜化合物26的脱羧烯基化反应, 合成了一系列烯丙胺化合物27 (Scheme 6).在可见光氧化还原体系下α-氨基酸经历脱羧过程形成了α-氨甲基自由基, 其与烯基砜加成释放出芳基砜自由基并且得到烯基化产物, 而释放的芳基砜自由基氧化Ir(Ⅱ)至Ir(Ⅲ), 从而实现整个可见光催化循环.这种具有优异几何构型的反应体系被成功地应用于各种天然产物和医药分子的合成.
1, 2-二取代的烯烃化合物是一类重要的有机合成子, 吴杰课题组[29]报道了可见光催化剂高氯吖啶盐与钴催化剂的协同催化体系下烷基羧酸的Heck型交叉偶联反应(Scheme 7), 通过控制实验和DFT理论计算, 他们认为反应涉及自由基历程, 而通过钴与可见光结合的协同作用避免了额外氧化剂的使用, 只产生氢气和二氧化碳副产物, 提升了反应的清洁性.该反应高效地制备了一系列烯基硅烷、烯基硼酸酯及三组分偶联烯基化合物, 有利于开发更多具有高附加值的二取代烯基化产物及药物分子后期的功能化修饰.那日松等[30]研究了可见光和过渡金属钯协同催化的烷基羧酸与苯乙烯类化合物的Heck偶联反应, 作者认为反应经历铱催化剂的能量转移和电子转移及钯催化β-H消除和插入等过程, 高效地制备了一系列具有专一Z-选择性的β-烷基化苯乙烯衍生物, 然而一级脂肪酸和α-氧烷基羧酸则不能适用于该催化体系. MacMillan课题组[31]研究了可见光和镍协同催化烷基羧酸与非活化炔烃的脱羧氢烷基化反应, 制备了各类功能化烯烃化合物.
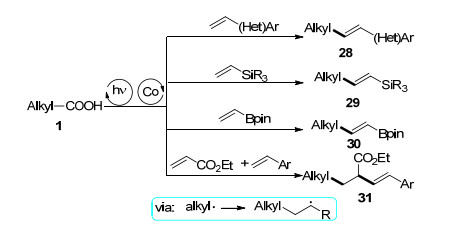
烷基N-羟基邻苯二甲酰亚胺酯作为烷基自由基前驱体的脱羧烯基化反应也受到了关注(Scheme 8), 段新华等[32]报道了在可见光条件下N-羟基邻苯二甲酰亚胺酯与不饱和烯基羧酸32经历双脱羧过程的偶联反应, 反应以fac-Ir(ppy)3作可见光催化剂, 添加剂Mg(ClO4)2能够显著提高反应的收率.通过自由基捕获和自由基钟实验, 他们认为反应体系可能涉及烷基自由基过程, 该反应具有良好的底物普适性, 适用于一级、二级和三级烷基羧酸酯.几乎同时, 徐坤等[33]报道了以Ru(bpy)3Cl2• 6H2O作可见光催化剂N-羟基邻苯二甲酰亚胺酯与烯基羧酸的双脱羧偶联反应, 1, 4-二氮杂二环[2.2.2]辛烷(DABCO)在反应中不仅作为碱, 还是单电子转移的共催化剂.傅尧等[34]发现Pd(PPh3)2Cl2与配体Xantphos在蓝色LED灯照射下能够高效地催化烷基N-羟基邻苯二甲酰亚胺酯与烯基芳烃34的脱羧自由基加成反应, 在光照条件下钯络合物通过单电子转移活化烷基N-羟基邻苯二甲酰亚胺酯, 同时抑制烷基钯中间体的β-H消除. Glorius等[35]以廉价Pd(PPh3)4作为可见光催化剂实现了烷基N-羟基邻苯二甲酰亚胺酯与烯烃36的脱羧Heck型偶联反应.金灿等[36]报道了在温和条件下可见光诱导烷基N-羟基邻苯二甲酰亚胺酯与香豆素38的区域选择性脱羧烷基化反应, 在无需额外氧化剂条件下以中等到优秀的收率合成了一系列3-烷基取代的香豆素衍生物.唐真宇等[37]报道了可见光促进的N-羟基邻苯二甲酰亚胺酯与末端炔烃40的脱羧偶联反应, 高选择性地得到了一系列Z-选择性的烯烃化合物41.鲍红丽等[38]研究了可见光与镍协同催化烷基二酰基过氧化物与末端炔烃的氢烷基化反应.
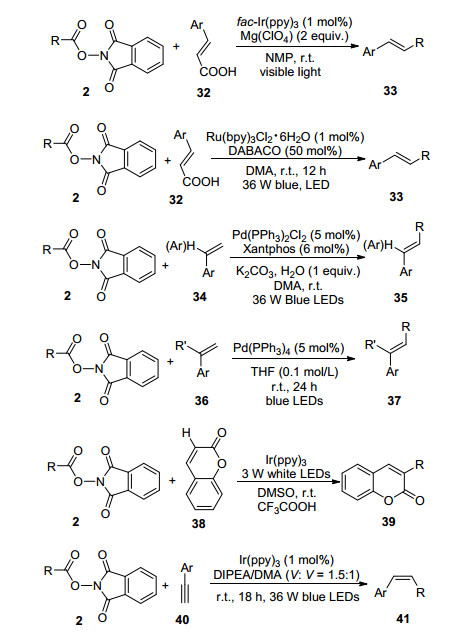
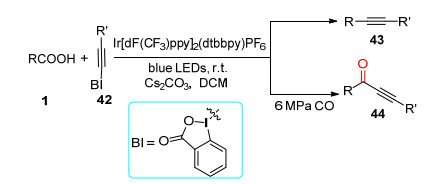
炔烃是有机合成中一类非常重要的化合物, 通过化学选择性C(sp3)—C(sp)键偶联反应能够高效地构建炔烃分子.其中, 自由基型C(sp3)—C(sp)键偶联反应能够高效地制备内炔化合物, 肖文精课题组[39]报道了可见光促进的烷基羧酸与炔基高碘化合物42的脱羧炔基化及羰基炔基化反应(Scheme 9). Ir[dF(CF3)ppy]2(dtbbpy)- PF6吸收可见光能量后进入到激发态, 烷基羧酸经历单电子氧化脱除二氧化碳得到烷基自由基, 随后发生自由基加成消除得到目标内炔化合物43, 若是在CO存在条件下能够得到羰基炔基化合物44.反应体系无需额外的氧化剂, 在温和反应条件能够以良好到优秀的产率得到一系列内炔及羰基炔化合物, 与此同时该催化体系被成功用于天然产物的后期功能化修饰.
陈以昀等[40]研究了可见光促进的烷基N-羟基邻苯二甲酰亚胺酯与炔基苯基砜45的还原交叉偶联反应(Eq. 6).在有机溶剂或中性水溶液条件下可以高效地制备烷基、芳基及硅基取代的炔烃化合物46, 值得注意的是该反应体系兼容各种官能团及生物分子, 并且对蛋白酶的活性没有任何影响.
|
|
(6) |
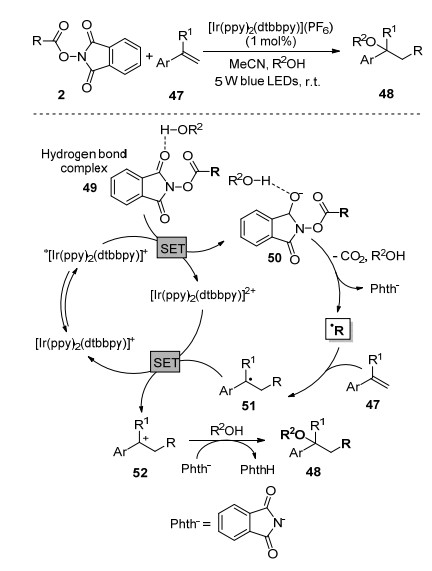
通过烯烃“一步”反应同时引入两个双官能团, 是有机反应中一种非常理想的合成策略. Glorius课题组[41]报道了可见光促进的N-羟基邻苯二甲酰亚胺酯、苯乙烯类化合物47与水或醇的多组分偶联反应, 通过烯烃的直接双官能团化高效地制备了一系列复杂的醇或醚化合物48.水或醇与N-羟基邻苯二甲酰亚胺酯通过氢键作用形成络合物49, 在可见光氧化下还原体系49得到一个电子形成物种50, 而中间体50经历N—O键断裂及脱羧形成烷基自由基, 随后烷基自由基与烯烃加成形成新的烷基自由基中间体51, 再失去一个电子得到碳正离子52, 最后在亲核试剂作用下生成目标化合物48.
2017年, 鲍红丽课题组[42]研究了可见光诱导的苯乙烯53与烷基二酰基过氧化物54的脱羧烷基酯化反应(Eq. 7).烷基二酰基过氧化物可以通过来源丰富的烷基羧酸简单制备, 在可见光氧化还原催化体系下能形成烷基自由基与烷基羧酸负离子, 在反应体系中既作为烷基自由基源, 还提供了烷基羧酸官能团, 从而实现了苯乙烯的双官能团化.在底物适用性方面, 多种苯乙烯类化合物及烷基二酰基过氧化物均能高效地转化得到目标产物, 而反应体系的唯一副产物是二氧化碳气体, 提高了反应的经济性.
|
|
(7) |
胺类化合物在有机合成中是一类非常重要的化合物, 不仅能够作为有机合成子, 其本身还具有独特的生物学活性, 从而被广泛地应用于医药领域.胺与烯烃的直接官能团化是目前构建功能化有机胺化合物强有力的合成手段, 2018年, 李金恒课题组[43]报道了可见光催化与路易斯酸协同作用下烷基N-羟基邻苯二甲酰亚胺酯、烯烃56与胺57的三组分1, 2-烷基胺化反应(Eq. 8).在反应体系中N-羟基邻苯二甲酰亚胺酯为烷基自由基源, 胺为末端亲核试剂, 通过碳-碳键和碳-氮键的高效构建制备了一系列官能团化的二级与三级胺化合物58.该反应体系具有良好的底物普适性, 适用于各类苯乙烯、烷基N-羟基邻苯二甲酰亚胺酯及胺化合物, 提供了一条高效制备具有高附加值胺类衍生物的合成路线.
|
|
(8) |
2018年, Glorius小组[44]在可见光条件下通过苯乙烯59氧化烷基化反应制备了一系列α-烷基苯乙酮衍生物60 (Eq. 9). N-羟基邻苯二甲酰亚胺酯在可见光氧化还原催化剂作用下脱羧形成烷基自由基, 而烷基自由基与苯乙烯发生加成得到稳定的苄基自由基, 随后其被氧化得到烷基正离子, 在二甲基亚砜(DMSO)存在下经历Kornblum氧化得到α-烷基苯乙酮, DMSO不仅作为反应的溶剂, 还是反应体系的氧源.与此同时, 叶松课题组[45]报道了通过类似的反应过程制备酮类化合物, 若在反应体系中加入三氟甲磺酸, 则形成Heck型产物.宋秋玲课题组[46]报道了可见光促进的N-羟基邻苯二甲酰亚胺酯与芳基烯基硅醚61制备烷基苯乙酮62的反应(Eq. 10).
|
|
(9) |
|
|
(10) |
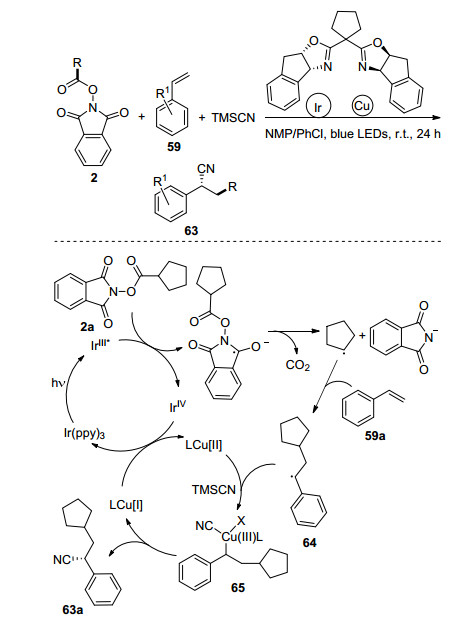
手性腈化合物在医药领域具有重要的作用, 2018年, 韩建林课题组[47]利用可见光与铜的协同催化作用实现了苯乙烯59的手性自由基氰烷基化反应, 高效地制备了一系列手性腈类化合物63 (Scheme 11).反应的推测机理如下: N-羟基邻苯二甲酰亚胺酯在可见光氧化还原催化体系中, 高效地形成环戊基自由基, 环戊基自由基与苯乙烯发生自由基加成得到关键的苄基自由基中间体64, 与此同时Cu(Ⅰ)经过氧化得到手性Box/ Cu(Ⅱ), 苄基自由基64氧化手性Box/Cu(Ⅱ)并与三甲基氰硅烷(TMSCN)反应得到手性中间体Box/Cu(Ⅲ)物种65, 最后经过还原消除得到目标化合物.
自由基串联环化反应在“一锅反应”中可以连续地形成多个化学键, 已经成为有机合成中提高反应效率的理想策略之一. Wang等[48]报道了可见光条件下有机染料荧光素催化的N-芳基甘氨酸与N-苯基马来酰亚胺66的氨烷基化反应(Eq. 11), 在可见光氧化还原催化剂作用下N-芳基甘氨酸形成的α-氨甲基自由基与N-苯基马来酰亚胺的双键加成, 随后经历环化/芳构化过程, 以良好的收率得到一系列类似天然化合物67.该氨甲基化反应条件温和, 分离产率良好, 适应于各类亲电性和亲核性的受体.
|
|
(11) |
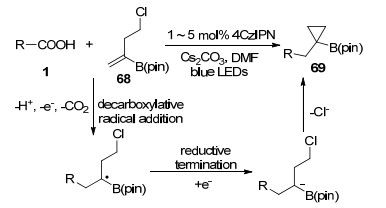
Aggarwal课题组[49]报道了可见光促进的脱羧自由基加成-极性环化反应构建功能化环丙烷化合物(Scheme 12), 作者认为烷基羧酸与缺电子烯烃68通过还原过程的自由基极性反转得到碳负离子中间体, 其与烷基氯代物发生分子内的烷基化反应, 得到环丙烷化合物69.该反应体系表现出良好的官能团容忍性, 适用于各类烷基羧酸和卤代烷基烯烃化合物, 提供了一条高原子经济性的制备各类环丙烷化合物的合成路线.
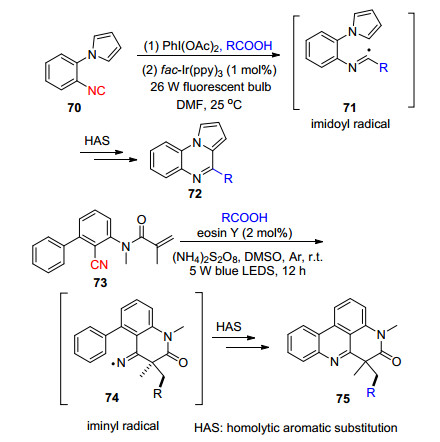
脱羧自由基环化反应可以简单高效地构建各类杂环化合物, Jamison等[50]开发了一种简单高效制备吡咯并[1, 2-α]喹喔啉衍生物72及含有喹喔啉骨架或类似物的方法.反应过程中烷基羧酸与PhI(OAc)2发生配体复分解形成PhI(OCOR)2, 其在可见光氧化还原体系形成烷基自由基, 随后发生自由基加成形成亚胺中间体自由基71.孙培培等[51]报道了烷基羧酸与芳基丙烯酰胺衍生物73的脱羧烷基化串联腈基插入/环化反应.烷基羧酸在有机染料催化体系中难以发生脱羧自由基过程, 而作者巧妙地将有机染料和过硫酸铵相结合, 实现了烷基羧酸的脱羧自由基, 从而发生随后的腈基插入及环化等过程, 得到烷基菲啶化合物75, 作者认为反应涉及亚胺氮自由基中间体74.由于使用有机染料与过硫酸铵的结合体系, 避免了昂贵可见光金属催化剂的使用, 提高了反应的经济性.
3, 3-二烷基吲哚酮化合物吲哚酮广泛存在于天然产物和具有生物学活性的药物分子之中, 朱成建课题组[52]采用可见光诱导的丙烯酰胺76与烷基羧酸的串联自由基环化反应, 构筑了一系列具有四级碳中心的吲哚酮化合物77 (Eq. 12), 该反应体系底物普适性良好, 适应于各种一级、二级及三级烷基羧酸.程辟课题组[53]报道了可见光条件下以N-羟基邻苯二甲酰亚胺酯作为烷基自由基前驱体的串联自由基环化反应, 高效制备了3, 3-二烷基吲哚酮化合物77 (Eq. 13), 该反应条件温和, 避免了具有易爆性的有机过氧化物类氧化剂的使用, 但是仅适用于三级烷基自由基, 对于一级、二级烷基自由基则不适用.
|
|
(12) |
|
|
(13) |
潘毅课题组[54]研究了可见光促进的不饱和羧酸的烷基化/内酯化反应, 高效地合成了具有生物学活性的烷基内酯化合物80 (Eq. 14). N-羟基邻苯二甲酰亚胺酯经历脱羧过程形成烷基自由基, 随后与不饱和炔酸发生自由基加成内酯化反应, 反应体系中的质子酸和水均起到重要的作用, 提供了一条由不饱和羧酸出发简单高效制备烷基取代内酯化合物的合成路线.肖文精课题组[55]报道了可见光促进的环烷醇取代的苯乙烯与N-羟基邻苯二甲酰亚胺酯经历烷基自由基加成与半频哪醇重排反应, 合成了一系列环酮化合物82 (Eq. 15). Reiser等[56]利用同样方法合成一系列螺丁烯酸内酯化合物及2, 3-端环化呋喃化合物.
|
|
(14) |
|
|
(15) |
郭丽娜等[57]以乙烯基叠氮化合物83与烷基N-羟基邻苯二甲酰亚胺酯为原料, 制备了一系列烷基取代的菲啶化合物84 (Eq. 16).在可见光条件下N-羟基邻苯二甲酰亚胺酯脱羧形成烷基自由基, 烷基自由基与乙烯基叠氮的烯基加成脱氮形成氮自由基, 随后发生环化得到目标产物.该串联自由基环化反应体系适用于一级、二级和三级N-羟基邻苯二甲酰亚胺酯, 以中等到良好的收率得到菲啶化合物, 反应体系无需过渡金属催化剂和氧化剂, 反应的经济性高.
|
|
(16) |
除了氮杂和氧杂环化合物, 有机磷杂环化合物在有机化学中也具有重要作用.周永波等[58]报道了一种简单高效地制备苯并[b]磷杂环化物86的合成路线(Eq. 17).在可见光作用下N-羟基邻苯二甲酰亚胺酯与炔基膦氧化物发生自由基加成及环化过程, 高效地构建了带有各种官能团的苯并[b]磷杂环化物.与传统方法相比, 该反应体系不需要使用任何金属催化剂, 因此无需考虑因为膦配位能力而导致的过渡金属残留, 同时反应无需使用强氧化剂, 大大提高了底物的官能团容忍性.
|
|
(17) |
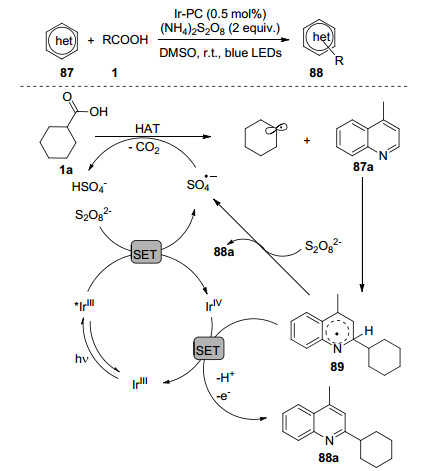
含氮杂环化合物广泛存在于天然产物、医药化学和农药分子之中, 因此氮杂环化合物的直接碳氢键官能团化反应一直受到广大科研工作者的关注.其中Minisci型反应是非常经典的氮杂环官能团化反应, 近来可见光氧化还原体系的Minisci型反应得到了快速发展. 2017年, Glorius课题组[59]研究了可见光促进的芳杂环与烷基羧酸的脱羧烷基化反应(Scheme 14).反应可能历程如下:在蓝光照射下Ir(Ⅲ)形成具有长寿命和强还原性的激发态*Ir(Ⅲ), 其还原过硫酸根负离子得到Ir(Ⅳ)和硫酸负离子自由基, 环己羧酸与硫酸负离子自由基之间发生氢原子转移得到环己基自由基, 环己基自由基与4-甲基喹啉加成得到中间体89, 89随后经历去质子单电子氧化得到目标化合物88.该反应体系具有良好地官能团容忍性, 适用于(异)喹啉、吡啶、2, 3-二氮杂萘及苯并噻唑等杂环衍生物及氨基酸和各种脂肪酸化合物, 由于反应的原料来源丰富, 该方法极大地便利了相关的各种具有药学活性分子的快速合成.由于可见光铱催化剂价格相对昂贵, 陆续有科研工作者开发了替代的方案. Ackermann课题组[60]将吖啶高氯酸盐与钴协同催化体系结合, 实现了金刚烷羧酸与杂环的直接脱羧烷基化反应, 使用钴催化剂能够再生吖啶高氯酸盐, 从而避免了额外氧化剂的使用, 但作者仅仅研究了金刚烷羧酸作为烷基自由基源.不久, 李洋等[61]报道了钴/铱协同催化的Minisci烷基化反应, 反应中不需要大量碱, 同时适用于各种烷基羧酸.王磊课题组[62]利用可见光催化剂吖啶高氯酸盐与空气中的氧分子相结合, 也高效地实现了杂环的直接烷基化反应.张博等[63]开发了无需额外可见光催化剂和酸添加剂的反应体系, 在可见光作用下三氟乙酸碘苯与烷基羧酸原位形成的高碘物种发生均裂得到烷基自由基, 从而实现杂环的直接烷基化.这些催化体系的开发都极大地丰富了杂环化合物烷基化的合成方法学.
基于可见光与Brønsted酸协同催化体系, 傅尧课题组[64, 65]报道了烷基N-羟基邻苯二甲酰亚胺酯与芳杂环的脱羧烷基化反应(Eq. 18), 其中可见光催化促进烷基自由基的形成, Brønsted酸增加杂环化合物的电子云密度, 实现了烷基自由基与芳杂环的交叉偶联.反应具有良好的底物普适性, 适用于各类烷基羧酸酯、天然/非天然氨基酸酯及二肽/三肽酯化合物, 提供了一种简单高效合成α-氨甲基及烷基取代的吡啶和喹啉等杂环化合物的方法.可见光催化自由基的不对称合成反应是光催化领域具有挑战性课题之一, Phipps课题组[66]通过可见光与手性Brønsted协同催化芳杂环的脱羧烷基化反应, 实现了手性α-氨甲基杂环化合物92的高效合成(Eq. 19).反应机理与傅尧等报道的催化体系相一致[64], 作者认为手性来源于手性磷酸与α-氨甲基自由基以及芳杂环之间的强烈氢键作用.该反应体系具有非常优秀的对映选择性和官能团兼容性, 从简单原料出发合成了一系列具有高附加值的化合物, 同时可用于各种药物分子的后期功能化修饰.江智勇等[67]研究了非金属可见光催化体系下α-氨甲基自由基与异喹啉加成合成手性α-氨甲基异喹啉化合物94 (Eq. 20). Sherwood等[68]报道了以4-CzIPN作可见光催化剂, 烷基羧酸为烷化试剂的“一锅两步”的Minisci反应. Opatz等[69]研究了Ru(bpy)3Cl2作可见光催化剂的类似反应体系.
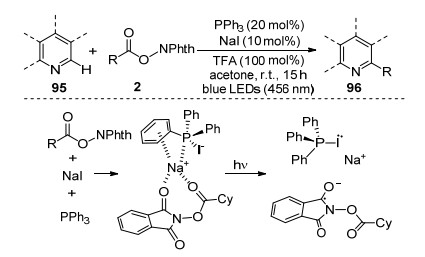
2019年, 傅尧课题组[70]首次实现了以三苯基膦和碘化钠复合体系作催化剂的芳杂环化合物的烷基化反应(Scheme 15).通常可见光催化剂与N-羟基邻苯二甲酰亚胺酯发生单电子转移形成烷基自由基, 而该复合体系是通过与N-羟基邻苯二甲酰亚胺酯发生分子内电荷转移得到烷基自由基物种.这种复合催化体系首次成功地将分子间电荷转移用于光氧化还原催化体系, 而且引入手性磷酸催化剂后能够高效地实现对映立体选择性.廉价三苯基膦和碘化钠的催化体系具有良好的官能团兼容性及优秀的对映选择性, 将在有机合成及药物分子开发等领域发挥重要的作用.
|
|
(18) |
|
|
(19) |
|
|
(20) |
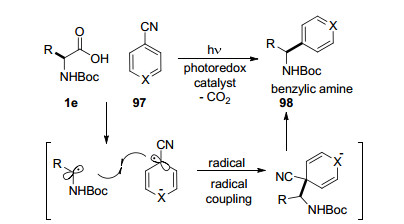
自由基之间交叉偶联反应已经成为构建新型碳-碳键强有力的手段之一, 但是两种不同活性自由基之间的选择性偶联依然面临挑战. 2014年, MacMillan课题组[71]研究了可见光促进的α-氨基酸与苯腈的脱羧自由基偶联反应, 以良好的收率得到了一系列苄胺类化合物98 (Scheme 16).在可见光氧化还原催化剂的作用下α-氨基酸脱羧得到烷基自由基, 苯腈得到芳烃自由基负离子, 随后两种自由基之间发生交叉偶联及腈基消除得到目标化合物.该方法适用于各类α-氨基酸化合物, 能够一步将生物质转化为药物分子.
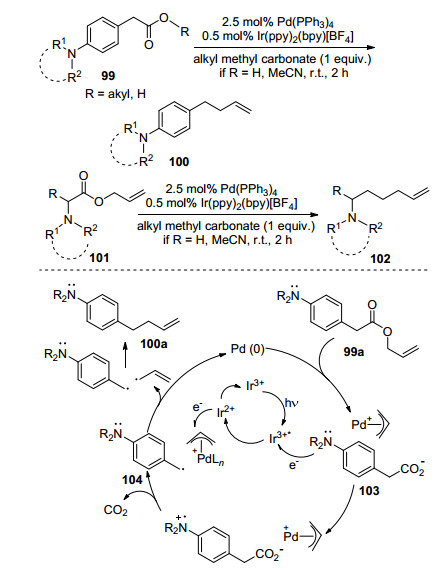
Tunge等[72]报道了可见光与钯协同催化体系中氨基烷基羧酸(酯)的脱羧烯基化反应(Scheme 17), 在Pd(0)的作用下烯基酯化合物形成Pd-π烯丙基物种与羧酸盐负离子103, 在可见光氧化还原体系下羧酸盐负离子103氧化后脱羧形成稳定的苄基自由基104, 而烯丙基钯物种被还原形成烯丙基自由基, 随后发生自由基偶联得到脱羧烯基化产物, 若氨基烷基羧酸作底物, 则反应体系需要加入烯丙基甲基碳酸酯, 从而原位形成羧酸酯.过渡金属与可见光协同催化体系拓宽了脱羧自由基烷基化反应的应用范围.
单氟烯基化分子在医药、农药及材料等领域有非常重要的应用. 2017年, 付华课题组[73]报道了可见光条件下α-氨基酸与偕二氟烯烃化合物105的自由基偶联反应, 制备了单氟烯基化合物106 (Eq. 21). α-氨基酸是自然界中最常见的具有生物学活性的有机分子, 在可见光氧化还原催化剂和碱的作用下, α-氨基酸脱除二氧化碳得到α-氨甲基自由基, 与此同时偕二氟烯烃分子得到一个电子脱去氟负离子形成单氟烯基自由基, 两种自由基高选择性地发生偶联生成α-氨基单氟烯基化合物, 可见光催化体系完成循环再生.但当作者选用烷基羧酸作为自由基前驱体时, 偶联反应收率很低.最近, 安光辉等[74]使用4-CzIPN作催化剂, 6 ewquiv.的碳酸铯作碱, 成功地实现了烷基羧酸与偕二氟烯烃化合物的脱羧自由基偶联.该反应在太阳光作用下也能顺利进行, 而且可以高效地进行复杂分子的后期功能化.
|
|
(21) |
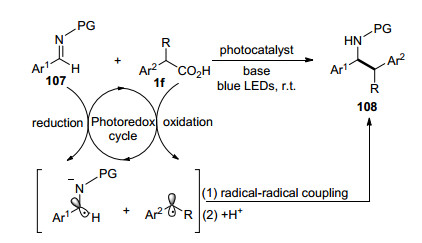
2018年, 翁江等[75]报道了可见光促进的亚胺107与苯乙酸类化合物的脱羧苄基化反应(Scheme 18).简单的一级、二级和三级芳基乙酸作为理想的苄基自由基源, 在可见光氧化还原体系中苯乙酸被氧化得到苄基自由基, 而相应的亚胺被还原成亚胺自由基负离子, 二者相互结合得到目标偶联化合物.该反应体系与之前报道的亚胺苄基化反应相比, 避免了底物预官能团化及α-位含杂原子化合物的使用, 提供了一条简单高效、操作方便且条件温和的制备β-芳基乙胺化合物108的合成路线.
2018年, 丛欢等[76]报道了可见光条件下N-芳基四氢异喹啉109与N-羟基邻苯二甲酰亚胺酯的脱羧烷基化反应(Eq. 22).以有机染料敏化半导体材料作为可见光催化剂, 它能够氧化N-芳基四氢异喹啉去质子化得到苄基自由基, 还原N-羟基邻苯二甲酰亚胺酯得到烷基自由基, 两种自由基之间快速结合得到烷基取代的四氢异喹啉化合物110.这种廉价的有机敏化半导体材料催化体系避免了贵重金属的使用, 降低了反应的成本, 提高了反应的经济性.
|
|
(22) |
2018年, 许兆青课题组[77]报道了可见光促进的甘氨酸和多肽酯化物的脱羧烷基化反应(Eq. 23).作者开发了一种新型的廉价铜与配体相结合的可见光催化体系, 该催化体系中铜配体络合物[Cu(Ⅰ)L]能够吸收可见光到达激发态[Cu(Ⅰ)L]*, 还原N-羟基邻苯二甲酰亚胺酯形成烷基自由基和[Cu(Ⅱ)L], 而[Cu(Ⅱ)L]氧化甘氨酸衍生物得到自由基阳离子, 随后在碱的作用下得到稳定的α-氨甲基自由基, 两种自由基交叉偶联得到目标产物, 铜催化剂还原再生.该反应体系具有良好的底物普适性, 成功地实现了多种非天然氨基酸的制备和多肽化合物的烷基化修饰.
|
|
(23) |
在可见光氧化还原体系中, 烷基羧酸及衍生物是一类理想的烷基自由基前驱体, 而过渡金属催化体系中过渡金属与卤代烃能够发生氧化加成得到M-R物种, 其与自由基结合发生还原消除能够构建新的碳-碳键.基于此思路, 过渡金属与可见光协同催化卤代烃与烷基羧酸的直接交叉偶联反应受到了广泛关注.
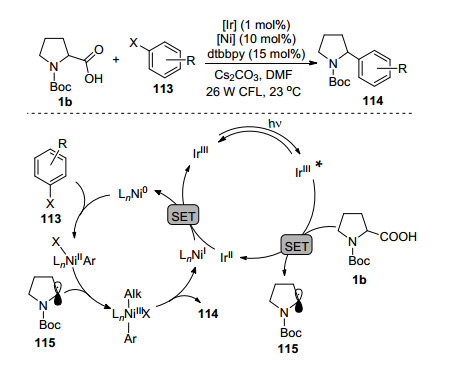
2014年, MacMillan课题组[78]首次报道了过渡金属镍和可见光协同催化的α-氨基羧酸与芳基卤化物113直接交叉偶联构建新的C(sp3)—C(sp2)键(Scheme 19).反应的可能历程如下:在可见光照射下铱催化剂吸收能量到达激发态, 在Ir(Ⅲ)*与碱作用下烷基羧酸被氧化脱羧得到烷基自由基115, 同时芳基卤代物在Ni(0)的作用下氧化加成得到Ni(Ⅱ)中间体, Ni(Ⅱ)物种迅速捕捉体系中的自由基得到加合物Ni(Ⅲ), 最后经历还原消除得到目标产物.随后, 他们[79]将芳基卤代物拓展到烷基溴代物, 在温和条件下通过简单丰富的烷基羧酸及烷基卤化物的偶联反应构建C(sp3)—C(sp3)键. MacMillan与Fu课题组[80]合作开发了镍与可见光协同催化体系的非对称合成, 从自然普遍存在的氨基酸和芳基卤化物出发, 制备了一系列具有高对映选择性的苄胺化合物. Oderinde等[81]研究了分子氧、溶剂及光源对协同催化体系的影响. Zhang等[82]用4-CzIPN作可见光催化剂来替代昂贵的钌和铱光催化剂. Scaiano等[83]报道了TiO2和二甲氧基乙烷氯化镍非均相催化体系中烷基羧酸与芳基碘化物的偶联反应. ElMarrouni等[84]利用该协同催化体系简单快速地合成了非核苷类逆转录酶抑制剂候选药doravirine类似物.此类过渡金属镍与可见光氧化还原协同催化体系很好地解决了传统合成方法中C(sp3)—C(sp2)或(sp3)—C(sp3)键不易构建的问题.
基于铜与可见光协同催化体系, MacMillan课题组[85]研究了烷基羧酸与Togni’s试剂116的脱羧三氟甲基化反应(Eq. 24), 反应体系具有良好的官能团容忍性, 包括杂环、烯烃、醇及张力环, 含有烷基羧酸的天然产物及药物分子都可以利用该策略制备相应的三氟甲基化产物, 该方法能够实现复杂分子的后期三氟甲基官能团化, 在生物医药领域具有重要的应用前景.付华课题组[86]报道了铜与可见光协同催化体系中α-氨基酸衍生物与末端炔烃的脱羧烷基化反应(Eq. 25), 在简单温和条件下以优秀至良好的收率制备了一系列丙炔胺化合物119.研究表明, α-氨基酸衍生物在可见光催化体系中转化为α-氨甲基自由基, α-甲基自由基继续被氧化成亚胺阴离子, 与此同时末端炔与碘化铜形成炔铜物种, 随后发生亲核进攻得到目标化合物.
|
|
(24) |
|
|
(25) |
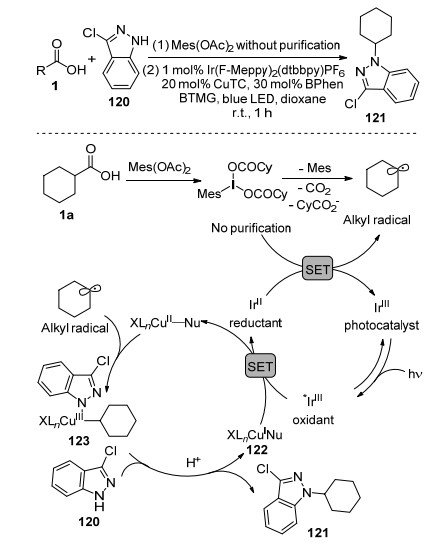
胡喜乐课题组[87]利用铜与可见光协同催化体系, 实现了芳胺与N-羟基邻苯二甲酰亚胺酯的脱羧偶联反应, 制备了一系列烷基胺化合物, 但是他们仅仅研究了芳香胺类亲核试剂.而MacMillan课题组[88]报道了在铜与可见光协同催化体系中烷基羧酸与各类胺化合物交叉偶联构建碳-氮键(Scheme 20).反应的可能历程: Cu(Ⅰ)与亲核试剂胺120发生取代反应得到Nu—Cu(Ⅰ)物种122, Ir光敏剂将Nu—Cu(Ⅰ)的电子转移给活化的羧酸形成环己基自由基, 与此同时Nu—Cu(Ⅱ)物种捕获环己基自由基得到中间体123, 其经过还原消除构建了碳-氮键得到目标产物.胡喜乐课题组[87]的工作认为是Nu—Cu(Ⅰ)物种捕获烷基自由基形成Nu—Cu(Ⅱ)—R中间体.该反应具有非常好的普适性, 各种氮杂环胺、芳香胺、酰胺等胺试剂与各种一级、二级与三级烷基羧酸都能够顺利发生偶联反应, 重要的是该反应体系能够用于天然产物和药物分子的后期官能化.
含氟有机化合物在医药、农药及材料等领域具有广泛的应用, 在有机分子中引入氟原子能够显著改变其本身的物理性质、化学性质和生物活性.在可见光条件下烷基羧酸的氟化反应也受到了越来越多的关注, Sammis等[89]首次实现了以Ru(bpy)3Cl2作可见光催化剂, 选择性氟试剂作氟源的苯氧乙酸的脱羧氟化反应(Eq. 26).瞬态吸收光谱表明, Ru(Ⅱ)催化剂与选择性氟试剂之间的单电子转移过程是反应体系的关键步骤.此反应体系相较之前的紫外光作用下的脱羧氟化反应具有重要的突破, 但仅仅苯氧乙酸适用该体系限制了其应用范围.随后, MacMillan及合作者[90]研究了[Ru(bpz)3](PF6)2或[Ir{dF(CF3)ppy}2(dtbbpy)]PF6作为光催化剂, 能够适用于各种烷基羧酸的脱羧氟化反应(Eq. 27, condition A).而叶金星等[91]报道了有机染料催化的一级、二级和三级烷基羧酸与选择性氟的脱羧氟化反应(Eq. 27, condition B), 制备了一系列具有高附加值的有机氟化合物126.由此可见, 可见光促进的脱羧自由基氟化反应能够高效地将来源广泛的烷基羧酸转化为一系列烷基氟化物, 这进一步丰富了有机氟化合物的合成方法学.
|
|
(26) |
|
|
(27) |
烷基胺化合物是一类重要的有机含氮化合物, 通过可见光促进的烷基羧酸与碳氮双键或氮氮双键的自由基加成反应能够高效构建烷基胺化合物, Tunge等[92]报道了可见光条件下烷基羧酸与偶氮二甲酸二异丙酯127的脱羧自由基加成反应, 制备了一系列烷基胺化合物128 (Eq. 28), 而烷基胺化合物在碱性条件下发生氮-氮键断裂能够得到氨基甲酸酯化合物. Leonori等[93]则报道了温和条件下烷基羧酸与亚硝基芳烃129的直接脱羧自由基加成反应, 合成了一系列烷基芳基羟胺化合物130 (Eq. 29), 该反应体系被成功地应用于各种复杂烷基酸的功能化修饰.
|
|
(28) |
|
|
(29) |
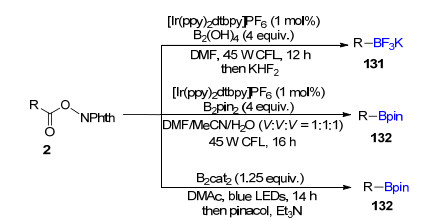
烷基硼化合物是有机合成中一类非常重要的有机合成子, 李鹏飞课题组[94]报道了一种在可见光条件下简单高效合成烷基硼酸酯的方法, 以N-羟基邻苯二甲酰亚胺酯与硼酸酯/硼酸为起始原料, [Ir(ppy)2-(dtbpy)]PF6作光催化剂, 在可见光作用下能够高效地生成三氟硼酸盐131及一级、二级烷基硼酸酯132 (Scheme 21).随后, Aggarwal课题组[95]报道了温和条件下无需使用额外可见光催化剂, 烷基N-羟基邻苯二甲酰亚胺酯与双联邻苯二酚硼酸酯的脱羧硼化反应, 经过频哪醇和三乙胺的简单后处理得到稳定的频哪醇硼酸酯132 (Scheme 21).通常烷基N-羟基邻苯二甲酰亚胺酯需要在可见光氧化还原催化剂作用下形成烷基自由基, 作者通过自由基钟和相关控制实验, 认为在该反应体系中N-羟基邻苯二甲酰亚胺酯、双联邻苯二酚硼酸酯及N, N-二甲基乙酰胺(DMAc)溶剂形成了三元杂化络合物, 这种络合物在可见光条件下通过分子内的电子转移直接形成烷基自由基, 从而完成脱羧硼化反应.
付华课题组[96]研究了在可见光条件下N-羟基邻苯二甲酰亚胺酯与二硒化合物133的脱羧偶联反应(Eq. 30), 在温和条件下以蛋白氨基酸、L-天冬氨酸及谷氨酸的衍生物N-羟基邻苯二甲酰亚胺酯作烷基自由基源以及手性源, 高效地制备了一系列手性α-硒氨基酸衍生物134, 利用该方法将有效地开发出更多具有生物和医药活性的非天然手性α-氨基酸分子.基于以上思路, 他们[97]报道了N-羟基邻苯二甲酰亚胺酯与芳基硫酚135的脱羧偶联反应(Eq. 31), 作者认为N-羟基邻苯二甲酰亚胺酯与碳酸铯能够结合形成在可见光范围内有吸收的络合物, 而这种络合物充当反应体系的光敏剂, 从而不需要额外的光催化剂.
|
|
(30) |
|
|
(31) |
综述了可见光促进的烷基羧酸及其衍生物脱羧偶联反应的研究进展, 对脱羧自由基反应类型进行了归纳和总结.可见光促进的脱羧偶联反应已经被应用于各类碳-碳及碳-杂键的高效构建, 实现了许多传统方法难以完成的反应过程.由于此类反应通过可见光提供能源且一般是在室温条件下进行, 使得反应更绿色环保, 在有机合成化学中具有重要应用.
尽管可见光促进的烷基羧酸及其衍生物的脱羧烷基化偶联反应取得了快速的发展, 该领域仍然存在诸多挑战.例如:提高脱羧偶联反应中烷基自由基物种的化学选择性和反应活性, 实现烷基自由基中间体的精准高效转化, 有效调控自由基烷基化反应的立体选择性, 开发新型可见光催化体系及深入系统地研究反应机理.此外, 可见光促进的脱羧烷基化反应在连续流动化学的发展能够加快药物开发.相信随着自由基化学的蓬勃发展及广大科研工作者的不断努力, 可见光促进的自由基烷基化反应将会得到长远的发展和应用.
Lunbderg, H.; Tinnis, F.; Selander, N.; Adolfsson, H. Chem. Soc. Rev. 2014, 43, 2714. doi: 10.1039/C3CS60345H
Morrill, L. C.; Smith, A. D. Chem. Soc. Rev. 2014, 43, 6214. doi: 10.1039/C4CS00042K
Straathof, A. J. J. Chem. Rev. 2014, 114, 1871. doi: 10.1021/cr400309c
(a) Sivaguru, P.; Wang, Z.; Zanoni, G.; Bi, X. Chem. Soc. Rev. 2019, 48, 2615.
(b) Wang, F.; Chen, P.; Liu, G. S. Acc. Chem. Res. 2018, 51, 2036.
(c) Yi, H.; Zhang, G.; Wang, H.; Huang, Z.; Wang, J.; Singh, A.; Lei, A. Chem. Rev. 2017, 117, 9016.
(d) Zhao, Y.; Liu, Z. Chin. J. Chem. 2018, 36, 455.
(e) Tan, F.; Yin, G. Chin. J. Chem. 2018, 36, 545.
(f) Cao, Y.; He, X.; Wang, N.; Li, H.; He, L.-N. Chin. J. Chem. 2018, 36, 644.
(a) Wang, H.; Wu, P.; Zhao, X.; Zeng, J.; Wan, Q. Acta Chim. Sinica 2019, 77, 231 (in Chinese).
(王浩, 吴品儒, 赵祥, 曾静, 万谦, 化学学报, 2019, 77, 231.)
(b) Chen, Y.; Chang, L.; Zuo, Z. Acta Chim. Sinica 2019, 77, 794 (in Chinese).
(陈奕霖, 常亮, 左智伟, 化学学报, 2019, 77, 794.)
(c) Ye, S.; Wu, J. Acta Chim. Sinica 2019, 77, 814 (in Chinese).
(叶盛青, 吴劼, 化学学报, 2019, 77, 814.)
(d) Zhang, H.-H.; Yu, S. Acta Chim. Sinica 2019, 77, 832 (in Chinese).
(张洪浩, 俞寿云, 化学学报, 2019, 77, 832.)
(e) Ren, L.; Ran, M.; He, J.; Qian, Y.; Yao, Q. Chin. J. Org. Chem. 2019, 39, 1583 (in Chinese).
(任林静, 冉茂刚, 何佳芯, 钱燕, 姚秋丽, 有机化学, 2019, 39, 1583.)
(a) Zhao, B.; Tan, H.; Chen, C.; Jiao, N.; Shi, Z. Chin. J. Chem. 2018, 36, 995.
(b) Yuan, Y.; Dong, W.; Gao, X.; Xie, X.; Curran, D.; Zhang, Z. Chin. J. Chem. 2018, 36, 1035.
(c) Li, C.; Qi, Z.; Yang, Q.; Qiang, X.-Y.; Yang, S.-D. Chin. J. Chem. 2018, 36, 1052.
(d) An, X.; Zhang, H.; Xu, Q.; Yu, L.; Yu, S. Chin. J. Chem. 2018, 36, 1147.
(e) Xu, W.; Dai, X.; Xu, H.; Weng, J. Chin. J. Org. Chem. 2018, 38, 2807 (in Chinese).
(徐雯秀, 戴小强, 徐涵靖, 翁建全, 有机化学, 2018, 38, 2807.)
(f) Ruan, L; Chen, C.; Zhang, X.; Sun, J. Chin. J. Org. Chem. 2018, 38, 3155 (in Chinese).
(阮利衡, 陈春欣, 张晓欣, 孙京, 有机化学, 2018, 38, 3155.)
Sun, A.; C.; McAtee, R. C.; McClain, E. J.; Stephenson, C. R. J. Synthesis 2019, 51, 1063. doi: 10.1055/s-0037-1611658
Murarka, S. Adv. Synth. Catal. 2018, 360, 1735. doi: 10.1002/adsc.201701615
Xuan, J.; Zhang, Z. G.; Xiao, W. J. Angew. Chem., Int. Ed. 2015, 54, 15632. doi: 10.1002/anie.201505731
Chu, L.; Ohta, C.; Zuo, Z.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 10886. doi: 10.1021/ja505964r
Miyake, Y.; Nakajima, K.; Nishibayashi, Y. Chem. Commun. 2013, 49, 7854. doi: 10.1039/c3cc44438d
Millet, A.; Lefebvre, Q.; Rueping, M. Chem.-Eur. J. 2016, 22, 13464. doi: 10.1002/chem.201602257
Ramirez, N.; Gonzalez-Gomez, J. Eur. J. Org. Chem. 2017, 2154.
Chinzei, T.; Miyazawa, K.; Yasu, Y.; Koike, T.; Akita, M. RSC Adv. 2015, 5, 21297. doi: 10.1039/C5RA01826A
Bloom, S.; Liu, C.; Kölmel, D.; Qiao, J.; Zhang, Y.; Poss, M.; Ewing, W.; MacMillan, D. W. C. Nat. Chem. 2018, 10, 205. doi: 10.1038/nchem.2888
Kölmel, D. K.; Loach, R. P.; Knauber, T.; Flanagan, M. E. ChemMedChem 2018, 13, 2159. doi: 10.1002/cmdc.201800492
Chen, J.-Q.; Chang, R.; Wei, Y.-L.; Mo, J.-N.; Wang, Z.-Y.; Xu, P.-F. J. Org. Chem. 2018, 83, 253. doi: 10.1021/acs.joc.7b02628
Xiao, T.; Li, L.; Zhou, L. J. Org. Chem. 2016, 81, 7908. doi: 10.1021/acs.joc.6b01620
Noble, A.; Mega, R. S.; Pflästerer, D.; Myers, E.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2018, 57, 2155. doi: 10.1002/anie.201712186
Guo, T.; Zhang, Y.; Fang, Y.; Jin, X.; Li, Y.; Li, R.; Li, X.; Cen, W.; Liu, X.; Tian, Z. Adv. Synth. Catal. 2018, 360, 1352. doi: 10.1002/adsc.201701285
Yin, Y.; Dai, Y.; Jia, H.; Li, J.; Bu, L.; Qiao, B.; Zhao, X.; Jiang, Z. J. Am. Chem. Soc. 2018, 140, 6083. doi: 10.1021/jacs.8b01575
Okada, K.; Okamoto, K.; Morita, N.; Okubo, K.; Oda, M. J. Am. Chem. Soc. 1991, 113, 9401. doi: 10.1021/ja00024a074
Schnermann, N.; Overman, L. E. Angew. Chem., Int. Ed. 2012, 51, 9576. doi: 10.1002/anie.201204977
Pratsch, G.; Lackner, G. L.; Overman, L. E. J. Org. Chem. 2015, 80, 6025. doi: 10.1021/acs.joc.5b00795
Hu, C.; Chen, Y. Org. Chem. Front. 2015, 2, 1352. doi: 10.1039/C5QO00187K
Schwarz, J.; König, B. Green Chem. 2016, 18, 4743. doi: 10.1039/C6GC01101B
Jin, Y.; Yang, H.; Fu, H. Org. Lett. 2016, 18, 6400. doi: 10.1021/acs.orglett.6b03300
Noble, A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 11602. doi: 10.1021/ja506094d
Cao, H.; Jiang, H.; Feng, H.; Kwan, J.; Liu, X.; Wu, J. J. Am. Chem. Soc. 2018, 140, 16360. doi: 10.1021/jacs.8b11218
Zheng, C.; Chen, W.; Li, H.; Na, R.; Shang, R. Org. Lett. 2018, 20, 2559. doi: 10.1021/acs.orglett.8b00712
Till, N. A.; Smith, R. T.; MacMillan, D. W. C. J. Am. Chem. Soc. 2018, 140, 5701. doi: 10.1021/jacs.8b02834
Zhang, J.; Yang, J.; Guo, L.; Duan, X. Chem.-Eur. J. 2017, 23, 10259. doi: 10.1002/chem.201702200
Xu, K.; Tan, Z.; Zhang, H.; Liu, J.; Zhang, S.; Wang, Z. Chem. Commun. 2017, 53, 10719. doi: 10.1039/C7CC05910H
Wang, G.; Shang, R.; Fu, Y. Org. Lett. 2018, 20, 888. doi: 10.1021/acs.orglett.8b00023
Koy, M.; Sandfort, F.; Tlahuext-Aca, A.; Quach, L.; Daniliuc, C.; Glorius, F. Chem.-Eur. J. 2018, 24, 4552. doi: 10.1002/chem.201800813
Jin, C.; Yan, Z.; Sun, B.; Yang, J. Org. Lett. 2019, 21, 2064. doi: 10.1021/acs.orglett.9b00327
Dai, G.; Lai, S.; Luo, Z.; Tang, Z. Org. Lett. 2019, 21, 2269. doi: 10.1021/acs.orglett.9b00558
Li, Y.; Ge, L.; Qian, B.; Babu, K. R.; Bao, H. Tetrahedron Lett. 2016, 57, 5677. doi: 10.1016/j.tetlet.2016.11.020
Zhou, Q.; Guo, W.; Ding, W.; Wu, X.; Chen, X.; Lu, L.; Xiao, W. Angew. Chem., Int. Ed. 2015, 54, 11196. doi: 10.1002/anie.201504559
Yang, J.; Zhang, J.; Qi, L.; Hu, C.; Chen, Y. Chem. Commun. 2015, 51, 5275. doi: 10.1039/C4CC06344A
Tlahuext-Aca, A.; Garza-Sanchez, R.; Glorius, F. Angew. Chem., Int. Ed. 2017, 56, 3708. doi: 10.1002/anie.201700049
Ge, L.; Li, Y.; Jian, W.; Bao, H. Chem.-Eur. J. 2017, 23, 11767. doi: 10.1002/chem.201702385
Quyang, X.; Li, Y.; Song, R.; Li, J. Org. Lett. 2018, 20, 6659. doi: 10.1021/acs.orglett.8b02670
Tlahuext-Aca, A.; Garza-Sanchez, A.; Schäfer, M.; Glorius, F. Org. Lett. 2018, 20, 1546. doi: 10.1021/acs.orglett.8b00272
Xia, Z.; Zhang, C.; Gao, Z.; Ye, S. Org. Lett. 2018, 20, 3496. doi: 10.1021/acs.orglett.8b01268
Kong, W.; Yu, C.; An, H.; Song, Q. Org. Lett. 2018, 20, 349. doi: 10.1021/acs.orglett.7b03587
Sha, W.; Deng, L.; Ni, S.; Mei, H.; Han, J.; Pan, Y. ACS Catal. 2018, 8, 7489. doi: 10.1021/acscatal.8b01863
Chen, L.; Chao, C.; Pan, Y.; Dong, S.; Teo, Y.; Wang, J.; Tan, C. Org. Biomol. Chem. 2013, 11, 5922. doi: 10.1039/c3ob41091a
Shu, C.; Mega, R. S.; Andreassen, B. J.; Noblem A.; Aggarwal, V. K. Angew. Chem., Int. Ed. 2018, 57, 15430. doi: 10.1002/anie.201808598
He, Z.; Bae, M.; Wu, J.; Jamison, T. F. Angew. Chem., Int. Ed. 2014, 53, 14451. doi: 10.1002/anie.201408522
Yu, Y.; Yuan, W.; Huang, H.; Cai, Z.; Liu, P.; Sun, P. J. Org. Chem. 2018, 83, 1654. doi: 10.1021/acs.joc.7b03080
Xie, J.; Xu, P.; Li, H.; Xue, Q.; Jin, H.; Cheng, Y.; Zhu, C. Chem. Commun. 2013, 49, 5672. doi: 10.1039/c3cc42672f
Tang, Q.; Liu, X.; Liu, S.; Xie, H.; Liu, W.; Zeng, J.; Cheng, P. RSC Adv. 2015, 5, 89009. doi: 10.1039/C5RA17292F
Sha, W.; Ni, S.; Han, J.; Pan, Y. Org. Lett. 2017, 19, 5900. doi: 10.1021/acs.orglett.7b02899
Yao, S.; Zhang, K.; Zhou, Q.; Zhao, Y.; Shi, D.; Xiao, W. Chem. Commun. 2018, 54, 8096. doi: 10.1039/C8CC04503H
Kachkovskyi, G.; Faderl, C.; Reiser, O. Adv. Synth. Catal. 2013, 355, 2240. doi: 10.1002/adsc.201300221
Yang, J.; Zhang, J.; Zhang, J.; Duan, X.; Gao, L. J. Org. Chem. 2018, 83, 1598. doi: 10.1021/acs.joc.7b02861
Liu, L.; Dong, J.; Yan, Y.; Yin, S.; Han, L.; Zhou, Y. Chem. Commun. 2019, 55, 233. doi: 10.1039/C8CC08689C
Garza-Sanchez, A.; Tlahuext-Aca, A.; Glorius, F. ACS Catal. 2017, 56, 12336.
Koeller, J.; Gandeepan, P.; Ackermann, L. Synthesis 2019, 51, 1284. doi: 10.1055/s-0037-1611633
Tian, W.-F.; Hu, C.-H.; He, K.-N.; He, X.-Y.; Li, Y. Org. Lett. 2019, 21, 6930. doi: 10.1021/acs.orglett.9b02539
Wang, B.; Li, P.; Miao, T.; Zou, L.; Wang, L. Org. Biomol. Chem. 2019, 17, 115. doi: 10.1039/C8OB02476F
Zhang, X.-Y.; Weng, W.-Z.; Liang, H.; Yang, H.; Zhang, B. Org. Lett. 2018, 20, 4686. doi: 10.1021/acs.orglett.8b02016
Chen, W.; Shang, R.; Fu, Y. ACS Catal. 2017, 7, 907. doi: 10.1021/acscatal.6b03215
Cheng, W.-M.; Shang, R.; Fu, M.-C.; Fu, Y. Chem.-Eur. J. 2017, 23, 2537. doi: 10.1002/chem.201605640
Proctor, R. S. J.; Davis, H. J.; Phipps, R. J. Science 2018, 360, 419. doi: 10.1126/science.aar6376
Liu, X.; Liu, Y.; Chai, G.; Qiao, B.; Zhao, X.; Jiang, Z. Org. Lett. 2018, 20, 6298. doi: 10.1021/acs.orglett.8b02791
Sherwood, T. C.; Li, N.; Yazdani, A. N.; Murali Dhar, T. G. J. Org. Chem. 2018, 83, 3000. doi: 10.1021/acs.joc.8b00205
Kammer, L. M.; Rahman, A.; Opatz, T. Molecules 2018, 23, 764. doi: 10.3390/molecules23040764
Fu, M.-C.; Shang, R.; Zhao, B.; Wang, B.; Fu, Y. Science 2019, 363, 1429. doi: 10.1126/science.aav3200
Zuo, Z.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 5257. doi: 10.1021/ja501621q
Lang, S.; O'Nele, K.; Tunge, J. J. Am. Chem. Soc. 2014, 136, 13606. doi: 10.1021/ja508317j
Li, J.; Lefebvre, Q.; Yang, H.; Zhao, Y.; Fu, H. Chem. Commun. 2017, 53, 10299. doi: 10.1039/C7CC05758J
Yang, H.; Tian, C.; Qiu, D.; Tian, H.; An, G.; Li, G. Org. Chem. Front. 2019, 6, 2365. doi: 10.1039/C9QO00495E
Guo, J.; Wu, Q.; Xie, Y.; Weng, J.; Lu, G. J. Org. Chem. 2018, 83, 12559. doi: 10.1021/acs.joc.8b01849
Ren, L.; Cong, H. Org. Lett. 2018, 20, 3225. doi: 10.1021/acs.orglett.8b01077
Wang, C.; Guo, M.; Qi, R. Shang, Q.; Liu, Q.; Wang, S.; Zhao, L.; Wang, R.; Xu, Z. Angew. Chem., Int. Ed. 2018, 26, 15841.
Zuo, Z.; Ahneman, D. T.; Chu, L.; Terrett, J. A.; Doyle, A. G.; MacMillan, D. W. C. Science 2014, 345, 437.
Johnston, C. P.; Smith, R. T.; Allmendinger, S.; MacMillan, D. W. C. Nature 2016, 536, 322. doi: 10.1038/nature19056
Zuo, Z.; Cong, H.; Li, W.; Choi, J.; Fu, G. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 1832. doi: 10.1021/jacs.5b13211
Oderinde, M. S.; Varela-Alvarez, A.; Aquila, B.; Robbins, D. W.; Johannes, J. W. J. Org. Chem. 2015, 80, 7642. doi: 10.1021/acs.joc.5b01193
Luo, J.; Zhang, J. ACS Catal. 2016, 6, 873. doi: 10.1021/acscatal.5b02204
McTiernan, C. D.; Leblanc, X.; Scaiano, J. C. ACS Catal. 2017, 7, 2171. doi: 10.1021/acscatal.6b03687
Suen, L. M.; Wang, C.; Hunter, D. N.; Mitchell, H. J.; Cnverso, A.; ElMarrouni, A. Synthesis 2018, 50, 3177. doi: 10.1055/s-0037-1610155
Kautzky, J. A.; Wang, T.; Evans, R. W.; MacMillan, D. W. C. J. Am. Chem. Soc. 2018, 10, 6522.
Zhnag, H.; Zhang, P.; Jiang, M.; Yang, H.; Fu, H. Org. Lett. 2017, 19, 1016. doi: 10.1021/acs.orglett.6b03888
Mao, R.; Frey, A.; Balon, J.; Hu, X. Nat. Catal. 2018, 1, 120. doi: 10.1038/s41929-017-0023-z
Liang, Y.; Zhang, X.; MacMillan, D. W. C. Nature 2018, 559, 83. doi: 10.1038/s41586-018-0234-8
Rueda-Becerril, M.; Mahe, O.; Drouin, M.; Majewski, M. B.; West, J. G.; Wolf, M. O.; Sammis, G. M.; Paquin, J. F. J. Am. Chem. Soc. 2014, 136, 2637. doi: 10.1021/ja412083f
Ventre, S.; Petronijevi, F. R.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 5654. doi: 10.1021/jacs.5b02244
Wu, X.; Meng, C.; Yuan, X.; Jia, X.; Qian, X.; Ye, J. Chem. Commun. 2015, 51, 11864. doi: 10.1039/C5CC04527D
Lang, S. B.; Cartwright, K. C.; Welter, R. S.; Locascio, T. M.; Tunge, J. A. Eur. J. Org. Chem. 2016, 3331.
Davies, J.; Angelini, L.; Alkhalifah, M. A.; Sanz, L. M.; Sheikh, N. S.; Leonori, D. Synthesis 2018, 50, 821. doi: 10.1055/s-0036-1591744
Hu, D.; Wang, L.; Li, P. Org. Lett. 2017, 19, 2770. doi: 10.1021/acs.orglett.7b01181
Fawcett, A.; Pradeilles, J.; Wang, Y.; Mutsuga, T.; Myers, E.; Aggarwal, V. K. Science 2017, 357, 283. doi: 10.1126/science.aan3679
Jiang, M.; Yang, H.; Fu, H. Org. Lett. 2016, 18, 1968. doi: 10.1021/acs.orglett.6b00489
Jin, Y.; Yang, H.; Fu, H. Chem. Commun. 2016, 52, 12909. doi: 10.1039/C6CC06994K

图式1 烷基羧酸及其衍生物脱羧过程
Scheme 1 Decarboxylative of alkyl carboxylic acids and their derivatives

图式 7 烷基羧酸的Heck型脱羧偶联
Scheme 7 Decarboxylative Heck-type coupling of aliphatic carboxylic acids

图式 8 烷基N-羟基邻苯二甲酰亚胺酯脱羧烯基化反应
Scheme 8 Decarboxylative coupling of N-(acyloxy)phthalimide

图式 15 三苯基膦/碘化钠介导的光催化脱羧烷基化反应
Scheme 15 Photocatalytic decarboxylative alkylations mediated by triphenylphosphine and sodium iodide

图式 17 可见光与钯协同催化脱羧烯基化反应
Scheme 17 Decarboxylative allylation via photoredox and palladium catalysis

图式 19 可见光/镍协同催化脱羧偶联反应
Scheme 19 Photoredox-nickel catalyzed decarboxylative cross- coupling reaction

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:































 下载:
下载:














