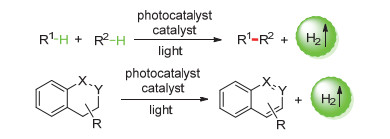
图式 1.
光催化放氢反应简介
Scheme 1.
Introduction of photocatalytic hydrogen-evolution

过去一个世纪, 光介入催化(light-mediated cataly- sis)的发展和应用使得有机化学中的各种非传统成键反应成为可能[1].最近, 光催化合成开始复兴, 通过一系列新的激活模式为导向, 引入了各种形式的新的成键方式和合成策略[2~4].光催化合成通过将光能选择性地转移到特别设计的光催化剂(photocatalyst)中, 被光激发的光催化剂能够引导底物、试剂或者催化剂参与到传统的热控制反应无法实现的独特的反应路径里[5~10].
开发绿色高效、原子经济性高的C—C、C—X(杂原子)键构筑策略是有机合成化学领域重要的研究内容之一.在光催化领域, 通过以光能为直接能量源, 用以脱氢来形成如C—C、C—X键的反应和芳构化的反应受到了人们的关注.传统的构建C—C、C—X键的反应一般需要化学计量的金属(准金属)试剂和有机卤化物作为原料, 并且伴随着化学计量的盐作为副产物[11~17].而在有机光合成领域, 近几年发展的一类基于光催化的C—H官能化和芳构化的反应受到了人们关注.以光能为直接能量源, 光催化剂/催化剂构建的双催化体系催化C—H键脱氢形成H2作为副产物, 这类反应无需外加氧化剂, 也不需要化学计量的牺牲剂, 可以实现C—C, C—X键的生成或芳构化, 具有原子经济高、高效和环境友好等优势(Schemes 1, 2).在这篇综述里, 我们将讨论这些具有广泛应用前景的光催化反应, 以显示光催化碳氢活化放氢反应的独特性和新颖性.
C—H活化因其原子经济性和步骤经济性而得到广泛关注, C—H活化官能团化反应在构建C—C键和C—X键方面是最直接、高效和绿色的途径[11~17].但由于sp3-碳的反应惰性等限制使得C(sp3)—H活化具有很大的难度和挑战性.光催化氧化偶联放氢反应是实现活化sp3-碳参与偶联反应的有效策略.通过光催化剂对底物上的杂原子(O、N原子)或是富电子基团氧化, 可以实现α-sp3-碳的活化, 从而可以与亲核试剂反应生成偶联产物, 反应中只生成H2作为副产物(Scheme 3).
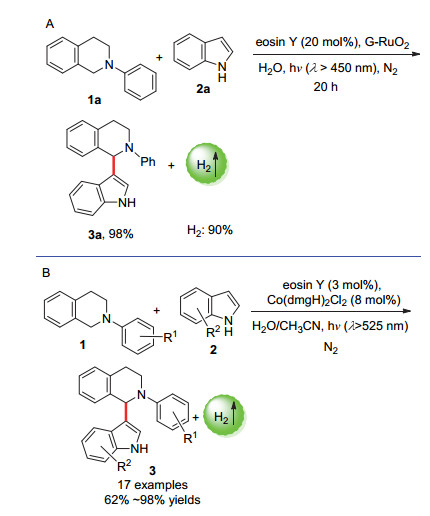
2013年, 吴骊珠课题组[21]报道了基于[曙红Y] (eosin Y)光催化剂和负载于石墨烯的RuO2纳米复合材料(G-RuO2)的双催化体系催化N-苯基-1, 2, 3, 4-四氢异喹啉(1)和吲哚(2)之间的交叉偶联放氢反应.在λ>450 nm高压汞灯下反应20 h, 可以以高达98%的收率得到偶联产物3a, H2是唯一的副产物(Scheme 4A).
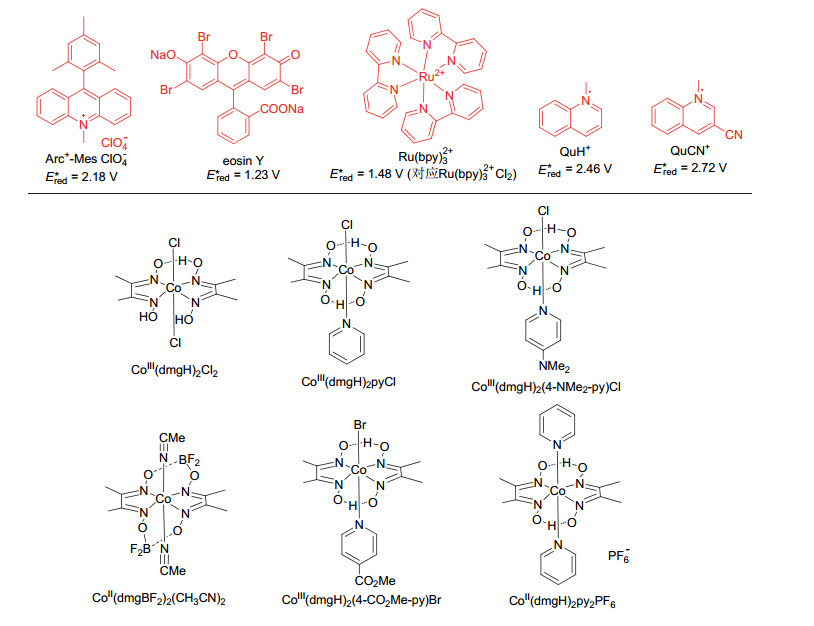
在该光催化偶联放氢反应体系中, 以RuO2纳米复合材料(G-RuO2)为非均相催化剂一定程度上限制了该合成方式的适用性.通过使用双乙二醛肟配体与氯化钻或醋酸钻反应可以合成一系列的钴肟催化剂[22]. Scheme 2列出了一部分较常见的钴肟催化剂.由于钴肟催化剂成本低廉, 稳定性好, 同时拥有理想可调节的氧化还原电位, 因而被作为质子还原催化剂广泛用于光致产氢体系中[23~25]. Artero和Peters课题组通过电化学催化产氢研究发现, 钴肟配合物在催化产氢过程中可以形成活性Co(Ⅰ)物种, 该物种可以捕获质子并通过均裂或异裂放出氢气[26~28].钴肟催化剂独特的特性使其具有用于有机合成的潜力.
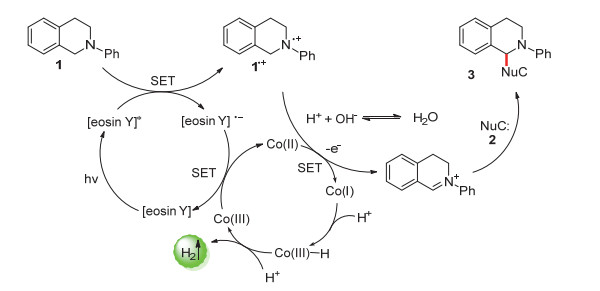
2014年, 吴骊珠课题组[29]通过使用钴肟催化剂[Co(dmgH)2Cl2]代替了贵金属催化剂G-RuO2以捕获因底物C—H键断裂产生的电子和质子(Scheme 4B), 以[eosin Y]为光敏催化剂成功实现了均相体系的N-苯基四氢异喹啉和吲哚之间的交叉偶联放氢反应.研究表明有机溶剂和水对反应的进行都很重要, 偶联产物收率最高达98%.
该反应机理如Scheme 5所示.光激发下, [eosin Y]被光激发为单重激发态后快速转变成三重激发态.叔胺底物可与三重激发态的[eosin Y]*进行单电子转移从而生成胺阳离子自由基中间体1•+, 并伴随着[eosin Y]•-的生成.同位素标定实验发现生成的H2质子主要来源于水, 证实了水也参与了反应过程. [eosin Y]•-可与Co催化剂通过电子交换再生到初始状态, 中间体1•+可以被Co(Ⅱ)进一步氧化生成亚胺鎓中间体并放出质子, 亚胺鎓中间体可以有效地与亲核试剂反应以提供偶联产物3.在反应中, Co催化剂通过将光催化剂和1•+氧化并对H质子还原实现了反应循环.
吴骊珠等[30]通过进一步研究发现, 通过向反应体系里加入质子捕获剂(硝基苯), 可以利用生成的H2以一锅法原位生成苯胺, 同时对原反应收率基本没有影响.在这里, 钴肟催化剂同时作为了氢化试剂(hydrogena- tion catalyst)还原了硝基.
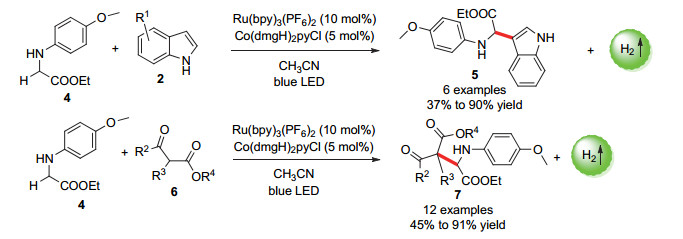
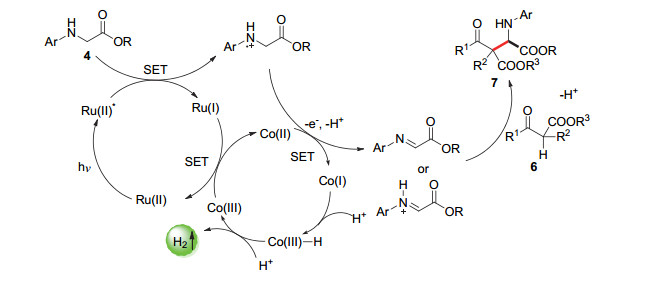
2015年, 吴骊珠课题组[31]报道了一种对α-氨基酸衍生物直接C—H活化的交叉偶联放氢反应, 但由于反应中使用的是仲胺底物而非叔胺底物, 活化需要具备更高氧化能力的光催化剂.此外, 反应体系必须无水以防止反应中的亚胺中间体的分解.最后, 作者使用了[Ru(bpy)3(PF6)2]光催化剂和[Co(dmgH)2pyCl]催化剂的双催化体系, 成功实现了含对甲氧基苯基结构的氨基酸衍生物和吲哚类底物的偶联反应, 最高偶联产物收率达90% (Scheme 6).底物扩展表明, 氨基酸底物上的对甲氧基苯基的存在对反应的进行是必须的, 此外将吲哚类底物换成β-酮酸酯底物, 也能以高收率得到偶联产物.机理研究表明, 该反应产生的H2的质子来源于参与反应的两个底物, 该反应也是通过3个单电子转移过程完成了底物的活化以及光催化剂/催化剂的再生(Scheme 7).
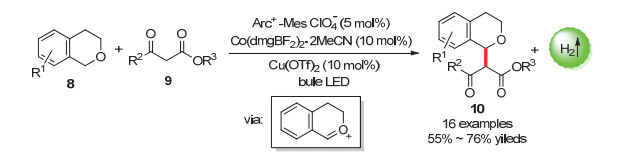
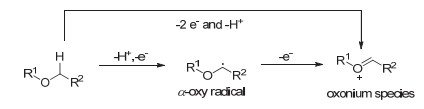
氧的α-sp3-碳的氧化电位较高, 因而相较于氮的α-碳的氧化难度更大[32~34], 为了实现这种类型的C(sp3)—H活化, 通常需要化学计量的氧化剂例如2, 3-二氯-5, 6-二氰基-1, 4-苯醌(DDQ)或加入过氧化物来除去醚α-碳的电子和氢原子, 从而形成α-氧自由基或氧鎓中间体用于随后的转化(Scheme 8)[35~37].
2015年, 吴骊珠课题组通过使用强氧化性吖啶盐光催化剂[Acr+-Mes ClO4-]与[Co(dmgBF2)2(MeCN)2]催化剂的双催化体系, 实现了含富电子芳环结构的醚的α-sp3-碳官能化, 偶联产物最高收率达到89% (Scheme 9)[38].机理研究表明, 该反应中醚底物中的芳烃被激发的光催化剂氧化后, 可以形成关键的苄基阳离子自由基.该阳离子自由基通过去质子化过程可以形成氧鎓中间体.加入的Cu盐在该体系中可以活化亲核试剂, 活化的亲核试剂可以进攻氧鎓中间体从而生成目标产物.
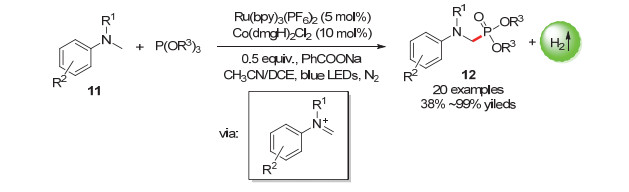
含磷化合物在医药上具有重大应用价值, 开发用于高效合成各种含磷化合物的方法十分重要[39, 40].雷爱文课题组[41]在2018年报道了使用[Ru(bpy)3(PF6)2]光催化剂和[Co(dmgH)2Cl2]催化剂对氮的相邻C(sp3)—H活化生成α-氨基膦酸盐12的反应, 由于体系不含氧化剂, 这避免了磷的氧化副反应的发生(Scheme 10).各种N-烷基苯胺衍生物, 包括N, N-二烷基苯胺、环胺和N-苯基四氢异喹啉, 可以以良好以上收率膦酰化生成一系列α-氨基膦酸盐.
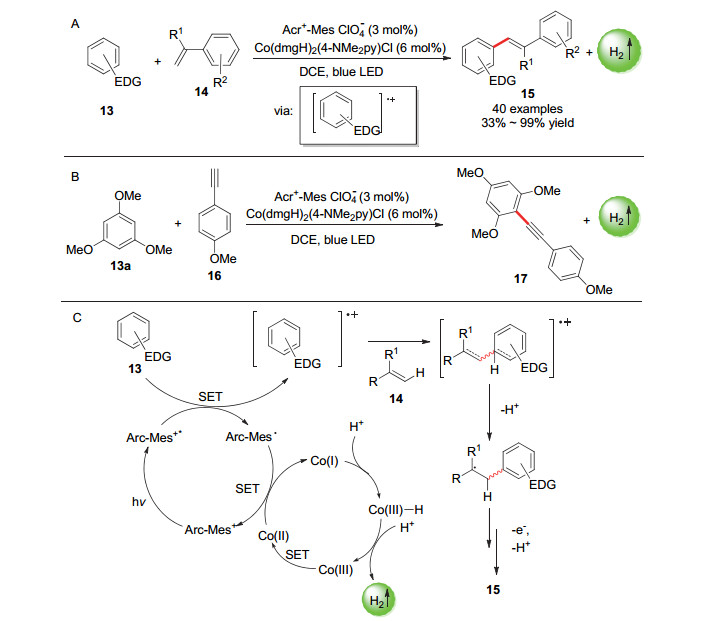
对sp2-碳进行光催化氧化活化使之参与到偶联反应也是光催化偶联放氢反应的一个很重要的应用领域. 2018年, 雷爱文课题组[42]使用[Arc+-Mes ClO4-]光催化剂和[Co(dmgH)2(4-NMe2py)Cl]催化剂实现了富电子芳烃和苯乙烯衍生物之间的交叉偶联放氢反应, 偶联产物收率最高达99% (Scheme 11A).将苯乙烯类底物改为苯丙炔类底物, 反应也能进行(Scheme 11B), 但是脂肪族烯烃类底物则无法参与反应.机理研究表明, 相较于苯乙烯底物, 富电子芳烃底物更容易失去电子, 因而富电子芳烃底物首先被激发态的光催化剂氧化生成阳离子自由基中间体(Scheme 11C).该中间体通过进攻苯乙烯的烯烃键然后脱去质子生成自由基中间体.该中间体通过脱去质子和电子形成了目标偶联产物.
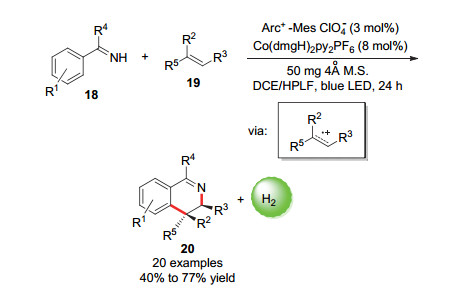
同年, 雷爱文课题组[43]报道了使用双催化体系实现了芳香族亚胺底物和苯乙烯衍生物的光催化放氢[4+2]环化生成3, 4-二氢异喹啉衍生物的反应.该方式可以以高区域选择性和非对映选择性合成一系列3, 4-二氢异喹啉(Scheme 12).研究表明反应中生成了关键的烯烃阳离子自由基中间体.通过进一步研究, 雷爱文等人发现该反应体系也可以推广到苯乙烯衍生物和富电子炔烃合成多取代的芳香族化合物的反应[44].
含氮分子广泛存在于天然产物、合成中间体和功能材料中, 超过90%的药物分子具有胺基或氮杂环结构[45].工业上制备含氮类化合物, 如苯胺, 芳基唑类化合物等会产生大量的废料, 污染环境[46].因此, 以经济可持续的方式制备这些有价值的化合物始终处于有机合成的研究领域前沿.通过一步反应直接将简单芳烃(Ar—H)与唑类、仲胺类, 甚至是NH3的交叉偶联生成C-N化合物的方法具有十分重要的经济价值.
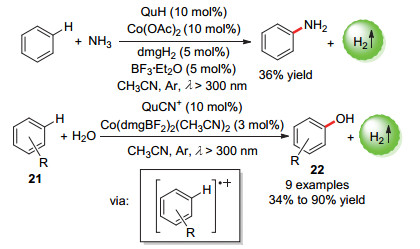
2016年, 吴骊珠课题组[47]通过有机光催化剂和钴肟催化剂的组合, 使用氨气在室温和λ>300 nm光照条件下将成功苯一步胺化为苯胺, 且不使用任何牺牲氧化剂(Scheme 13), 在最佳条件下苯胺收率达36%.研究发现在该反应中没有形成多胺基取代苯副产物, 作者猜测这是由于反应中生成的苯胺阳离子自由基会发生快速的背电子转移回到苯胺从而抑制反应的进一步进行.此外, 作者发现该反应体系也可以进一步拓展到苯与水的直接羟基化生成苯酚的反应, 收率高达90%.含吸电子取代的底物也能参与反应得到取代苯酚, 同样的, 多羟基取代的副产物也不会生成.由于无氧气和牺牲剂参与, 这种交叉偶联放氢反应对于氧化敏感的底物的特别有用.
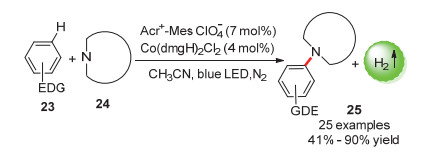
芳基唑类化合物是非常普遍的含氮化合物, 在农业化学和生物制药领域具有特殊地位, 在温和条件下直接使用简单芳烃(Ar—H)与唑类化合物偶联无疑是最经济的方法[48]. 2017年, 雷爱文课题组[49]使用吖啶盐光催化剂和金属钴肟催化剂的组合, 成功实现了富电子芳烃的无氧化剂C(sp2)—H胺化, 偶联产物最高收率达99%, 且芳烃的sp3-碳键不受影响(Scheme 14).该方法可在无氧化剂的条件下合成各种芳基唑类化合物.机理研究表明, 该反应也是通过生成芳烃阳离子自由基中间体进行的, 生成的不同阳离子自由基的比例决定了胺化产物的选择性.
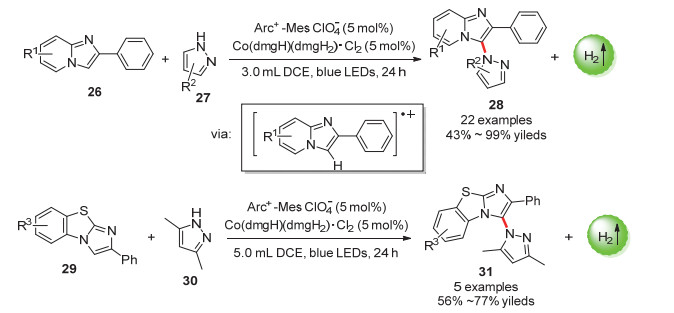
咪唑并吡啶是重要的稠合双环5~6元杂环结构, 在药物化学中被广泛应用[50, 51].雷爱文课题组[52]通过使用吖啶盐光催化剂和钴肟催化剂的双催化体系, 实现了对咪唑杂环的直接和区域选择性官能化(Scheme 15).一系列唑类衍生物和咪唑并吡啶底物都能在该反应体系中以极高收率得到产物.同时, 苯并[d]-咪唑并[2, 1-b]噻唑底物也可以以中等收率得到目标产物.该反应中, 激发的光催化剂可以通过氧化咪唑杂环生成阳离子自由基中间体,该中间体进一步参与反应.
通过使用强氧化性光催化剂氧化芳环生成阳离子自由基这种策略, 雷爱文课题组[53]还报道了富电子芳烃(甲基芳烃, 茴香醚, 多环芳烃, 杂芳族化合物, 苯胺)的C(sp2)—H键膦酰化反应.该反应不需要过量的膦酰化试剂, 进一步展现了这种催化策略的应用潜力.
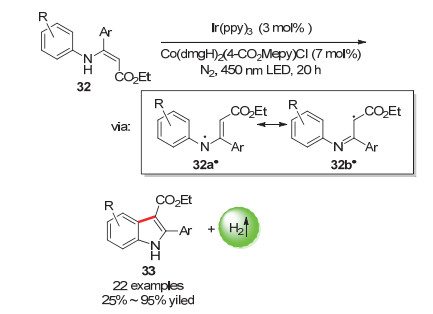
吲哚是一种重要的杂环化合物, 广泛存在于天然产物、药物、农用化学品中[54~57].吴骊珠课题组[58]在2016年报道了使用催化量的光催化剂[Ir(ppy)3]和钴肟催化剂[Co(dmgH)2(4-CO2Mepy)Cl]的双催化体系将N-芳基烯胺底物分子转化为吲哚33的反应(Scheme 16).该反应条件温和, 底物范围广, 为各种取代吲哚的合成提供了新的思路.在该反应里, 底物32上的N原子通过光催化氧化并失去质子生成中间体32a•, 该中间体具有共振体32b•, 这使得烯烃上的sp2碳活化, 发生环化反应.
通过对杂原子(O、N、S)进行光催化氧化从而生成具备亲电性的杂原子自由基, 可以实现分子内的偶联放氢反应.同样的, 这类反应不需要添加任何氧化剂.
由于金属与氧元素的HOMO和LUMO之间较高的能垒, C—H官能化形成C—O键在金属催化中受到的关注较少[59].近年来, 过渡金属催化的羧酸的氧化脱氢交叉偶联技术推进了C—O键的构建, 但必须加入氧化剂和牺牲剂限制了反应的适用性[60, 61]. 2018年, 朱成建课题组[62]使用了含[Arc+-Mes BF4-]光催化剂和[Co-(dmgH)2pyCl]催化剂的双催化体系, 成功实现了无氧化剂和牺牲剂条件下苯的C(sp2)—H官能化形成内酯的反应(Scheme 17).该合成方式具有好的底物适应性、官能团耐受性和高产率的优点, 这给无氧化剂体系构建C—O键提供了新的策略.
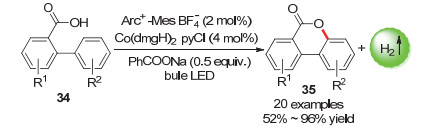
机理研究表明, 在紧凑型荧光灯(CFL Light)产生的可见光照射下, 光催化剂[Arc+-Mes BF4-]可以产生长寿命激发态[Acr-Mes]+*, 底物在存在碱(苯甲酸钠)的情况下氧化峰电位得到降低, 因而可以通过与[Acr-Mes]+*发生单电子转移(SET)过程以生成芳酰氧基自由基(Scheme 18).芳酰氧基自由基再经历: (1)分子内自由基加成, (2)与Co(Ⅲ)发生单电子转移, (3)脱去氢质子.最终生成了产物.
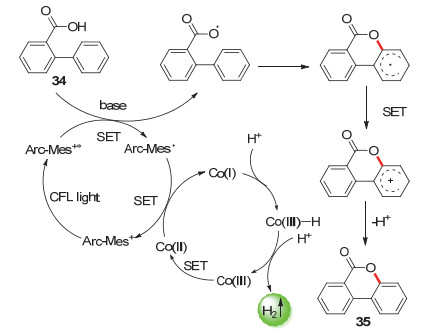
C—S键常见于重要的药物和天然生物活性化合物中[63]. 2015年, 吴骊珠等[64]报道了芳香族化合物的分子内偶联放氢生成硫醚的反应.类似的, 通过在体系内加入合适的碱可以活化底物, 谷氨酸钠对收率的提升特别有效.各种类型的硫代苯甲酰苯胺衍生物底物36可以以良好的收率生成苯并噻唑类产物, 并且仅产生H2作为副产物(Scheme 19).而通过向反应体系中加入硝基苯, 可以以一锅法利用产生的H2同时生成苯胺.这种偶联放氢反应可以应用于带有苯并噻唑结构(可用于抗肿瘤药物)的生物活性分子的合成.
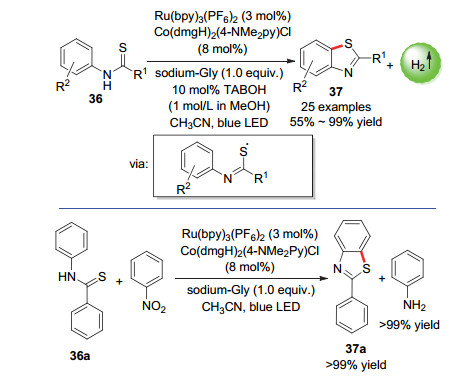
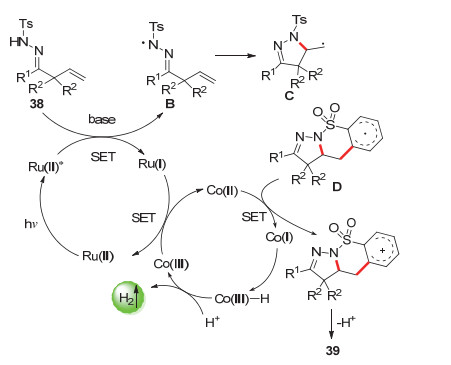
内磺酰胺结构是许多具有生物活性的天然产物和药物相关化合物中常见的杂环组成部分, 因此能以简单的方法合成新颖的含内磺酰胺结构的化合物十分有必要[65]. 2016年, 肖文精课题组[66]报道了一种在无外部氧化剂条件下的以可见光光催化β, γ-不饱和腙化合物38生成5元exo-β, γ-不饱和腙39的方法.可以合成各种生物学上有用的二氢吡唑稠合的苯并内磺酰胺类化合物.各种β, γ-不饱和腙底物都可以被很好地耐受(Scheme 20).
机理研究表明, 该反应是由可见光诱导的N自由基引发的级联C—N/C—C偶联反应(Scheme 21).反应中首先生成的N-自由基中间体B通过连续2次自由基加成反应生成了中间体D.中间体D再通过与Co(Ⅱ)的单电子转移-质子脱去过程, 生成目标产物.
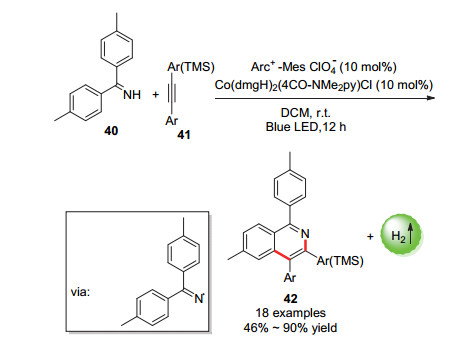
2018年, 李洋课题组[67]报道了通过可见光光催化氧化使得双(4-甲基苯基)甲亚胺的N—H键裂解形成亚胺基自由基, 用于合成异喹啉和相关的多芳烃化合物的反应(Scheme 22).机理研究表明, 该反应是由亚胺基自由基引发的级联C—N/C—C偶联反应.
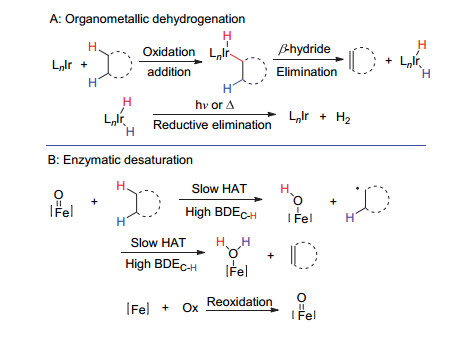
烯烃广泛存在于各类化合物中, 且可用于复杂天然产物和药物衍生合成[68, 69].饱和烷基的脱氢可以使得不饱和键的引入位置具有灵活选择性.传统的脱氢策略需要贵金属催化, 如Bergman[70]和Graham[71]等开发了使用低价铱配合物氧化加成C—H键的体系(Scheme 23A).天然去饱和酶体系也可以在酶活性温度下进行氢原子的转移, 但是这种转化需要化学计量的氧化剂, 如天然铁基去饱和酶系统引发的底物氢原子转移反应[72~74] (Scheme 23B).
2015年, Sorensen等[75]使用四丁基铵十聚钨酸盐(TBADT)和钴肟催化剂的双催化体系, 近紫外光照射下成功实现了一系列烷烃类底物和醇类底物的无受体脱氢, 这是有所报道的烷烃室温条件无受体脱氢的第一个例子.
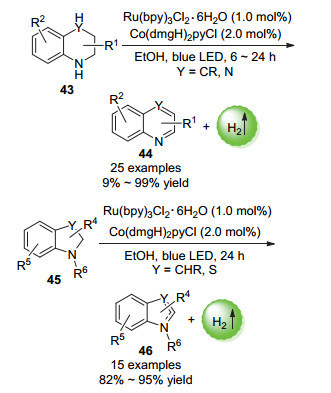
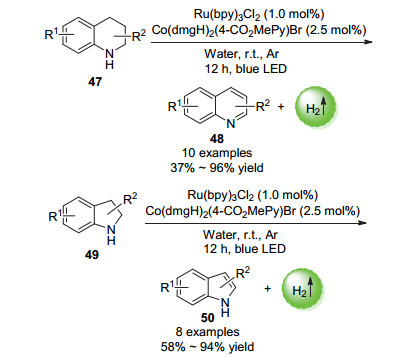
2017年, 李洋课题组[76]报道了以Ru光催化剂和钴肟催化剂的双催化体系催化的N-杂环化合物的无受体芳构化放氢反应(Scheme 24).在蓝色LED照射下, 收率最高可达99%, 该方法具有较高的通用性.
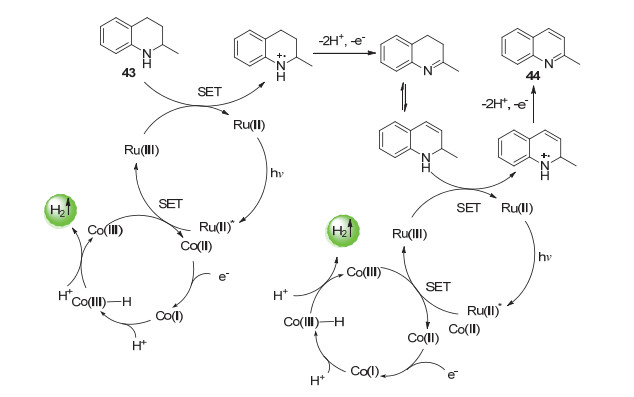
机理研究表明, 芳构化放氢的反应的机理类似于交叉偶联放氢反应, 也是通过三个单电子转移过程完成了底物的放氢和光催化剂/催化剂的再生过程.不同的是, Co(Ⅱ)淬灭激发态光催化剂的能力比N-杂环化合物更强.因此在这里, 反应主要通过Ru(bpy)32+的氧化淬灭途径进行而非偶联放氢反应中常见的还原淬灭途径. N-杂环化合物可通过与Ru(bpy)33+进行电子转移而活化(Scheme 25).
Balaraman等[77]通过研究发现, 将上述芳构化放氢的反应体系改为纯水体系后, N-杂环化合物也能以非常好的收率得到芳构化产物.这给合成各种N-杂环芳烃如喹啉、喹喔啉、吖啶和吲哚衍生物提供了更加绿色环保的策略(Scheme 26).
2017年, Kojima等[78]开发了一种可用于N-杂环化合物和四氢化萘的无受体脱氢的混合催化体系.通过使用吖啶盐光催化剂和过渡金属Pd催化剂的双催化体系, 可以使得N-杂环化合物脱氢芳构化.体系中额外加入一定量的KSbF6后, 1-苯基四氢异喹啉(51)可以在最佳反应条件下以最高96%的收率得到芳构化产物(Scheme 27A).该体系也可以应用于2, 3-二氢吲哚衍生物的芳构化反应.
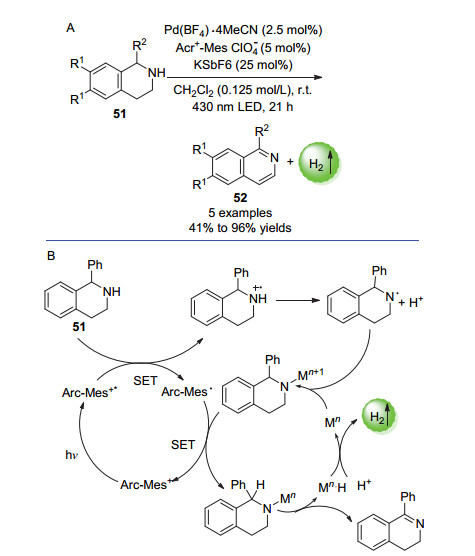
在该反应里, 被光催化剂氧化生成的自由基中间体可被金属催化剂(Mn)捕获以产生带有n+1价的金属胺中间体(Mn+1), 该中间体可与光催化剂发生电子交换, 最后经历还原消除步骤以生成单烯烃产物.该烯烃产物可重复上述步骤从而生成芳构化产物(Scheme 27B).
而通过向反应体系中加入硫代磷酰亚胺有机催化剂形成三元混合催化体系后, 该方法甚至可以实现四氢化萘的脱氢反应, 最高收率可达82% (Scheme 28).机理研究表明, 加入的硫醇55 (RSH)可以与激发的光催化剂发生单电子转移(SET)过程生成硫自由基RS•, 四氢化萘可与该自由基(RS•)反应产生苄位自由基.类似的, 苄位自由基可与Pd金属配位.该反应条件温和, 该反应对不同底物具有较高的耐受性, 具有进一步发展的潜力.
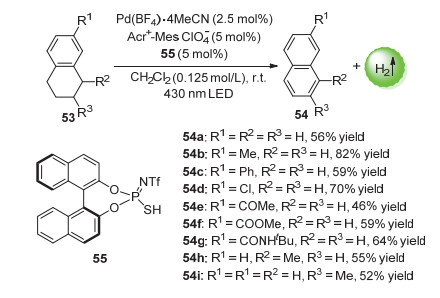
值得一提的是, Kojima等[79]通过进一步研究发现, 将反应体系改为吖啶盐光催化剂、Ni(Ⅱ)催化剂和硫代磷酸有机催化剂的三元催化体系, 也能实现四氢化萘甚至是富电子环己烷、富电子环己烯的直接芳构化反应.这是报道的于可见光温和条件下的非贵金属催化烷烃类无受体脱氢芳构化的第一个例子.
通过使用可见光介入的光催化偶联/芳构化放氢这种合成策略, 可以实现一系列不同的、热力学上有难度的官能化反应.这类反应的反应条件温和, 不需要加入化学计量的氧化剂和电子牺牲剂, 具有绿色、高效、安全等优势, 这为合成一些医药、功能材料提供了新的方法.虽然目前该合成策略仍存在一些局限性, 如难以活化惰性的碳或杂原子, 合成方式的适用性较差.同时, 对于复杂的分子以及天然产物, 该合成方法仍然处于研究初级阶段.挑战虽然存在, 但是由于该催化方式的独特性和潜力, 相信该合成策略相信能够得到进一步的优化和拓展, 能用于更多的催化反应中, 适用于更多种类的底物.
Shaw, M. H.; Twilton, J.; MacMillan, D. W. J. Org. Chem. 2016, 81, 6898. doi: 10.1021/acs.joc.6b01449
Karkas, M. D.; Porco, J. A., Jr.; Stephenson, C. R. Chem. Rev. 2016, 116, 9683. doi: 10.1021/acs.chemrev.5b00760
Shvydkiv, O.; Nolan, K.; Oelgemoller, M. Beilstein J. Org. Chem. 2011, 7, 1055. doi: 10.3762/bjoc.7.121
Xuan, J.; Xiao, W. -J. Angew. Chem. Int. Ed. 2012, 51, 6828. doi: 10.1002/anie.201200223
Luo, S. -P.; Chen, N.-Y.; Sun, Y.-Y.; Xia, L.-M.; Wu, Z.-C. Junge, H.; Beller, M.; Wu, Q.-A. Dyes Pigm. 2016, 134, 580. doi: 10.1016/j.dyepig.2016.07.040
Takeda, H.; Ishitani, O., Coord. Chem. Rev. 2010, 254, 346 doi: 10.1016/j.ccr.2009.09.030
陈嘉欣, 苗媛媛, 天然气化工(C1化学与化工), 2019, 44, 116.Chen J.-X.; Miao, Y.-Y. Nat. Gas Chem. Ind. 2019, 44, 116 (in Chinese).
Xie, J.; Jin, H.; Xu, P.; Zhu, C. Tetrahedron Lett. 2014, 55, 36. doi: 10.1016/j.tetlet.2013.10.090
Chen, J.; Cen, J.; Xu, X.; Li, X. Catal. Sci. Technol. 2016, 6, 349. doi: 10.1039/C5CY01289A
Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075. doi: 10.1021/acs.chemrev.6b00057
Heck, R. F.; Nolley, J. P. J. Org. Chem. 1972, 37, 2320. doi: 10.1021/jo00979a024
Tamao, K. K. Y.; Sumitani, K. J. Am. Chem. Soc. 1972, 26, 9268.
Hiyama, T.; Sawahata, M.; Obayashi, M. Chem. Informationsdienst 1984, 15.
张文曼, 戴建军, 许华建, 有机化学, 2015, 35, 1820. http://sioc-journal.cn/Jwk_yjhx//CN/abstract/abstract345062.shtmlZhang, W.; Dai, J.; Xu, H. Chin. J. Org. Chem. 2015, 35, 1820 (in Chinese). http://sioc-journal.cn/Jwk_yjhx//CN/abstract/abstract345062.shtml
曹莎莎, 化学通报, 2019, 82, 684.Cao, S.-S. Chem. Bull. 2019, 82, 684 (in Chinese).
袁定重, 张庆华, 廖世军, 熊文文, 元利刚, 蔡奇胜, 杨梦梅, 李雄, 蒋烨佳, 刘妍, 李萍, 徐贞帅, 孙盼盼, 耿会玲, 有机化学, 2015, 35, 961. doi: 10.6023/cjoc201407022Yuan, D.; Zhang, Q.; Liao, S.; Xiong, W.; Yuan, L.; Cai, Q.; Yang, M.; Li, X.; Jiang, Y.; Liu, Y.; Li, P.; Xu, Z.; Sun, P.; Geng, H. Chin. J. Org. Chem. 2015, 35, 961 (in Chinese). doi: 10.6023/cjoc201407022
Girard, S. A.; Knauber, T.; Li, C. J. Angew. Chem., Int. Ed. 2014, 53, 74. doi: 10.1002/anie.201304268
Tang, S.; Zeng, L.; Lei, A.-W. J. Am. Chem. Soc. 2018, 140, 13128. doi: 10.1021/jacs.8b07327
Chen, B.; Wu, L.-Z.; Tung, C.-H. Acc. Chem. Res. 2018, 51, 2512. doi: 10.1021/acs.accounts.8b00267
钟建基, 孟庆元, 陈彬, 佟振合, 吴骊珠, 化学学报, 2017, 75, 34. doi: 10.3969/j.issn.0253-2409.2017.01.006Zhong, J.-J.; Meng, Q.-Y.; Chen, B.; Tung, C.-H.; Wu, L.-Z. Acta Chim. Sinica 2017, 75, 34 (in Chinese). doi: 10.3969/j.issn.0253-2409.2017.01.006
Meng, Q.-Y.; Zhong, J.-J.; Liu, Q.; Gao, X.-W.; Zhang, H.-H.; Lei, T.; Li, Z.-J.; Feng, K.; Chen, B.; Tung, C.-H.; Wu, L.-Z. J. Am. Chem. Soc. 2013, 135, 19052.
张盼, 博士论文, 大连理工大学, 大连, 2011.Zhang, P., Ph.D. Dissertation, Dalian University of Technology, Dalian, 2011 (in Chinese).
Schrauzer, G. N. Acc. Chem. Res. 1968, 1, 97. doi: 10.1021/ar50004a001
Connolly, P.; Espenson, J. H. Inorg. Chem. 1986, 25, 2684. doi: 10.1021/ic00236a006
Pantani, O.; Naskar, S.; Guillot, R.; Millet, P.; Anxolabéhère- Mallart, E.; Aukauloo, A. Angew. Chem., Int. Ed. 2008, 47, 9948. doi: 10.1002/anie.200803643
Razavet, M.; Artero, V.; Fontecave, M. Inorg. Chem. 2005, 44, 4786. doi: 10.1021/ic050167z
Baffert, C.; Artero, V.; Fontecave, M., Inorg. Chem. 2007, 46, 1817. doi: 10.1021/ic061625m
Hu, X.; Brunschwig, B. S.; Peters, J. C. J. Am. Chem. Soc. 2007, 129, 8988. doi: 10.1021/ja067876b
Zhong, J.-J.; Meng, Q. Y.; Liu, B.; Li, X.-B.; Gao, X.-W.; Lei, T.; Wu, C.-J.; Li, Z.-J.; Tung, C.-H.; Wu, L.-Z. Org. Lett. 2014, 16, 1988. doi: 10.1021/ol500534w
Zhong, J. J.; Wu, C.-J.; Meng, Q. Y.; Gao, X.-W.; Lei, T.; Tung, C. H.; Wu, L.-Z. Adv. Synth. Catal. 2014, 356, 2846. doi: 10.1002/adsc.201400588
Gao, X. W.; Meng, Q.-Y.; Li, J.-X.; Zhong, J.-J.; Lei, T.; Li, X.-B.; Tung, C.-H.; Wu, L.-Z. ACS Catal. 2015, 5, 2391. doi: 10.1021/acscatal.5b00093
Liu, C.; Zhang, H.; Shi, W.; Lei, A.-W. Chem. Rev. 2011, 111, 1780. doi: 10.1021/cr100379j
Sun, C.-L.; Li, B.-J.; Shi, Z.-J. Chem. Rev. 2011, 111, 1293. doi: 10.1021/cr100198w
Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215. doi: 10.1021/cr100280d
Zhang, S.-Y.; Zhang, F.-M.; Tu, Y.-Q. Chem. Soc. Rev. 2011, 40, 1937. doi: 10.1039/c0cs00063a
Li, Z.; Yu, R.; Li, H. Angew. Chem., Int. Ed. 2008, 120, 7607. doi: 10.1002/ange.200802215
Liu, D.; Liu, C.; Li, H.; Lei, A.-W. Chem. Commun. 2014, 50, 3623. doi: 10.1039/c4cc00867g
Xiang, M.; Meng, Q.-Y.; Li, J.-X.; Zheng, Y.-W.; Ye, C.; Li, Z.-J.; Chen, B.; Tung, C. H.; Wu, L.-Z. Chemistry 2015, 21, 18080 doi: 10.1002/chem.201503361
Rezaei, Z.; Firouzabadi, H.; Iranpoor, N.; Ghaderi, A.; Jafari, M. R.; Jafari, A. A.; Zare, H. R. Eur. J. Med. Chem. 2009, 44, 4266. doi: 10.1016/j.ejmech.2009.07.009
Turski, L.; Schneider, H. H.; Neuhaus, R. Restor. Neurol. Neurosci. 2000, 17, 45.
Niu, L.; Wang, S.; Liu, J.; Yi, H.; Liang, X.-A.; Liu, T.; Lei, A.-W. Chem. Commun. 2018, 54, 1659. doi: 10.1039/C7CC09624K
Hu, X.; Zhang, G.-D.; Bu, F.-X.; Luo, X.; Yi, K.-B.; Zhang, H.; Lei, A.-W. Chem. Sci. 2018, 9, 1521. doi: 10.1039/C7SC04634K
Hu, X.; Zhang, G.; Bu, F.; Lei, A. Angew. Chem., Int. Ed. 2018, 57, 1286. doi: 10.1002/anie.201711359
Zhang, G.; Lin, Y.; Luo, X.; Hu, X.; Chen, C.; Lei, A.-W. Nat. Commun. 2018, 9, 1225. doi: 10.1038/s41467-018-03534-z
Schlogl, R. Angew. Chem., Int. Ed. 2003, 42, 2004. doi: 10.1002/anie.200301553
Karam, A. R.; Catarí, E. L.; López-Linares, F.; Agrifoglio, G.; Albano, C. L.; Díaz-Barrios, A.; Lehmann, T. E. Appl. Catal. 2005, 280, 165. doi: 10.1016/j.apcata.2004.10.047
Zheng, Y.-W.; Chen, B.; Ye, P.; Feng, K.; Wang, W.; Meng, Q.-Y.; Wu, L.-Z.; Tung, C.-H. J. Am. Chem. Soc. 2016, 138, 10080. doi: 10.1021/jacs.6b05498
Vicentini, C. B.; Romagnoli, C.; Andreotti, E. Agric. Food Chem. 2007, 55, 10331. doi: 10.1021/jf072077d
Niu, L.; Yi, H.; Wang, S.; Liu, T.; Liu, J.; Lei, A.-W. Nat. Commun. 2017, 8, 14226. doi: 10.1038/ncomms14226
Marson, C. M. Chem. Soc. Rev. 2011, 40, 5514. doi: 10.1039/c1cs15119c
Baviskar, A. T.; Amrutkar, S. M.; Trivedi, N.; Chaudhary, V.; Nayak, A.; Guchhait, S. K.; Banerjee, U. C.; Bharatam, P. V.; Kundu, C. N. ACS Med. Chem. Lett. 2015, 6, 481. doi: 10.1021/acsmedchemlett.5b00040
Chen, H.; Yi, H.; Tang, Z.; Bian, C.; Zhang, H.; Lei, A.-W. Adv. Synth. Catal. 2018, 360, 3220. doi: 10.1002/adsc.201800531
Niu, L.; Liu, J.; Yi, H.; Wang, S.; Liang, X.-A.; Singh, A. K.; Chiang, C.-W.; Lei, A.-W. ACS Catal. 2017, 7, 7412. doi: 10.1021/acscatal.7b02418
Janicki, S. Z.; Schuster, G. B. J. Am. Chem. Soc. 1995, 117, 8524. doi: 10.1021/ja00138a005
Somei, M.; Yamada, F. J. Nat. Prod. 2004, 21, 278. doi: 10.1039/b212257j
Somei, M.; Yamada, F. J. Nat. Prod. 2005, 22, 73. doi: 10.1039/b316241a
Kochanowska-Karamyan, A. J.; Hamann, M. T. Chem. Rev. 2010, 110, 4489. doi: 10.1021/cr900211p
Wu, C.-J.; Meng, Q.-Y.; Lei, T.; Zhong, J.-J.; Liu, W.-Q.; Zhao, L.-M.; Li, Z.-J.; Chen, B.; Tung, C.-H.; Wu, L.-Z. ACS Catal. 2016, 6, 4635. doi: 10.1021/acscatal.6b00917
Petersen, A. R.; Taylor, R. A.; Vicente-Hernandez, I.; Mallender, P. R.; Olley, H.; White, A. J.; Britovsek, G. J. J. Am. Chem. Soc. 2014, 136, 14089. doi: 10.1021/ja5055143
Zhang, C.; Tang, C.; Jiao, N. Chem. Soc. Rev. 2012, 41, 3464. doi: 10.1039/c2cs15323h
Liu, C.; Yuan, J.; Gao, M.; Tang, S.; Li, W.; Shi, R.; Lei, A.-W. Chem. Rev. 2015, 115, 12138. doi: 10.1021/cr500431s
Zhang, M.-L.; Ruzi, R.; Li, N.; Xie, J.; Zhu, C. Org. Chem. Front. 2018, 5, 749. doi: 10.1039/C7QO00795G
Sun, Q, W. R.; Cai, S. J. Med. Chem. 2011, 54, 1126. doi: 10.1021/jm100912b
Zhang, G.; Liu, C.; Yi, H.; Meng, Q.; Bian, C.; Chen, H.; Jian, J.-X.; Wu, L.-Z.; Lei, A.-W. J. Am. Chem. Soc. 2015, 137, 9273. doi: 10.1021/jacs.5b05665
Welsch, M. E.; , Snyder, S. A., Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347. doi: 10.1016/j.cbpa.2010.02.018
Zhao, Q.-Q.; Hu, X.-Q.; Yang, M.-N.; Chen, J.-R.; Xiao, W.-J. Chem. Commun. 2016, 52, 12749. doi: 10.1039/C6CC05897C
Tian, W.-F.; Wang, D.-P.; Wang, S.-F.; He, K.-H.; Cao, X.-P.; Li, Y. Org. Lett. 2018, 20, 1421. doi: 10.1021/acs.orglett.8b00193
Dobereiner, G. E.; Crabtree, R. H. Chem. Rev. 2010, 110, 681. doi: 10.1021/cr900202j
Voica, A. F.; Mendoza, A.; Gutekunst, W. R.; Fraga, J. O.; Baran, P. S. Nat. Chem. 2012, 4, 629. doi: 10.1038/nchem.1385
Janowicz, A. H.; Bergman, R. G. J. Am. Chem. Soc. 1982, 104, 352. doi: 10.1021/ja00365a091
Hoyano, J. K.; Graham, W. A. G. J. Am. Chem. Soc. 1982, 104, 3723. doi: 10.1021/ja00377a032
Buist, P. H. Nat. Prod. Rep. 2004, 21, 249. doi: 10.1039/b302094k
Breslow, R.; Baldwin, S.; Flechtner, T.; Kalicky, P.; Liu, S.; Washburn, W. J. Am. Chem. Soc. 1973, 95, 3251. doi: 10.1021/ja00791a031
Bigi, M. A.; Reed, S. A.; White, M. C. Nat. Chem. 2011, 3, 216. doi: 10.1038/nchem.967
West, J. G.; Huang, D.; Sorensen, E. J. Nat. Commun. 2015, 6, 10093. doi: 10.1038/ncomms10093
He, K.-H.; Tan, F.-F.; Zhou, C.-Z.; Zhou, G.-J.; Yang, X.-L.; Li, Y. Angew. Chem., Int. Ed. 2017, 56, 3080. doi: 10.1002/anie.201612486
Sahoo, M. K.; Balaraman, E. Green Chem. 2019, 21, 2119. doi: 10.1039/C9GC00201D
Kato, S.; Saga, Y.; Kojima, M.; Fuse, H.; Matsunaga, S.; Fukatsu, A.; Kondo, M.; Masaoka, S.; Kanai, M. J. Am. Chem. Soc. 2017, 139, 2204. doi: 10.1021/jacs.7b00253
Fuse, H.; Kojima, M.; Mitsunuma, H.; Kanai, M. Org. Lett. 2018, 20, 2042. doi: 10.1021/acs.orglett.8b00583

图式 2 常用于光催化放氢反应的光催化剂和钴肟催化剂
Scheme 2 Photocatalysts and Co catalysts commonly used in photocatalytic hydrogen-evolution coupling reaction

图式 4 (A) 以G-RuO2为催化剂的光催化偶联放氢反应和(B)以Co(dmgH)2Cl2为催化剂时的光催化偶联放氢反应
Scheme 4 (A) Photocatalytic hydrogen-evolution coupling with G-RuO2 as catalyst, and (B) photocatalytic hydrogen-evolution coupling with Co(dmgH)2Cl2 as catalyst

图式 6 α-氨基酸衍生物的光催化偶联放氢反应
Scheme 6 Photocatalytic hydrogen-evolution coupling reaction of α-amino acid derivatives

图式 9 醚底物的光催化偶联放氢反应
Scheme 9 Photocatalytic hydrogen-evolution coupling reaction of ethers

图式 10 光催化偶联放氢反应制备α-氨基膦酸盐
Scheme 10 Preparation of α-aminophosphonates by photocatalytic hydrogen-evolution coupling reaction

图式 11 富电子芳烃和苯乙烯/苯乙炔类物质的光催化偶联放氢反应以及反应机理
Scheme 11 Photocatalytic hydrogen-evolution coupling reaction of electron-rich aromatic hydrocarbons and styrenes/phenylacetylenes

图式 12 光催化偶联合成3, 4-二氢异喹啉
Scheme 12 Photocatalytic hydrogen cross-coupling synthesis of 3, 4-dihydroisoquinolines

图式 13 苯的直接光催化偶联放氢合成苯胺/苯酚
Scheme 13 Direct photocatalytic hydrogen-evolution coupling of benzenes to anilines/phenols

图式 14 光催化偶联放氢合成芳基唑类化合物
Scheme 14 Photocatalytic hydrogen-evolution coupling synthesis of aryl azoles

图式 15 光催化偶联放氢反应合成咪唑并吡啶
Scheme 15 Synthesis of imidazopyridines by photocatalytic hydrogen-evolution coupling reaction

图式 16 光催化偶联放氢反应制备吲哚
Scheme 16 Preparation of rutheniums by photocatalytic hydrogen-evolution coupling reaction

图式 17 羧酸的光催化偶联放氢反应
Scheme 17 Photocatalytic hydrogen-evolution coupling reaction of carboxylic acids

图式 18 羧酸的分子内光催化偶联放氢反应机理
Scheme 18 Mechanism of intramolecular photocatalytic hydrogen-evolution coupling reaction of carboxylic acids

图式 19 光催化偶联放氢反应合成硫醚类物质
Scheme 19 Photocatalytic hydrogen-evolution coupling synthesis of sulfides

图式 20 5元exo-β, γ-不饱和腙化合物的合成
Scheme 20 Synthesis of 5-element exo-β, γ-unsaturated rutheniums

图式 22 异喹啉以及相关的多芳烃化合物的合成
Scheme 22 Synthesis of isoquinolines and related polyaromatic compounds

图式 24 N-杂环化合物的光催化无受体芳构化放氢反应
Scheme 24 Photocatalytic non-receptor hydrogen-evolution aromatization reaction of N-heterocyclic compounds

图式 25 N-杂环化合物的光催化芳构化放氢反应机理
Scheme 25 Photocatalytic hydrogen-evolution aromatization mechanism of N-heterocyclic compounds

图式 26 纯水体系N-杂环化合物的光催化无受体芳构化放氢反应
Scheme 26 Photocatalytic non-receptor hydrogen-evolution aromatization reaction of N-heterocyclic compounds in water

图式 27 N-杂环化合物的光催化析氢芳构化反应以及反应机理
Scheme 27 Photocatalytic hydrogen-evolution aromatization reaction of N-heterocyclic compounds and reaction mechanism

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:






