

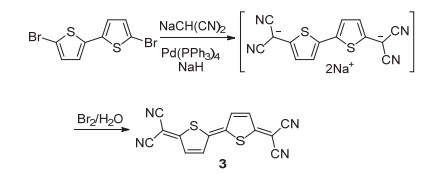
图式 1.
Takahashi反应合成二氰基亚甲基醌式噻吩
Scheme 1.
Synthesis of dicyanomethylene quinoidal thiophene by Takahashi reaction

近几十年来, 醌式杂环化合物因其具有结构平面刚性、最低未占据分子轨道(LUMO)能级低、最高占据分子轨道(HOMO)//LUMO能级带隙窄及吸收强度大等特点, 作为有机半导体材料而受到广泛关注[1~3].从结构上看, 可将醌式分子分为醌式杂环母体和端基两部分.其中, 醌式杂环母体提供醌式分子的主要性质, 如分子平面性和吸收光特性等; 端基则起到稳定醌式状态及阻止醌式结构恢复芳香结构的作用.常见的端基包括二氰基亚甲基、(烷氧基)羰基氰基亚甲基、酰基氰基亚甲基、二苯基亚甲基、亚芴基和苯醌基等.通常吸电性端基较供电性端基更有利于稳定醌式杂环结构.当端基无法稳定醌式结构时, 醌式结构往往以双自由基形式存在.醌式结构的双自由基物质同样也受到众多科学家的关注.鉴于此, 本文根据醌式杂环结构的特点, 将其分为二氰基亚甲基、(烷氧基)羰基氰基亚甲基、酰基氰基亚甲基、二苯基亚甲基、亚芴基和苯醌基等醌式杂环化合物, 对各类醌式杂环化合物的研究进展进行了综述.
二氰基亚甲基醌式杂环化合物是以二氰基亚甲基为端基的一类醌式分子, 可以看作是四氰基对二甲基苯醌(TCNQ, 1)的衍生物, 是最早出现的一类醌式杂环化合物.基于TCNQ单晶的场效应晶体管的电子迁移率可达0.5 cm2•V-1•s-1, 显示出了TCNQ作为n型半导体材料的巨大潜力[4].但TCNQ及其衍生物存在溶解性差、结构难于扩展和制膜困难等缺点, 进而催生出二氰基亚甲基醌式噻吩衍生物.
|
|
最初, 制备二氰基亚甲基醌式噻吩衍生物的方法是以二溴代噻吩为底物, 四氰基环氧乙烷为端基化试剂, 在1, 2-二溴乙烷中回流一定时间(Eq. 1)[5~8].采用该方法可以制备具有1~3个噻吩单元的简单醌式分子.不过, 该方法存在反应温度高(>130 ℃)、反应时间长、收率低(10%~40%)和底物拓展受限等缺点, 未能得到广泛应用.
|
|
(1) |
1984年, Takahashi课题组[9]报道了丙二腈负离子在钯催化剂的作用下与卤代芳香化合物作用生成二氰基甲基芳香化合物的碳-碳偶联反应, 该反应也被称之为Takahashi反应.随后, Ogura等[6]应用Takahashi反应在二溴代联噻吩上对称地接入二氰基甲基, 再在溴水的作用下经氧化反应生成二氰基亚甲基醌式联噻吩化合物3 (Scheme 1).与以四氰基环氧乙烷为端基化试剂的合成方法相比, 采用Takahashi反应的合成方法极大地提高了反应收率(>60%), 且底物拓展适用性良好.
另外, 针对二噻吩并吡咯为母体的二氰基亚甲基醌式化合物, 文献还报道了另一种方法(Scheme 2). 2008年, Rasmussen等[10]在研究三氰基乙烯基化合物时发现, 二噻吩并[3, 2-b:2', 3'-d]吡咯在与四氰基乙烯(TCNE)反应时, 除生成主要的三氰基乙烯基化合物外, 还得到了部分二氰基亚甲基醌式化合物4(收率10%). Marder等[11]在此基础上采用富电性底物以29%的收率得到醌式化合物5, 使得该法也成为一种构建二氰基亚甲基醌式结构的可选途径.
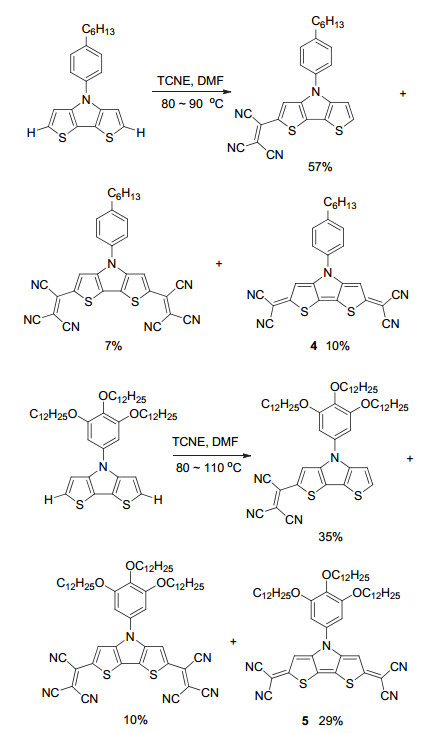
氰基由于强吸电子能力可以很好地稳定醌式结构, 二氰基亚甲基醌式杂环结构可以较容易地进行结构衍生.同时, 该类醌式分子普遍具有较低的LUMO能级(<-4.0 eV), 使其在n型有机场效应晶体管领域受到青睐.目前, Takahashi反应及氧化反应已成为构建二氰基亚甲基醌式杂环分子的主要方法, 分子拓展的方式更多地集中于设计醌式杂环母体.二氰基亚甲基醌式噻吩衍生物的母体拓展方法主要体现在以下四个方面.
增加噻吩单元的数量用以研究拓展的醌式π共轭体系与性能之间的关系[12~20]. 2003年, Frisbie课题组[12, 13]报道了以3', 4'-二丁基-2, 2':5', 2''-联三噻吩为母体的二氰基亚甲基醌式化合物6, 并首次将醌式噻吩化合物应用于有机场效应晶体管中, 取得0.02 cm2•V-1•s-1的电子迁移率. Otsubo课题组[15]以5, 5-二丁氧基-4H-环戊烷并[c]噻吩为噻吩单元, 成功地将醌式噻吩体系拓展至六个醌式噻吩单元(7a~7f).丁氧基有效克服了醌式分子随体系增大而溶解性大幅降低的缺点.随着噻吩单元的增加, 化合物7a~7f在溶液中的吸收波长由可见光区红移至近红外光区域.同时发现在醌式分子7e和7f中分别有2.8%和29%的分子以双自由基形式存在, 表明此时二氰基亚甲基已难以维持稳定的醌式结构.到目前为止, 六个醌式寡噻吩单元已是二氰基亚甲基醌式噻吩体系的极限.为研究苯环对稳定醌式基态结构的作用, 2018年, Aso课题组[20]报道了基于苯并[c]噻吩结构单元的醌式噻吩化合物8.经计算, 化合物8的单重态双自由基特征值(y)为0.56, 而不含苯环的相似物的y值为0.75.其最大吸收波长为978 nm, 摩尔消光系数达到2.6×105 L•mol-1•cm-1, 作者将该物质的强吸收特性归因于苯环对于醌式结构的稳定作用.化合物8的HOMO/LUMO能级为-4.88/-4.26 eV.基于化合物8的场效应晶体管表现出双极性电荷传输特征(μe=8.9× 10-4 cm2•V-1•s-1, μh=3.1×10-2 cm2•V-1•s-1).
|
|
将醌式噻吩化合物中的噻吩单元替换为硒吩[21, 22]、吡咯[23]、呋喃[24]等杂环结构.例如, 分子中引入硒吩通常有助于增强分子间作用力. 2009年, Takimiya课题组[21]报道了含有硒吩单元的化合物9, 其HOMO/ LUMO能级为-5.5/-4.1 eV.基于化合物9的场效应晶体管表现出双极性电荷传输特征(μe=1.6×10-2 cm2• V-1•s-1, μh=7.0×10-3 cm2•V-1•s-1).分子中采用吡咯结构则便于引入烷基链, 可以较好解决醌式杂环化合物溶解性不佳的问题. 2012年, 朱道本课题组[23]报道了含有吡咯结构的化合物10, 单晶结构分析结果表明, 该化合物分子仅含顺式结构.其LUMO能级约为-4.30~-4.39 eV, HOMO能级约为-5.48~-5.56 eV.基于化合物10a~10c的场效应晶体管的电子迁移率最高为0.014 cm2•V-1•s-1. 2016年, 李洪祥课题组[24]将醌式联三噻吩中的中间噻吩单元替换为呋喃, 设计合成了化合物11, 其HOMO/ LUMO能级为-5.72/-4.15 eV.作者发现化合物11具有很强的分子间π-π及CN…H(噻吩)相互作用.基于该化合物的有机场效应晶体管的电子迁移率最高达到7.7 cm2•V-1•s-1.
|
|
向分子中引入吡咯并[3, 4-c]吡咯-1, 4-二酮[25~30]或噻吩并[3, 4-c]吡咯-4, 6-二酮[31~34]结构, 也是设计高性能醌式杂环化合物的有效方法. 2012年, 朱道本课题组[25]报道了基于吡咯并[3, 4-c]吡咯-1, 4-二酮结构的化合物12, 其LUMO能级低至-4.51 eV. 2014年, 朱道本课题组[27]进一步将噻吩并噻吩结构引入该体系中, 合成了具有更大共轭体系的化合物13, 其LUMO能级进一步降低至-4.61 eV. 2019年, 朱晓张课题组[30]则设计合成了同时含有噻吩并[3, 4-b]噻吩和吡咯并[3, 4-c]吡咯-1, 4-二酮单元的醌式化合物14, 其LUMO能级为-4.37 eV.基于化合物12~14的有机场效应晶体管的电子迁移率分别达到0.55、0.22和0.13 cm2•V-1•s-1.在应用噻吩并吡咯二酮结构方面, 2014年, 朱晓张课题组[31]采用噻吩并[3, 4-c]吡咯-4, 6-二酮和噻吩并[3, 4-b]噻吩结构作为醌式结构单元, 设计合成了新型2D醌式化合物15和16.分子间相互作用因共轭体系的增大而增大, 而分子内氧-硫和硫-硫等弱相互作用则增加了醌式结构的稳定性.其中, 基于化合物15的有机场效应晶体管的电子迁移率达到3.0 cm2•V-1•s-1.而对化合物15的烷基链进一步修饰后(化合物17), 器件的电子迁移率提升至5.2 cm2•V-1•s-1[32].
|
|
稠环类杂环母体的应用及其体系拓展.由于稠环类杂环母体的醌式化合物不存在顺反异构问题, 因此利于对其进行结构表征、改善器件成膜效果和提高器件效率等[35~43]. 2014年, Takimiya课题组[39]以萘并[1, 2-b:5, 6-b']二噻吩为母体, 设计合成了化合物18, 其HOMO/ LUMO能级分别为-5.8/-4.6 eV.基于化合物18的有机场效应晶体管的电子迁移率为0.1 cm2•V-1•s-1.李荣金和李洪祥等[40]则以并四噻吩为母体, 设计合成了化合物19, 其LUMO能级为-4.3 eV, 基于该化合物的有机场效应晶体管的电子迁移率达到0.9 cm2•V-1•s-1.李洪祥课题组[41]进一步设计合成了以二噻吩并[2, 3-d; 2', 3'- d']苯并[1, 2-b; 4, 5-b']二噻吩为母体的并五环醌式噻吩化合物20, 并发现虽然20a、20b和20c的HOMO/LUMO能级均为-5.68/-4.39 eV, 但基于三者的有机场效应晶体管却随着烷基链侧链位置的改变而呈现从单极性电子传输向双极性电荷传输特征的转变趋势. Baumgarten课题组[42]以二噻吩并[2, 3-d; 2', 3'-d]苯并[2, 1-b; 3, 4-b']二噻吩为母体设计合成了化合物21, 并证明该化合物以稳定的双自由基形式存在.金铁男课题组[43]则采用拓展噻吩并吡咯体系的方法成功构建了具有并六环及并七环结构的醌式化合物22和23, 其中化合物23的HOMO/LUMO能级为-5.18/-4.22 eV, 能级带隙仅为0.96 eV.
|
|
在二氰基亚甲基醌式噻吩分子体系中, 随着醌式体系的增大, 其溶解性会大幅下降.为增加合成操作的便捷性, 通常需要在醌式噻吩母体上引入很长的烷基链, 但这会影响分子的固体排列状态进而降低器件效率. 2010年, Takimiya课题组[44]设计合成了一系列以(烷氧基)羰基氰基亚甲基为端基的醌式噻吩化合物24~28 (Scheme 3).以(烷氧基)羰基替代氰基的思路具有以下优点: (烷氧基)羰基仍具有一定的吸电性, 能够保证醌式分子的稳定性; 便于进行烷基链修饰, 增加醌式分子的溶解性; 烷基链沿醌式共轭长轴方向延伸, 有利于醌式分子在成膜状态的有序排列.作者采用了两种方法来进行合成:第一种方法以氯甲基联噻吩为起始原料, 分步引入氰基和(烷氧基)羰基, 再经N-氯代丁二酰亚胺(NCS)氧化得到目标产物, 但总收率低于2% (Scheme 3).第二种方法以溴代噻吩为原料, 经Takahashi反应和氧化反应直接合成目标产物.该方法不仅合成步骤大幅缩减, 总收率得到提升, 且中间体更为稳定, 易于实际操作(Scheme 3).因此, 作者进一步拓展了该体系的母体结构.聚四噻吩等寡噻吩及并三噻吩等稠环母体均适用于该体系(化合物25~28).由于(烷氧基)羰基的吸电性弱于氰基, 该系列醌式噻吩化合物的LUMO能级略高于二氰基亚甲基醌式噻吩化合物, 但其LUMO值仍略低于-4.0 eV, 符合成为在空气氛围中能够稳定存在的n型半导体材料的基本要求.基于该系列醌式化合物的场效应晶体管的电子迁移率最高为0.015 cm2•V-1• s-1.
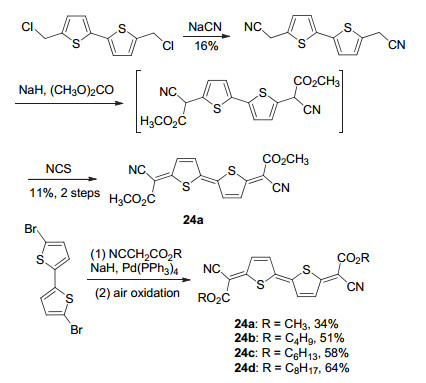
|
|
关于(烷氧基)羰基氰基亚甲基醌式噻吩衍生物的报道较少[45]. 2015年, 朱晓张课题组[46]在研究醌式噻吩化合物的荧光性质时采用了(烷氧基)羰基氰基亚甲基作为醌式化合物的端基.由于醌式分子较易产生第一单重激发态(S1态)到三重激发态(T1态)的隙间穿越, 醌式噻吩化合物的荧光发射强度普遍极其微弱[35].作者在(烷氧基)羰基氰基亚甲基醌式联噻吩的母体结构上引入并噻吩环和苯环, 合成了化合物29a~29g, 成功将醌式化合物的荧光发射能力[47, 48]加强了25~100倍.
|
|
受(烷氧基)羰基氰基亚甲基醌式噻吩衍生物的启发, Takimiya课题组[49]还设想采用酰基氰基亚甲基作为醌式杂环化合物的端基.酰基氰基碳负离子因易于异构形成烯醇式结构, 使其反应活性受到限制, 导致无法通过Takahashi反应制备关键中间体30 (Scheme 4).因此, 作者采用分步合成策略向分子中引入氰基和酰基.当先引入氰基再接入酰基时, 酰基化易于过度反应而得到二取代酰基化产物31.最后, 采用先引入酰基再接入氰基的策略, 成功得到了目标产物32.电化学结果表明, 酰基的引入同样可以有效降低醌式化合物的LUMO能级(约-4.2 eV), 使其成为有效的n型有机半导体材料.经测试, 基于该类物质的有机场效应晶体管的电子迁移率最高为0.06 cm2•V-1•s-1.由于合成路线较为复杂, 尚未见该类醌式化合物的结构拓展及应用报道.
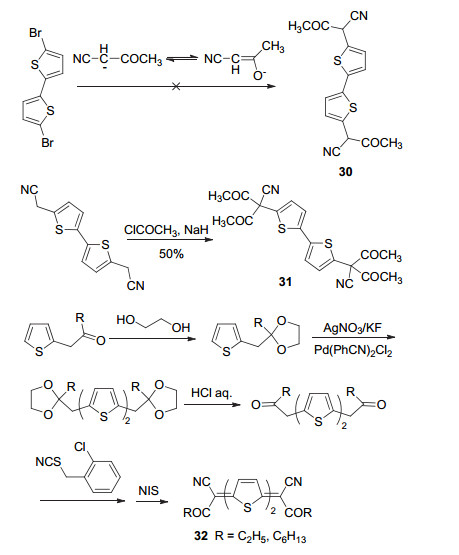
二苯基亚甲基醌式杂环化合物的出现源自于对齐齐巴宾化合物(Chichibabin hydrocarbon)的研究.齐齐巴宾存在双自由基形式与稳定醌式结构形式之间的互变特性, 活泼的双自由基形态往往导致齐齐巴宾异常不稳定.为获得稳定的齐齐巴宾衍生物, 1991年, Nakayama课题组[50]采用噻吩代替苯环设计合成了二苯基亚甲基醌式噻吩化合物33 (Scheme 5).该合成路线首先合成二苯基羟基甲基噻吩中间体, 随后在高氯酸作用下脱去两个羟基, 再在金属锌的作用下得到醌式产物.采用该方法还可以得到醌式呋喃、醌式硒吩和醌式吡咯等衍生物, 也可以使用噻吩基代替苯基得到二噻吩基亚甲基醌式噻吩化合物.
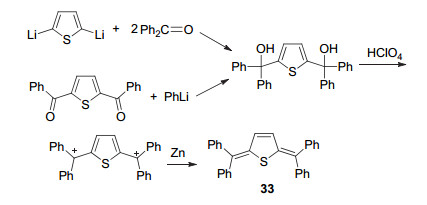
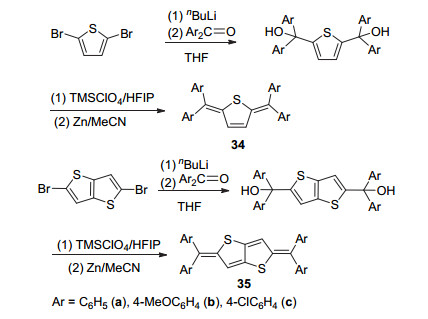
然而, 上述合成路线中的正离子中间体的稳定性较差, 以致不易分离.为克服该缺点, Takeda等[51]在2015年对该合成路线进行了改进(Scheme 6).在新路线中, 作者改用路易斯酸TMSClO4来代替高氯酸进行脱羟基反应, 原位获得正离子中间体后再将TMSClO4蒸除, 加入还原剂Zn得到产物.采用该路线可将制备化合物34和35的最后一步反应收率从6%大幅提升至99%.由于给电子效应显著, 二苯基亚甲基难以维持具有较大体系的醌式噻吩结构, 该类醌式化合物的母体仅限于噻吩、噻吩并噻吩和联噻吩[52, 53].
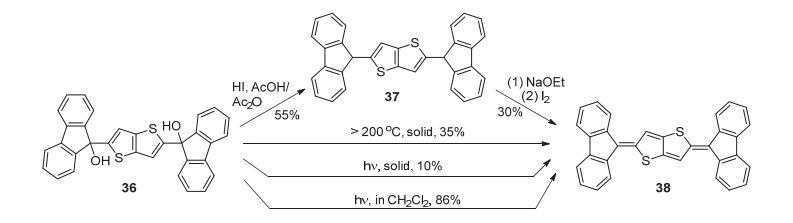
与二苯基亚甲基醌式噻吩相比, 亚芴基醌式噻吩具有更高的结构稳定性.已报道的衍生物包括亚芴基醌式噻吩、亚芴基醌式噻吩并噻吩及亚芴基醌式联噻吩等. 1991年, Kobayashi等[54]采用了多种方法来合成该类物质.以二(9-羟芴基)噻吩并噻吩(36)为原料合成目标化合物38时, 既可利用氢碘酸在醋酸-醋酸酐体系中进行回流脱除羟基得到中间体37, 再经脱氢氧化反应得到目标产物, 也可由原料直接经加热或光照得到(Scheme 7).
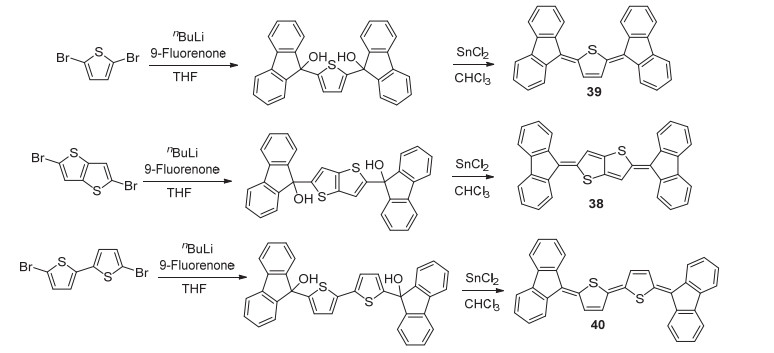
单线态裂分材料是指物质受激发至第一单重激发态(S1)后, 裂分成两个三重激发态(T1)载流子, 这样便可以在理论上将光伏器件的外量子效率提高近两倍.成为单线态裂分材料需要在能量上满足E(S1)≥2E(T1)及在空间上达到足够近的分子间距离.研究表明, 部分醌式杂环化合物能够满足成为单线态材料的要求. 2016年, Kido课题组[55]在得到2-(9-羟芴基)噻吩衍生物后采用氯仿为溶剂、二氯化锡为脱羟基试剂, 合成了亚芴基醌式噻吩衍生物38~40 (Scheme 8), 并将其作为电子给体材料用于有机太阳能电池中, 虽然电池的光电转换效率只有约1.1%, 但却验证了二亚芴基醌式噻吩化合物作为单线态材料在有机太阳能电池中应用的可行性.
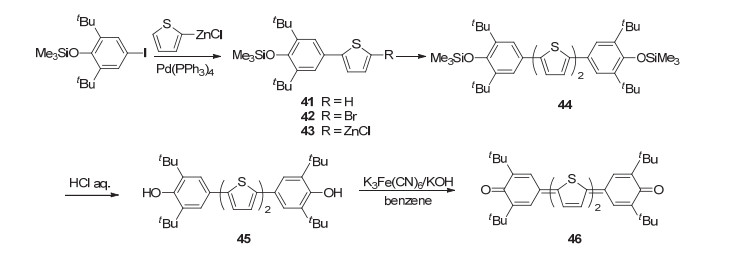
早在1989年, Takahashi课题组[56]就报道了以2, 6-二叔丁基苯醌基为端基的醌式噻吩化合物.以醌式联噻吩化合物46为例, 合成路线如Scheme 9所示.以3, 5-二叔丁基-4-三甲基硅氧基碘苯为起始原料, 采用Negishi反应和溴代反应制备中间体42, 并制备锌试剂43, 再将中间体42和43通过Negishi偶联反应合成中间体44, 随后使用盐酸脱甲硅基, 并采用铁氰化钾氧化, 即得到目标产物46.采用类似的合成路线可以合成具有1~4个噻吩单元的二苯醌基醌式杂环化合物.化合物46的最大吸收波长为680 nm, 摩尔消光系数达到2.05×105 L•mol-1•cm-1. Keivanidis等[57]利用化合物46具有强烈吸收特征将其添加进P3HT/PCBM本体异质结有机太阳能电池中, 将电池的光电转换效率提升47%.
与其它类型的醌式杂环化合物相似, 苯醌基醌式杂环化合物也存在着醌式结构形式与双自由基形式的平衡关系.双自由基形式会导致顺反异构问题加剧, 结构不稳定性增加以及器件效率降低.随着醌式噻吩单元的增加, 双自由基形式所占比例越来越大, 极大地限制了苯醌基醌式杂环化合物在器件中的应用[58, 59]. Bertarelli课题组[60~62]设想通过在固定长度苯醌基醌式噻吩结构上调节醌式母体上的取代基来调节醌式结构形式与双自由基形式的平衡.他们设计并合成了醌式化合物47~54.结果表明, 在该类醌式联噻吩结构的噻吩单元上引入供电子基团或在端基苯醌结构上引入吸电子基团, 有利于增强醌式结构的稳定性, 而在端基苯醌单元上引入供电子基团更易诱发产生双自由基.
|
|
靛吩咛是指以2-吲哚酮为端基的醌式联噻吩化合物.以靛红和噻吩为原料, 在浓硫酸作用下生成蓝色靛吩咛的反应, 被称之为“靛吩咛反应”.早期, 该反应被用于鉴别噻吩, 但人们并不清楚所得蓝色物质的精确结构.直到1993年, Cava等[63]使用N-庚烷基靛红为原料合成了易溶于有机溶剂且能分离的N, N'-二庚烷基靛吩咛衍生物55, 才确认靛吩咛为含有六个顺反异构形式的醌式联噻吩化合物(Eq. 2).
2015年, Kim课题组[64]采用靛吩咛反应制备了N, N'-二(十二烷基)靛吩咛(56)及其硒吩衍生物57, 并将其作为半导体材料应用于有机场效应晶体管, 发现该类物质具有空穴流动性和电子流动性的双极性传输特征.与二氰基亚甲基醌式化合物相比, 靛吩咛的HOMO/ LUMO能级较高.化合物56的HOMO/LUMO能级为-5.25/-3.76 eV, 化合物57的HOMO/LUMO能级为-5.33/-3.73 eV.较高的HOMO能级可能是靛吩咛能够产生空穴流动特征的原因.
|
|
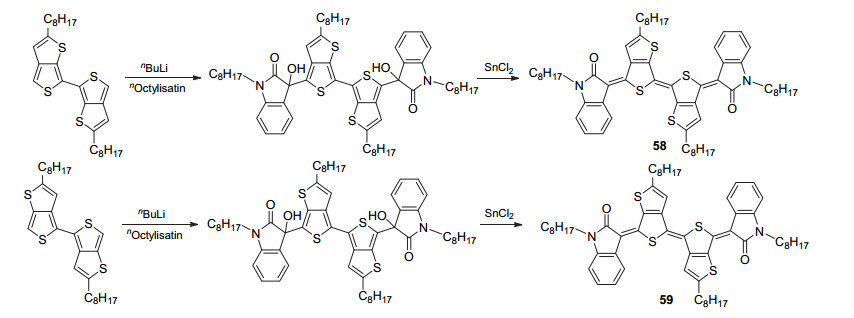
为开发基于靛吩咛结构的p型半导体材料, 朱晓张课题组[65]采用噻吩并[3, 4-b]噻吩结构替代靛吩咛中的噻吩结构, 设计了二噻吩并靛吩咛衍生物58和59.为保证分子中噻吩并噻吩结构的取向, 作者采用了如Scheme 10所示的合成路线来合成化合物58和59.核磁共振波谱数据表明该类分子结构并不存在异构现象.化合物58的HOMO/LUMO能级为-5.10/-3.58 eV, 而化合物59的HOMO/LUMO能级为-5.13/-3.49 eV, 基于化合物58和59的有机场效应晶体管的空穴迁移率分别为0.22和0.055 cm2•V-1•s-1.基于此, 作者还将化合物58和59作为电子给体材料用于有机太阳能电池中, 分别取得3.41%和4.07%的光电转换效率.
|
|
(2) |
为实现靛吩咛结构的单极性电子流动, 李玉宁课题组[66]采用间氯过氧苯甲酸将靛吩咛衍生物氧化, 得到化合物60a~60c.氧化过程使得靛吩咛的六种异构形式全部转变为反式结构, 从而方便了结构表征以及促进固体状态的分子有序排列.经过氧化的三只靛吩咛衍生物60a~60c的HOMO/LUMO能级分别下降至-5.85/-3.99、-6.33/-4.12和-6.30/-4.18 eV.最终该类物质在有机场效应晶体管中的电子迁移率最高为0.11 cm2•V-1•s-1.
|
|
李玉宁课题组[67, 68]进一步将氧化靛吩咛结构作为共聚单元制备成“给体-受体”型聚合物, 并将其作为半导体材料应用在有机场效应晶体管中.基于化合物61和62的有机场效应晶体管的电子流动率分别为0.18和0.14 cm2•V-1•s-1.
|
|
2005年, Okada课题组[69]设计合成了以1, 4-二氮杂环戊二烯(1, 4-Diazacyclopentadienylidene, DACP)为端基的醌式杂环化合物63~65 (Scheme 11).以化合物63为例, 其合成路线以2, 5-二甲酰基噻吩为原料, 在醋酸铵-醋酸介质中与偶苯酰进行缩合反应, 得到相应中间体后, 采用铁氰化钾进行氧化, 得到最终的醌式杂环化合物.化合物63~65由于醌式π共轭体系较小, 因而具有较好的结构稳定性.
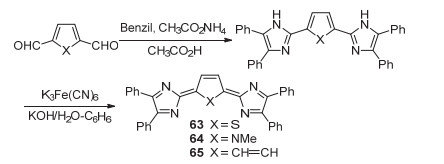
2006年, Okada课题组[70]又报道了以醌式联噻吩为母体的DACP型醌式杂环化合物66.作者采用了与化合物63相同的合成路线来制备化合物66.但与63相比, 化合物66已经表现出明显的双自由基特征.
|
|
Nakamura课题组[71]鉴于醌式杂环化合物良好的吸收特性, 尝试将以DACP为端基的醌式单噻吩衍生物用作电子给体材料, 应用于有机太阳能电池中.为提升给电子能力, 在DACP型醌式杂环分子上引入甲氧基, 设计合成了化合物67~69.虽然以该类物质制作的有机太阳能电池的光电转换效率还有待提升(仅有0.3%), 但其光电响应范围能够拓展至近红外区域(约890 nm), 为利用近红外区域的光能提供了有益的思路.
|
|
采用多环芳烃的结构设计方式也是构造醌式杂环分子的方法之一.大共轭体系的多环芳烃通常因具有较高的HOMO能级而使得结构稳定性不足.采用醌式杂环结构作为多环芳烃的内核可以有效降低HOMO能级而增加结构稳定性. 2014年, Haley等[72]设计合成了茚并醌式噻吩衍生物70~72, 其HOMO能级分别为-5.77、-5.66和-5.63 eV, LUMO能级分别为-3.84、-3.97和-4.03 eV. Chi课题组[73]研究表明, 随着醌式共轭体系的增大, 化合物73已含有一定量的双自由基形式, 使其表现出较高的活泼性.
|
|
2015年, Chi课题组[74]又报道了醌式噻吩化合物74, 在二氯甲烷中的最大吸收波长为682 nm (ε=1.07×105 L•mol-1•cm-1), HOMO/LUMO能级为-4.65/-3.46eV.基于化合物74的有机场效应晶体管的空穴迁移率为0.26 cm2•V-1•s-1.
|
|
2016年, Chi课题组[75]进一步设计合成了醌式化合物75~78, 并研究了该类物质的基态结构特征. 75~78的最大吸收波长分别为570、640、666和702 nm, HOMO能级范围为-4.39~-4.77 eV, LUMO能级范围为-3.06~-3.40 eV.随着醌式体系的增大, 基态时的双自由基特征指数则由化合物75的0增大至化合物78的38.2%.
|
|
通过构建“电子给体-电子受体”型(即“D-A”型)分子, 可以引入分子内电荷转移特征, 从而拓展分子的吸收光谱, 增强吸光能力.具有推拉电子体系的“D-A”型分子在有机太阳能电池、非线性光学材料及近红外染料领域均有广泛应用[76].在醌式结构中, 若分别采用给电子基团和吸电子基团作为端基, 即可形成典型的“D-A”分子.
1997年, Otsubo课题组[77]设计了以1, 3-二硫杂环戊烯和二氰基亚甲基为醌式杂环结构端基的“D-A”型醌式杂环化合物79~81.首先采用Takahashi反应将二氰基甲基连接到噻吩单元上, 再与2-甲硫基-1, 3-二硫杂环戊烯硫酸甲酯缩合即得到化合物79a.采用该方法还可以合成出硒吩、碲吩、呋喃衍生物以及联噻吩、噻吩并[3, 2-b]噻吩等衍生物.研究发现, 醌式单杂环衍生物79的最大吸收波长为533~561 nm, 醌式联噻吩(硒吩)衍生物80的最大吸收波长为693~739 nm, 醌式噻吩并噻吩衍生物81的最大吸收波长为622~625 nm. 2017年, Holzmüller等[78]以化合物80a为给体材料、以C60为受体材料制作了体相异质结有机太阳能电池, 取得1.9%的光电转换效率.
|
|
为增加“D-A”型醌式杂环化合物的溶解性, 2005年, Otsubo课题组[79]进一步设计合成了以5, 5-双(丁氧基甲基)-5, 6-二氢-4H-环戊烷并[c]噻吩为醌式π桥的醌式噻吩化合物82和83.与化合物79a (2.3×10-3 mol/L)和80a (<10-5 mol/L)相比, 82和83在氯仿中的溶解度(>10-2 mol/L)有了大幅提升.化合物82和83的吸收波长随着溶剂极性的增加而红移, 表明该电子推拉体系具有良好的分子内电荷转移特征.
|
|
2009年, Liu等[80]设计合成了醌式化合物84.他们采用缩合反应将二氰基亚甲基醌式联噻吩与四硫富瓦烯连接起来, 使二者成为一个完整的共轭体系.通过理论计算发现, HOMO状态的电子云主要分布于四硫富瓦烯结构上, 而LUMO状态的电子云则位于醌式噻吩母体上, 这表明化合物84也是典型的具有电子推拉效应的“D-A”型分子.
|
|
2015年, Jiang等[81]报道了以三苯胺为给体、二氰基亚甲基为受体、醌式噻吩为π桥的化合物85, 最大吸收波长为550 nm, 摩尔消光系数为5.8×104 L•mol-1• cm-1.量子化学计算结果发现, HOMO状态的电子云分布于整个分子的共轭体系上, 而LUMO状态的电子云则主要位于醌式噻吩及氰基上.以化合物85作为光敏染料制作的p型染料敏化太阳能电池的光电转换效率为0.5%, 其中, 短路电流密度达到8.2 mA•cm-2.
|
|
2019年, Gräßler等[82]进一步设计合成了分别以苯并噻唑与二氰基亚甲基为端基的醌式噻吩化合物86以及吡喃与二氰基亚甲基为端基的醌式噻吩化合物87.化合物86和87在二甲基亚砜(DMSO)中的最大吸收波长分别为544 (ε=5.1×104 L•mol-1•cm-1)和592 (ε=3.0×104 L•mol-1•cm-1) nm, HOMO/LUMO能级分别为-4.94/-3.20 eV和-5.08/-3.54 eV.
|
|
综述了各类型醌式杂环化合物的分子结构特点、设计合成方法及其应用性能.理解醌式杂环分子结构与光物理、电化学及应用性能之间的构效关系是进行醌式杂环分子设计的必要基础.从已报道的文献中可总结如下: (1)醌式分子平面刚性的结构特点有利于缩小分子间距离, 提升分子间电荷传输性能, 但却可能会大幅降低溶解性; 合理引入烷基链可以有效增加溶解性, 但却可能改变分子间排列方式. (2)醌式寡噻吩衍生物通常包含多种顺反异构体, 不利于进行结构表征及制膜应用等; 消除异构现象的手段包括引入分子内S…S或S…O等弱作用, 采用吡咯或呋喃等特殊结构单元, 以及采用稠杂环母体等. (3) HOMO/LUMO能级是醌式杂环化合物作为半导体材料的重要参数.醌式杂环分子通常具有较低的HOMO/LUMO能级, 其中, 绝大部分以氰基为端基的醌式分子的LUMO能级小于-4.0 eV, 符合成为在空气氛围中稳定的n型半导体材料的要求. (4)醌式杂环分子具有很窄的能级带隙, 且随着醌式共轭体系的增大而大幅缩小.这也是醌式杂环化合物在可见光区及近红外光区具有强吸收的重要原因, 使其具有作为光电材料或近红外材料的潜质. (5)本综述所列分子内端基相同的醌式化合物的HOMO/LUMO电子云均匀分布于整个醌式共轭体系上, 为引入分子内电荷传输特性, 设计“D-A”型醌式分子是有效方法.常见设计思路包括在醌式结构两端分别同时使用供电性端基和吸电性端基, 以及将整个醌式结构作为受体单元与富电性结构缩合. (6)醌式杂环化合物还是为数不多的可以制备稳定双自由基的物质来源, 同时作为单线态裂分材料也受到众多研究者的关注.醌式杂环化合物的发展仍然处于起步阶段.当前, 醌式杂环化合物已在有机场效应晶体管、有机太阳能电池、非线性光学材料、近红外材料和纺织染料[83, 84]等领域受到广泛关注.未来, 新结构的设计开发、性能研究及应用探索仍将是醌式杂环化合物的研究方向.
Casado, J.; Rocio, P. O.; Juan, T. N. Chem. Soc. Rev. 2012, 41, 5672. doi: 10.1039/c2cs35079c
Burrezo, P. M.; Zafra, J. L.; Navarrete, J. T. L.; Casado, J. Angew. Chem., Int. Ed. 2017, 56, 2250. doi: 10.1002/anie.201605893
王瑞豪, 乔小兰, 李洪祥, 科学通报, 2016, 61, 296.Wang, R.-H.; Qiao, X.-L.; Li, H.-X. Chin. Sci. Bull. 2016, 61, 296 (in Chinese).
Horowitz, G.; Kouki, F.; Spearman, P.; Fichou, D.; Nogues, C.; Pan, X.; Garnier, F. Adv. Mater. 1996, 8, 242. doi: 10.1002/adma.19960080312
Yui, K.; Ishida, H.; Aso, Y.; Otsubo, T.; Ogura, F. Chem. Lett. 1987, 16, 2339. doi: 10.1246/cl.1987.2339
Yui, K.; Aso Y.; Otsubo, T.; Ogura, F. Bull. Chem. Soc. Jpn. 1989, 62, 1539. doi: 10.1246/bcsj.62.1539
Yui, K.; Ishida, H.; Aso, Y.; Otsubo T.; Ogura, F.; Kawamoto, A.; Tanaka, J. Bull. Chem. Soc. Jpn. 1989, 62, 1547. doi: 10.1246/bcsj.62.1547
Higuchi, H.; Nakayama, T.; Koyama, H.; Ojima, J.; Wada, T.; Sasabe, H. Bull. Chem. Soc. Jpn. 1995, 68, 2363. doi: 10.1246/bcsj.68.2363
Uno, M.; Seto, K.; Takahashi, S. J. Chem. Soc., Chem. Commun. 1984, 932.
Pappenfus, T. M.; Hermanson, B. J.; Helland, T. J.; Lee, G. G. W.; Drew, S. M.; Mann, K. R.; McGee, K. A.; Rasmussen, S. C. Org. Lett. 2008, 10, 1553. doi: 10.1021/ol8002018
Getmanenko, Y. A.; Purcell, T. A.; Hwang, D. K.; Kippelen, B.; Marder, S. R. J. Org. Chem. 2012, 77, 10931. doi: 10.1021/jo3020006
Pappenfus, T. M.; Chesterfield, R. J.; Frisbie, C. D.; Mann, K. R.; Casado, J.; Raff, J. D.; Miller, L. L. J. Am. Chem. Soc. 2002, 124, 4184. doi: 10.1021/ja025553j
Chesterfield, R. J.; Newman, C. R.; Papperfus, T. M.; Ewbank, P. C.; Haukaas, M. H.; Mann, K. R.; Miller, L. L.; Frisbie, C. D. Adv. Mater. 2003, 15, 1278. doi: 10.1002/adma.200305200
Janzen, D. E.; Burand, M. W.; Ewbank, P. C.; Pappenfus, T. M.; Higuchi, H.; Filho, D. A. S.; Young, V. G.; Brédas, J. L.; Mann, K. R. J. Am. Chem. Soc. 2004, 126, 15295. doi: 10.1021/ja0484597
Takahashi, T.; Matsuoka, K.; Takimiya, K.; Otsubo, T.; Aso, Y. J. Am. Chem. Soc. 2005, 127, 8928. doi: 10.1021/ja051840m
Berlin, A.; Grimoldi, S.; Zotti, G.; Osuna, R. M.; Delgado, M. C. R.; Ortiz, R. P.; Casado, J.; Hernández, V.; Navarrete, J. T. L. J. Phys. Chem. B 2005, 109, 22308. doi: 10.1021/jp054204j
Handa, S.; Miyazaki, E.; Takimiya, K.; Kunugi, Y. J. Am. Chem. Soc. 2007, 129, 11684. doi: 10.1021/ja074607s
Wu, Q.-H.; Ren, S.-D.; Wang, M.; Qiao, X.-L.; Li, H.-X.; Gao, X.-K.; Yang, X.-D.; Zhu, D.-B. Adv. Funct. Mater. 2013, 23, 2277. doi: 10.1002/adfm.201202744
Nakano, M.; Osaka, I.; Takimiya, K. J. Mater. Chem. C 2015, 3, 283. doi: 10.1039/C4TC02164A
Yamamoto, K.; Ie, Y.; Nitani, M.; Tohnai, N.; Kakiuchi, F.; Zhang, K.; Pisula, W.; Asadi, K.; Blom, P. W. M.; Aso, Y. J. Mater. Chem. C 2018, 6, 7493. doi: 10.1039/C8TC01802B
Handa, S.; Miyazaki, E.; Takimiya, K. Chem. Commun. 2009, 3919.
Kunugi, Y.; Takimiya, K.; Toyoshima, Y.; Yamashita, K.; Aso, Y.; Otsubo, T. J. Mater. Chem. 2004, 14, 1367. doi: 10.1039/b401209g
Qiao, Y.-L.; Zhang, J.; Xu, W.; Zhu, D.-B. J. Mater. Chem. 2012, 22, 5706. doi: 10.1039/c2jm16700j
Xiong, Y.; Tao, J.-W.; Wang, R.-H.; Qiao, X.-L.; Yang, X.-D.; Wang, D.-L.; Wu, H.-Z.; Li, H.-X. Adv. Mater. 2016, 28, 5949. doi: 10.1002/adma.201600120
Qiao, Y.-L.; Guo, Y.-L.; Yu, C.-M.; Zhang, F.-J.; Xu, W.; Liu, Y.-Q.; Zhu, D.-B. J. Am. Chem. Soc. 2012, 134, 4084. doi: 10.1021/ja3003183
Zhong, H.-L.; Smith, J.; Rossbauer, S.; White, A. J. P. Adv. Mater. 2012, 24, 3205. doi: 10.1002/adma.201200859
Wang, C.; Zang, Y.-P.; Qin, Y.-K.; Zhang, Q., Sun, Y.-H.; Di, C.-A.; Xu, W.; Zhu, D.-B. Chem.-Eur. J. 2014, 20, 13755. doi: 10.1002/chem.201403037
Wang, C.; Qin, Y.-K.; Sun, Y.-H.; Guan, Y.-S.; Xu, W.; Zhu, D.-B. ACS Appl. Mater. Interfaces 2015, 7, 15978. doi: 10.1021/acsami.5b04082
Ray, S.; Sharma, S.; Salzner, U.; Patil, S. J. Phys. Chem. C 2017, 121, 16088. doi: 10.1021/acs.jpcc.7b04085
Ren, L.-B.; Yuan, D.-F.; Zhu, X.-Z. Chem.-Asian J. 2019, 14, 1717. doi: 10.1002/asia.201801737
Zhang, C.; Zang, Y.-P.; Gann, E.; McNeill, C. R.; Zhu, X.-Z.; Di, C.-A.; Zhu, D.-B. J. Am. Chem. Soc. 2014, 136, 16176. doi: 10.1021/ja510003y
Zhang, C.; Zang, Y.-P.; Zhang, F.-J.; Diao, Y.; McNeill, C. R.; Di, C.-A.; Zhu, X.-Z.; Zhu, D.-B. Adv. Mater. 2016, 28, 8456. doi: 10.1002/adma.201602598
Zhang, C.; Yuan, D.-F.; Wu, H.; Gann, E.; Thomsen, L.; McNeill, C.-R.; Di, C.-A.; Zhu, X.-Z.; Zhu, D.-B. J. Mater. Chem. C 2017, 5, 1935.
Zhang, C.; Rivero, S. M.; Liu, W.-Y.; Casanova, D.; Zhu, X.-Z.; Casado, J. Angew. Chem., Int. Ed. 2019, 58, 11291. doi: 10.1002/anie.201904153
Chonan, T.; Takahashi, K. Bull. Chem. Soc. Jpn. 2004, 77, 1487. doi: 10.1246/bcsj.77.1487
Kozaki, M.; Sugimura, K.; Ohnishi, H.; Okada, K. Org. Lett. 2006, 8, 5235. doi: 10.1021/ol061985d
Kashiki, T.; Miyazaki, E.; Takimiya, K. Chem. Lett. 2009, 38, 568. doi: 10.1246/cl.2009.568
Yanai, N.; Mori, T.; Shinamura, S.; Osaka, I.; Takimiya, K. Org. Lett. 2014, 16, 240. doi: 10.1021/ol403234q
Mori, T.; Yanai, N.; Osaka, I.; Takimiya, K. Org. Lett. 2014, 16, 1334. doi: 10.1021/ol5000567
Wu, Q.-H.; Li, R..; Hong, W.; Li, H.-X.; Gao, X.-K.; Zhu, D.-B. Chem. Mater. 2011, 23, 3138. doi: 10.1021/cm201326c
Li, J.; Qiao, X.-L.; Xiong, Y.; Li, H.-X.; Zhu, D.-B. Chem. Mater. 2014, 26, 5782. doi: 10.1021/cm502952u
Xia, D.-B.; Keerthi, A.; An, C.-B.; Baumgarten, M. Org. Chem. Front. 2017, 4, 18. doi: 10.1039/C6QO00543H
Jiang, H.; Oniwa, K.; Xu, Z. Q.; Bao, M.; Yamamoto, Y.; Jin, T.-N. Bull. Chem. Soc. Jpn. 2017, 90, 789. doi: 10.1246/bcsj.20170083
Suzuki, Y.; Miyazaki, E.; Takimiya, K. J. Am. Chem. Soc. 2010, 132, 10453. doi: 10.1021/ja103171y
Jiang, H.; Zhang, L.; Cai, J.-F.; Ren, J.-H.; Cui, Z.-H.; Chen, W.-G. Dyes Pigm. 2018, 151, 363. doi: 10.1016/j.dyepig.2018.01.017
Ren, L.-B.; Liu, F.; Shen, X.-X.; Zhang, C.; Yi, Y.-P.; Zhu, X.-Z. J. Am. Chem. Soc. 2015, 137, 11294. doi: 10.1021/jacs.5b03899
黄池宝, 陈会, 李福琴, 安思雅, 有机化学, 2019, 39, 2467. http://www.ccspublishing.org.cn/advancesearchHuang, C.-B.; Chen, H.; Li, F.-Q; An, S.-Y. Chin. J. Org. Chem. 2019, 39, 2467 (in Chinese). http://www.ccspublishing.org.cn/advancesearch
王金金, 戚少龙, 杜建时, 杨清彪, 宋岩, 李耀先, 高等学校化学学报, 2019, 40, 1397. doi: 10.7503/cjcu20190159Wang, J.-J.; Qi, S.-L.; Du, J.-S.; Yang, Q.-B.; Song, Y.; Li, Y.-X. Chem. J. Chin. Univ. 2019, 40, 1397 (in Chinese). doi: 10.7503/cjcu20190159
Suzuki, Y.; Shimawaki, M.; Miyazaki, E.; Osaka, I.; Takimiya, K. Chem. Mater. 2011, 23, 795. doi: 10.1021/cm102109p
Ishii, A.; Horikawa, Y.; Takaki, I.; Shibata, J.; Nakayama, J.; Hoshino, M. Tetrahedron Lett. 1991, 32, 4313. doi: 10.1016/S0040-4039(00)92158-0
Takeda, T.; Akutagawa, T. J. Org. Chem. 2015, 80, 2455. doi: 10.1021/acs.joc.5b00021
Kawase, T.; Ueno, N.; Oda, M. Tetrahedron Lett. 1992, 33, 5405. doi: 10.1016/S0040-4039(00)79106-4
Bhattacharyya, K.; Dey, D.; Datta, A. J. Phys. Chem. C 2019, 123, 4749. doi: 10.1021/acs.jpcc.9b00230
Mazaki, Y.; Murata, S.; Kobayashi, K. Tetrahedron Lett. 1991, 32, 4367. doi: 10.1016/S0040-4039(00)92172-5
Kawata, S.; Pu, Y. J.; Saito, A.; Kurashige, Y.; Beppu, T.; Katagiri, H.; Hada, M.; Kido, J. Adv. Mater. 2016, 28, 1585. doi: 10.1002/adma.201504281
Takahashi, K.; Suzuki, T. J. Am. Chem. Soc. 1989, 111, 5483. doi: 10.1021/ja00196a075
Kan, Z.-P.; Colella, L.; Canesi, E. V.; Lerario, G.; Kumar, R. S. S.; Bonometti, V.; Mussini, P. R.; Cavallo, G.; Terraneo, G.; Pattanasattayavong, P.; Anthopoulos, T. D.; Bertarelli, C.; Keivanidis, P. E. Sol. Energy Mater. Sol. Cells 2014, 120, 37. doi: 10.1016/j.solmat.2013.08.007
Agostinelli, T.; Caironi, M.; Natali, D.; Sampietro, M.; Dassa, G.; Canesi, E. V.; Bertarelli, C.; Zerbi, G.; Cabanillas-Gonzalez, J.; Silvestri, S. D.; Lanzani, G. J. Appl. Phys. 2008, 104, 114508. doi: 10.1063/1.3033376
Fazzi, D.; Canesi, E. V.; Negri, F.; Bertarelli, C.; Castiglioni, C. ChemPhysChem 2010, 11, 3685. doi: 10.1002/cphc.201000675
Canesi, E. V.; Fazzi, D.; Colella, L.; Bertarelli, C.; Castiglioni, C. J. Am. Chem. Soc. 2012, 134, 19070. doi: 10.1021/ja3072385
Colella, L.; Brambilla, L.; Nardone, V.; Parisini, E.; Castiglioni, C.; Bertarelli, C. Phys. Chem. Chem. Phys. 2015, 17, 10426. doi: 10.1039/C4CP05748A
Francesco, T.; Colella, L.; Maghsoumi, A.; Martí-Rujas, J.; Parisini, E.; Tommasini, M.; Bertarelli, C.; Barbon, A. J. Phys. Chem. C 2016, 120, 5732.
Tormos, G. V.; Belmore, K. A.; Cava, M. P. J. Am. Chem. Soc. 1993, 115, 11512. doi: 10.1021/ja00077a057
Hwang, H.; Khim, D.; Yun, J. M.; Jung, E.; Jang, S. Y.; Jang, Y. H.; Noh, Y. Y.; Kim, D. Y. Adv. Funct. Mater. 2015, 25, 1146. doi: 10.1002/adfm.201402758
Ren, L.-B.; Fan, H.-J.; Huang, D.-Z.; Yuan, D.-F.; Di, C.-A.; Zhu, X.-Z. Chem.-Eur. J. 2016, 22, 17136. doi: 10.1002/chem.201603112
Deng, Y.-F.; Sun, B.; Quinn, J.; He, Y.-H.; Ellard, J.; Guo, C.; Li, Y.-N. RSC Adv. 2016, 6, 45410. doi: 10.1039/C6RA06316K
Deng, Y.-F.; Quinn, J.; Sun, B.; He Y.-H.; Ellard, J.; Li, Y.-N. RSC Adv. 2016, 6, 34849. doi: 10.1039/C6RA03221D
Deng, Y.-F.; Sun, B.; He, Y.-H.; Quinn, J.; Guo, C.; Li, Y.-N. Angew. Chem., Int. Ed. 2016, 55, 3459. doi: 10.1002/anie.201508781
Kozaki, M.; Isoyama, A.; Akita, K.; Okada, K. Org. Lett. 2005, 7, 115. doi: 10.1021/ol0476927
Kozaki, M.; Isoyama, A.; Okada, K. Tetrahedron Lett. 2006, 47, 5375. doi: 10.1016/j.tetlet.2006.05.084
Ay, E.; Furukawa, S.; Nakamura, E. Org. Chem. Front. 2014, 1, 988. doi: 10.1039/C4QO00182F
Rudebusch, G. E.; Fix, A. G.; Henthorn, H. A.; Vonnegut, C. L.; Zakharov, L. N.; Haley, M. M. Chem. Sci. 2014, 5, 3627. doi: 10.1039/C4SC01432D
Shi, X.-L.; Burrezo, P. M.; Lee, S.-S.; Zhang, W.-H.; Zheng, B.; Dai, G.-L.; Chang, J.-J.; Navarrete, J. T. L.; Huang, K. W.; Kim, D. H.; Casado, J.; Chi, C. Y. Chem. Sci. 2014, 5, 4490. doi: 10.1039/C4SC01769B
Shi, X.-L.; Lee, S.-S.; Son, M.-J.; Zheng, B.; Chang, J.-J.; Jing, L.-Z.; Huang, K.-W.; Kim, D.-H.; Chi, C.-Y. Chem. Commun. 2015, 51, 13178. doi: 10.1039/C5CC04243G
Shi, X.-L.; Quintero, E.; Lee, S.-S.; Jing, L.-Z.; Herng, T. S.; Zheng, B.; Huang, K. W.; Navarrete, J. T. L.; Ding, J.; Kim, D. H.; Casado, J.; Chi, C.-Y. Chem. Sci. 2016, 7, 3036. doi: 10.1039/C5SC04706D
谭继华, 霍延平, 蔡宁, 籍少敏, 李宗植, 张力, 有机化学, 2017, 37, 2457. http://www.ccspublishing.org.cn/advancesearchTan, J.-H.; Huo, Y.-P.; Cai, N.; Ji, S.-M.; Li, Z.-Z.; Zhang, L. Chin. J. Org. Chem. 2017, 37, 2457 (in Chinese). http://www.ccspublishing.org.cn/advancesearch
Inoue, S.; Mikami, S.; Aso, Y.; Otsubo, T.; Wada, T.; Sasabe, H. Synth. Met. 1997, 84, 395. doi: 10.1016/S0379-6779(97)80799-0
Holzmüller, F.; Gräßler, N.; Sedighi, M.; Müller, E.; Knupfer, M.; Zeika, O.; Vandewal, K.; Koerner, C.; Leo, K. Org. Electron. 2017, 45, 198. doi: 10.1016/j.orgel.2017.03.009
Takahashi, T.; Takimiya, K.; Otsubo, T.; Aso, Y. Org. Lett. 2005, 7, 4313. doi: 10.1021/ol0513037
Guegano, X.; Kanibolotsky, A. L.; Blum, C.; Mertens, S. F. L.; Liu, S. X.; Neels, A.; Hagemann, H.; Skabara, P. J.; Leutwyler, S.; Wandlowski, T.; Hauser, A.; Decurtins, S. Chem.-Eur. J. 2009, 15, 63. doi: 10.1002/chem.200802011
Zhang, Q.-Q.; Jiang, K.-J.; Huang, J.-H.; Zhao, C.-W.; Zhang, L.-P.; Cui, X.-P.; Su, M.-J.; Yang, L.-M.; Song, Y.-L.; Zhou, X.-Q. J. Mater. Chem. A 2015, 3, 7695. doi: 10.1039/C5TA01348H
Gräßler, N.; Wolf, S.; Holzmüller, F.; Zeika, O.; Vandewal, K.; Leo, K. Eur. J. Org. Chem. 2019, 845.
蔡金芳, 陈维国, 崔志华, 江华, 纺织学报, 2018, 39, 81.Cai, J.-F.; Chen, W.-G.; Cui, Z.-H.; Jiang, H. J. Text. Res. 2018, 39, 81 (in Chinese).
Jiang, H.; Hu, Q.; Cai, J.-F.; Cui, Z.-H.; Zheng, J.-H.; Chen, W.-G. Dyes Pigm. 2019, 166, 130. doi: 10.1016/j.dyepig.2019.03.025

图式 1 Takahashi反应合成二氰基亚甲基醌式噻吩
Scheme 1 Synthesis of dicyanomethylene quinoidal thiophene by Takahashi reaction

图式 2 以四氰基乙烯为端基化试剂合成醌式噻吩衍生物
Scheme 2 Synthesis of dicyanomethylene quinoidal thiophene by using tetracyanoethylene as terminal reagent

图式 3 (烷氧基)羰基氰基亚甲基醌式联噻吩的合成路线
Scheme 3 Synthetic routes for (alkoxy)carbonylcyanomethyl- ene quinoidal bithiophene

图式 4 酰基氰基亚甲基醌式噻吩衍生物的分子结构及其合成路线
Scheme 4 Molecular structures and synthetic routes for acylcyanomethylene quinoidal thiophenes

图式 5 二苯基亚甲基醌式噻吩的合成路线
Scheme 5 Synthetic route for diphenylmethylene quinoidal thiophene

图式 6 改进后的二苯基亚甲基醌式噻吩的合成路线
Scheme 6 Modified synthetic route for diphenylmethylene quinoidal thiophenes

图式 7 2, 5-二(9-亚芴基)-2, 5-醌式噻吩并[3, 2-b]噻吩的合成路线
Scheme 7 Synthetic routes for 2, 5-bis(fluorene-9-ylidene)-2, 5-dihydrothieno[3, 2-b]thiophene

图式 8 醌式化合物38~40的分子结构及其合成路线
Scheme 8 Molecular structures and synthetic routes for quinoidal compounds 38~40

图式 9 二苯醌基醌式噻吩化合物46的分子结构及其合成路线
Scheme 9 Molecular structure and synthetic route for p-diphenoquinone 46

图式 10 噻吩并噻吩型靛吩咛衍生物的分子结构及合成路线
Scheme 10 Molecular structures and synthetic route of indophenine derivatives based on thienothiophene moiety

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:





















 下载:
下载:
