Figure 1.
Selected bioactive indazole compounds
Synthesis of 3-Carboxylate Indazoles via Ru(Ⅱ)-Catalyzed Annulation of Azobenzenes with Ethyl Glyoxalate
Xun Chen , Ying Wang , Shuojin Wang , Dulin Kong , Lijun Wen , Ruirui Zhai , Ke Zhao , Lili Bai , Youbin Li

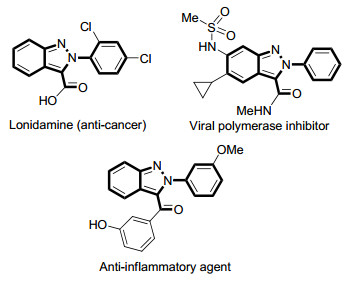
Indazole heterocycles have attracted significant interest among the medicinal chemistry community because of their promising pharmaceutical applications and biological activities.[1] In particular, the 3-carboxylate indazole scaffolds are constituents of key privileged molecules, acting as anti-cancer drug, viral polymerase inhibition and anti-inflammatory agent, etc. (Figure 1).[2] Thus, the development of robust synthetic approaches for affording substituted 3-carboxylate indazoles has been the focus of intensive research. The classical routes to 3-carboxylate indazoles involve direct lithiation at the C3-position of indazoles followed by the addition of electrophiles, but these strategies are limited by their low yields and harsh reaction conditions.[3] Therefore, there is a continued strong demand for constructing 3-carboxylate indazoles in simple operation from cheap starting materials.
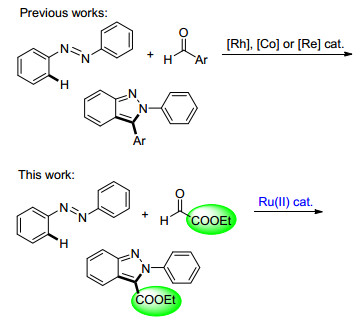
Transition-metal-catalyzed C—H bond functionaliza- tion has emerged a powerful strategy to the rapid assembly of various heterocycles.[4] In this regard, the recent progress has been focused on the synthesis of indazoles via C—H bond activation process under Rh(Ⅲ), Co(Ⅲ) and Re(Ⅰ) catalysts. For example, the pioneering work by Ellman took advantage of the directing character of azo group to enable a Rh(Ⅲ)-catalyzed C—H activation/cyc- lization of azobenzenes with aryl aldehydes for the synthesis of 2, 3-diaryl-2H-indazoles (Scheme 1).[5] Moreover, Ellman and Wang[6] also reported representative works on the synthesis of 2, 3-diaryl-2H-indazoles via the Co(Ⅲ)- or Re(Ⅰ)-catalyzed redox-neutral coupling of azobenzenes with aryl aldehydes, respectively. However, these methods are only limited to furnishing 3-aryl indazole under expensive catalysts. On the other hand, Ru(Ⅱ) complexes have been shown to efficiently catalyze arene C—H functionalization with outstanding catalytic efficiency.[7] In continuation of our effort in developing efficient Ru(Ⅱ)-catalyzed C—H functionlization for the assembly of various heterocycles, [8] we herein report an efficient Ru(Ⅱ)-catalyzed C—H bond [4+1] cycloaddition of azobenzenes with ethyl glyoxalate for the production of the desired 3-car- boxylate indazoles (Scheme 1).
Initially, we commenced our studies by screening reaction conditions for the envisioned C—H bond [4+1] cycloaddition of azobenzenes 1a with ethyl glyoxalate 2a (Table 1). To our delight, the desired 3-carboxylate indazole 3a was obtained in 24% yield by using [Ru(p-cy-mene)Cl2]2/AgSbF6 as the co-catalyst (Entry 1). It is well- known that the OAc source is important for the current C—H functionalization reactions. We next investigated the effects of additives for this reaction optimization. Further investigation revealed that additives such as NaOAc, KOAc, LiOAc, CsOAc and AgOAc led to a significantly increase in the product yield (38%~51%, Entries 2~6), whereas the reaction of 1a with 2a in the presence of acid additives such as AcOH and PivOH furnished the product 3a in slightly decreased yields (41% and 26%, Entries 6, 7). Subsequently, a variety of solvents were screened, and these results showed that 1, 2-dichloroethane (DCE) was the optimal solvent (Entries 8~12 vs 13). In addition, 3a was formed in 76% yield when the reaction temperature increased to 100 ℃ (Table 1, Entry 14). To our delight, we soon found that extending the reaction time could afford the improved reaction yield of 82% (Entries 15). Finally, various Ru complexes were tested, but only [Ru(p-cyme-ne)Cl2]2 was active. The other complexes, including Ru- Cl2(PPh3)4, Ru3(CO)12 and RuHCl(CO)(PPh3)3 were totally ineffective (Entries 16~18).
 下载:
导出CSV
下载:
导出CSV
 |
||||
| Entry | Catalyst | Additive | Solvent | Yieldb/% |
| 1 | [Ru(p-cymene)Cl2] | None | Dioxane | 24 |
| 2 | [Ru(p-cymene)Cl2] | NaOAc | Dioxane | 51 |
| 3 | [Ru(p-cymene)Cl2] | KOAc | Dioxane | 46 |
| 4 | [Ru(p-cymene)Cl2] | LiOAc | Dioxane | 47 |
| 5 | [Ru(p-cymene)Cl2] | CsOAc | Dioxane | 40 |
| 6 | [Ru(p-cymene)Cl2] | AgOAc | Dioxane | 38 |
| 7 | [Ru(p-cymene)Cl2] | PivOH | Dioxane | 41 |
| 8 | [Ru(p-cymene)Cl2] | AcOH | CH3CN | 26 |
| 9 | [Ru(p-cymene)Cl2] | NaOAc | Toluene | Trace |
| 10 | [Ru(p-cymene)Cl2] | NaOAc | THF | 48 |
| 11 | [Ru(p-cymene)Cl2] | NaOAc | TFE | 21 |
| 12 | [Ru(p-cymene)Cl2] | NaOAc | MeOH | Trace |
| 13 | [Ru(p-cymene)Cl2] | NaOAc | DCE | 64 |
| 14 | [Ru(p-cymene)Cl2] | NaOAc | DCE | 76c |
| 15 | [Ru(p-cymene)Cl2] | NaOAc | DCE | 82d |
| 16 | RuCl2(PPh3)4 | NaOAc | DCE | 0 |
| 17 | Ru3(CO)12 | NaOAc | DCE | 0 |
| 18 | RuHCl(CO)(PPh3)3 | NaOAc | DCE | 0 |
| a Unless otherwise noted, all the reactions were carried out using azobenzenes (1a, 0.20 mmol) and ethyl glyoxalate (2a, 0.40 mmol) with catalysts (5 mol%) in the presence of AgSbF6 (20 mol%) and additive (50 mol%) in solvent (2.0 mL) at 80 ℃ for 12 h under Ar in a sealed reaction tube, followed by flash chromatography on SiO2. b Isolated yield. c The reaction temperature is 100 ℃. d The reaction time is 24 h. TFE=2, 2, 2-trifluoroethanol. | ||||
With an optimized catalytic system in hand, we then turned our attention toward exploring scope and limitation of Ru(Ⅱ)-catalyzed C—H bond [4+1] cycloaddition of azobenzenes (1) with ethyl glyoxalate (2a). As shown in Table 2, the reactivity of substituted azobenzenes 1 was obviously dependent on the electronic properties of substitutents. Azobenzenes bearing electron-donating groups (Me, MeO) on the para-position afforded excellent yields of 3b (84% yield) and 3c (88% yield), respectively. In contrast, electron-withdrawing halogen group (Cl), ester group (COOEt) and cyano group (CN) slightly decreased the reaction conversion and produced 3d~3f in moderate to good yields (59%~77% yields), which indicated that C—H activation was involved in an electrophilic aromatic substitution process. Notably, this reaction protocol could also smoothly convert ortho- or meta-substituted azobenzenes to the desired 3g or 3h with excellent yields (80% and 83%) in spite of steric congestion. Moreover, 3, 5-demethyl-substituted azobenzene 1i could afford the desired product 3i with 78% yield, in which no significant effect of steric hindrance from methyl moiety was observed in this transformation. In addition, the unsymmetrical azobenzene 1j having both methy group and methoxy group on one aryl ring is also allowed for this transformation to assemble the corresponding indazole 3j in 76% yield. Beside, methyl 2-oxoacetate and 2-oxo-2-phenylace- taldehyde could also react with 1a to afford the corresponding products 3k and 3l, respectively. Unfortunately, other aldehydes such as acetaldehyde were not allowed for this transformation, and the starting materials were recovered completely. When we moved to employ 2-arylpyri- dine as an arene substrate (Eq. 1), indolizine products 5a~5c were also isolated in accepted yield under slightly modified reaction condition. Finally, we converted 3k to the corresponding product 6a in 78% yield via a hydrolysis and subsequent decarboxylation process (Eq. 2).
|
|
(1) |
|
|
(2) |
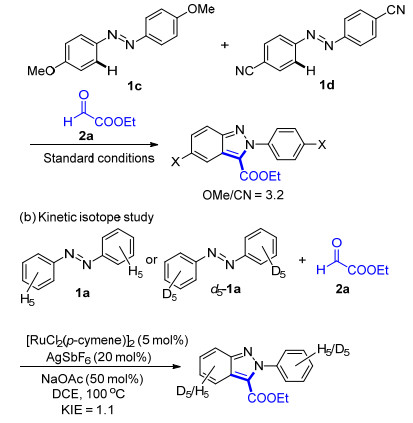
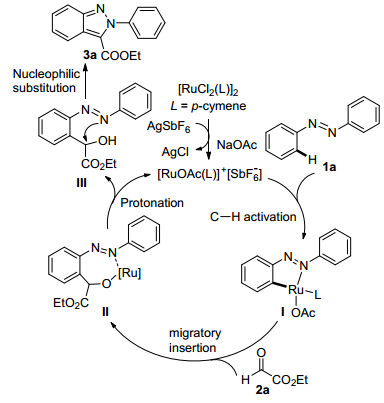
To gain more insights into the mechanism, some experiments were conducted (Scheme 2). A competition reaction was performed to determine the electronic prefe- rence of this transformation. When 1c and 1f were treated at the same time, the reaction favored the electron-rich azo-benzenes in 3.2:1 ratio (Scheme 2a), thus implying that the C—H activation step might be an electrophilic aromatic substitution process.[9] Subsequently, the intermolecular isotope effect (KH/KD=1.1) further indicated that C—H bond-breaking was not involved in the rate- limitting step of this transformation (Scheme 2b).[10] On the basis of the above-mentioned results, a plausible mechanism is proposed in Scheme 3. First, coordination of an azo group in azobenzene 1a to a cationic Ru species to form a five-membered cobaltacycle intermediate I. Subse- quently, an insertion of C=O to Ru—C bond of interme-diate A forms the seven-membered intermedaite II, which undergoes protonation to give the alcohol III with release of Ru catalyst. Finally, indazole 3a is obtained through an intramolecular nucleophilic substitution/cyclization process.
In conclusion, we have developed a novel Ru(Ⅱ)- catalyzed regioselective C—H bond cycloaddition of azobenzenes with ethyl glyoxalate for the rapid assembly of 3-carboxylate indazole. This catalytic system tolerates a wide range of azobenzene substrates with high reaction efficiency. Further synthetic applications to bioactive molecules are underway in sour laboratory.
The azobenzene substrates were prepared according to the previously reported procedure.[5] Solvents were treated with 4 Å molecular sieves or sodium and distilled prior to use. Purifications of reaction products were carried out by flash chromatography using silica gel (200~300 mesh). 1H NMR and 13C NMR spectra were recorded with tetramethylsilane (TMS) as internal standard at ambient temperature unless otherwise indicated on a Bruker Avance DPX 600 fourier Transform spectrometer operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. High resolution mass spectra (HRMS) were recorded on an IF-TOF spectrometer (Micromass).
A 10 mL of reaction tube was charged with azobenzenes 1 (0.2 mmol), [Ru(p-cymene)Cl2] (5 mol%), AgSbF6 (20 mol%), NaOAc (50 mol%) and DCE (1.5 mL) under Ar atmosphere. Then ethyl glyoxalate 2a (0.4 mmol) in DCE (0.5 mL) was added in one-pot under Ar and the mixture was stirred at 100 ℃ for 24 h. The corresponding reaction mixture was filtered through a pad of Celite, washed with DCE and concetrated under reduced pressure. The residue was purified by flash chromatography on silical gel using ethyl acetate/petroleum ether (V:V=1:8) as eluent to afford the products 3.
Ethyl 2-phenyl-2H-indazole-3-carboxylate (3a): Brown solid, 82% yield. m.p. 80~82 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.12 (d, J=8.3 Hz, 1H), 7.85 (d, J=8.5 Hz, 1H), 7.52 (s, 5H), 7.41 (t, J=7.1 Hz, 1H), 7.34 (t, J=7.4 Hz, 1H), 4.36 (q, J=13.1, 2H), 1.33 (t, J=6.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 159.5, 148.4, 141.0, 129.3, 128.6, 127.1, 126.4, 125.5, 125.1, 124.01, 121.6, 118.6, 61.1, 14.1; HRMS (ESI) calcd for C16H15N2O2 [M+H]+ 267.1132, found 267.1135.
Ethyl 5-methyl-2-(p-tolyl)-2H-indazole-3-carboxylate (3b): Brown solid, 84% yield. m.p. 68~70 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.85 (s, 1H), 7.73 (d, J=8.6 Hz, 1H), 7.39 (d, J=7.3 Hz, 2H), 7.30 (d, J=7.5 Hz, 2H), 7.24 (d, J=9.1 Hz, 1H), 4.36 (q, J=6.6 Hz, 2H), 2.50 (s, 3H), 2.44 (s, 3H), 1.34 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 159.7, 147.4, 139.2, 138.7, 135.3, 129.8, 129.2, 126.1, 124.4, 119.7, 118.2, 60.9, 22.2, 21.3, 14.2; HRMS (ESI) calcd for C18H19N2O2 [M+H]+ 295.1631, found 295.1636.
Ethyl 5-methoxy-2-(4-methoxyphenyl)-2H-indazole-3- carbo-xylate (3c): Brown solid, 88% yield. m.p. 88~90 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.64 (d, J=9.1 Hz, 1H), 7.35 (d, J=7.6 Hz, 2H), 7.26 (s, 1H), 7.01 (d, J=9.2 Hz, 1H), 6.93 (d, J=7.6 Hz, 2H), 4.30~4.21 (q, 2H), 3.83 (d, J=8.6 Hz, 3H), 3.80 (s, 3H), 1.25 (t, J=6.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 160.0, 159.8, 157.9, 145.0, 134.2, 127.4, 125.1, 124.1, 121.8, 119.9, 113.7, 97.9, 60.8, 55.6, 55.4, 14.2; HRMS (ESI) calcd for C18H19N2O4 [M+H]+ 327.1328, found 327.1332.
Ethyl 5-chloro-2-(4-chlorophenyl)-2H-indazole-3-car- boxylate (3d): Brown solid, 77% yield. m.p. 186~188 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.08 (s, 1H), 7.77 (d, J=9.1 Hz, 1H), 7.56~7.44 (m, 5H), 7.36 (d, J=9.0 Hz, 1H), 4.39 (q, J=7.0 Hz, 2H), 1.38 (t, J=7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 159.0, 146.9, 139.1, 135.6, 131.7, 129.4, 128.9, 127.6, 124.4, 124.2, 120.5, 120.1, 61.5, 14.2; HRMS (ESI) calcd for C16H13Cl2N2O2 [M+H]+ 335.0355, found 335.0359.
Diethyl 2-(4-(ethoxycarbonyl)phenyl)-2H-indazole-3, 5 - dicarb-oxylate (3e): Brown solid, 68% yield. m.p. 139~141 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.91 (s, 1H), 8.23 (d, J=7.3 Hz, 2H), 8.05 (d, J=8.9 Hz, 1H), 7.86 (d, J=8.8 Hz, 1H), 7.63 (d, J=7.8 Hz, 2H), 4.51~4.36 (m, 6H), 1.42 (m, 9H); 13C NMR (101 MHz, CDCl3) δ: 166.5, 165.6, 158.9, 149.8, 143.9, 131.6, 130.6, 130.1, 127.8, 127.3, 127.2, 126.3, 125.7, 123.4, 122.9, 118.5, 61.7, 61.4, 61.3, 14.3, 14.1; HRMS (ESI) calcd for C22H23N2O6 [M+H]+ 411.1501, found 411.1508.
Ethyl 5-cyano-2-(4-cyanophenyl)-2H-indazole-3-car- boxylate (3f): Brown solid, 59% yield. m.p. 192~194 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.81 (s, 1H), 8.07 (s, 1H), 7.93 (d, J=8.5 Hz, 1H), 7.84 (d, J=8.6 Hz, 1H), 7.24 – 7.03 (m, 3H), 4.40 (q, 2H), 1.42 (t, 3H); 13C NMR (101 MHz, CDCl3) δ: 162.5, 142.2, 141.1, 132.7, 131.4, 128.8, 127.7, 127.3, 121.9, 120.9, 119.9, 118.0, 117.9, 117.5, 117.3, 104.4, 59.6, 14.9; HRMS (ESI) calcd for C18H13N4O2 [M+H]+317.1064, found 317.1070.
Ethyl 7-methyl-2-(o-tolyl)-2H-indazole-3-carboxylate (3g): Brown solid, 80% yield. m.p. 74~76 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J=8.4 Hz, 1H), 7.42 (dd, J=10.5, 4.2 Hz, 1H), 7.38~7.26 (m, 4H), 7.18 (d, J=6.8 Hz, 1H), 4.30 (q, 2H), 2.69 (s, 3H), 2.01 (s, 3H), 1.27 (t, 3H); 13C NMR (101 MHz, CDCl3) δ: 159.4, 148.8, 140.8, 135.0, 130.5, 129.6, 128.9, 127.0, 126.3, 126.1, 125.7, 123.3, 119.0, 60.9, 17.1, 17.0, 14.0; HRMS (ESI) calcd for C18H19N2O2 [M+H]+ 295.1446, found 295.1453.
Ethyl 6-methyl-2-(m-tolyl)-2H-indazole-3-carboxylate (3h): Brown solid, 83% yield. m.p. 85~87 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.99 (d, J=8.6 Hz, 1H), 7.58 (s, 1H), 7.41~7.26 (m, 4H), 7.17 (d, J=8.6 Hz, 1H), 4.35 (q, J=7.0 Hz, 2H), 2.49 (s, 3H), 2.43 (s, 3H), 1.33 (t, J=7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 159.5, 149.0, 141.0, 138.7, 137.0, 130.0, 128.4, 128.3, 126.8, 124.9, 123.5, 122.4, 121.0, 116.8, 61.0, 22.1, 21.3, 14.2; HRMS (ESI) calcd for C18H19N2O2 [M+H]+295.1508, found 295.1514.
Ethyl 2-(3, 5-dimethylphenyl)-4, 6-dimethyl-2H-indazo- le-3-carboxylate (3i): Brown solid, 78% yield. m.p. 67~69 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.38 (s, 1H), 7.26 (s, 1H), 7.12 (s, 2H), 6.85 (s, 1H), 4.25 (q, J=7.1 Hz, 2H), 2.61 (s, 3H), 2.42 (s, 3H), 2.37 (s, 6H), 1.13 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 165.8, 142.0, 132.4, 131.2, 130.0, 123.3, 121.9, 119.1, 115.8, 104.3, 98.2, 60.8, 17.9, 17.5, 13.7, 11.0; HRMS (ESI) calcd for C20H23N2O2 [M+H]+ 323.1791, found 323.1795.
Ethyl 2-(3-methoxy-5-methylphenyl)-2H-indazole-3- carboxylate (3j): Brown solid, 76% yield. m.p. 95~97 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.73 (d, J=9.2 Hz, 1H), 7.33 (dt, J=30.7, 9.0 Hz, 5H), 7.09 (d, J=9.0 Hz, 1H), 4.38 (q, 2H), 3.92 (s, 3H), 2.45 (s, 3H), 1.32 (t, J=6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 159.8, 157.9, 145.1, 139.1, 138.7, 129.1, 126.0, 125.1, 121.9, 120.0, 97.9, 60.8, 55.4, 21.3, 14.2; HRMS (ESI) calcd for C18H19- N2O3 [M+H]+ 311.1338, found 311.1342.
Methyl 2-phenyl-2H-indazole-3-carboxylate (3k): Brown solid, 74% yield. m.p. 79~81 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.11 (d, J=8.2 Hz, 1H), 7.86 (d, J=8.3 Hz, 1H), 7.53 (s, 5H), 7.42 (t, J=7.2 Hz, 1H), 7.35 (t, J=7.2 Hz, 1H), 3.92 (s, 3H); 13C NMR (126 MHz, CDCl3) δ: 160.0, 148.4, 140.9, 129.4, 128.7, 127.1, 126.33, 125.6, 124.83 123.9, 121.5, 118.6, 52.0; HRMS (ESI) calcd for C15H13N2O2 [M+H]+ 253.1901, found 253.1908.
Phenyl(2-phenyl-2H-indazol-3-yl)methanone (3l): Bro- wn solid, 32% yield. m.p. 153~155 ℃; 1H NMR (400 MHz, ) δ: 7.89~7.85 (m, 3H), 7.60 (t, J=7.5 Hz, 1H), 7.55~7.52 (m, 2H), 7.47~7.35 (m, 7H), 7.19~7.16 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 168.9, 138.9, 132.5, 130.3, 126.9, 125.9, 124.0, 123.3, 123.2, 122.9, 121.7, 120.5, 120.1, 119.3, 116.5, 114.9; HRMS (ESI) calcd for C20H15N2O [M+H]+ 299.3208, found 299.3213.
Ethyl pyrido[2, 1-a]isoindole-6-carboxylate (5a): Yellow solid, 36% yield. m.p. 65~67 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.98 (d, J=6.6 Hz, 1H), 8.27 (d, J=8.5 Hz, 1H), 8.10 (ddt, J=12.0, 8.2, 1.1 Hz, 2H), 7.55 (m, 1H), 7.29~7.27 (m, 1H), 7.27~7.25 (m, 1H), 7.21 (td, J=6.9, 1.5 Hz, 1H), 4.51 (q, 2H), 1.52 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 150.1, 126.3, 124.8, 122.5, 122.1, 117.3, 116.6, 115.9, 115.6, 114.9, 114.0, 113.9, 103.3, 67.6, 31.9; HRMS (ESI) calcd for C15H14NO2 [M+H]+ 240.3609, found 240.3612.
Ethyl 8-methoxypyrido[2, 1-a]isoindole-6-carboxylate (5b): Yellow solid, 27% yield. m.p. 94~96 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.88 (s, 1H), 8.00~7.91 (m, 2H), 7.60 (s, 1H), 7.25 (m, 1H), 7.12 (td, J=7.0, 1.4 Hz, 1H), 6.90 (dd, J=8.8, 2.3 Hz, 1H), 4.49 (q, J=7.1 Hz, 3H), 3.95 (s, 3H), 1.51 (t, J=7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 150.1, 148.3, 126.5, 122.3, 117.8, 116.7, 113.4, 112.9, 111.1, 110.9, 103.1, 99.0, 67.5, 64.2, 31.9; HRMS (ESI) calcd for C16H16NO3 [M+H]+ 270.1681, found 270.1685.
Ethyl 8-acetylpyrido[2, 1-a]isoindole-6-carboxylate (5c): Yellow solid, 21% yield. m.p. 113~115 ℃; 1H NMR (400 MHz, CDCl3) δ: 9.90 (d, J=6.7 Hz, 1H), 8.85 (s, 1H), 8.11~8.07 (m, 2H), 7.79 (dd, J=8.6, 1.5 Hz, 1H), 7.33~7.27 (m, 1H), 7.24 (td, J=6.9, 1.5 Hz, 1H), 4.51 (q, J=7.1 Hz, 2H), 2.72 (s, 3H), 1.54 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 179.0, 149.7, 129.2, 125.7, 123.9, 122.1, 118.1, 117.6, 116.2, 115.7, 115.2, 114.6, 114.5, 104.1, 67.9, 41.4, 31.8; HRMS (ESI) calcd for C17H15NO3 [M+H]+ 282.1131, found 282.1134.
2-Phenyl-2H-indazole (6a): Yellow solid, 36% yield. m.p. 83~85 ℃; 1H NMR (500 MHz, CDCl3) δ: 8.43 (s, 1H), 7.93 (d, J=7.7 Hz, 2H), 7.83 (d, J=8.8 Hz, 1H), 7.73 (d, J=8.5 Hz, 1H), 7.55 (t, J=7.9 Hz, 2H), 7.42 (t, J=7.4 Hz, 1H), 7.38~7.33 (m, 1H), 7.15 (dd, J=8.0, 7.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 149.8, 140.5, 129.6, 127.9, 126.8, 122.8, 122.4, 121.0, 120.4, 120.4, 118.0; MS (ESI) m/z: 195.09.
Supporting Information The kinetic isotope effect (KIE) experiments and the 1H NMR and 13C NMR spectra of all products. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) De Angelis, M.; Stossi, F.; Carlson, K. A.; Katzenellenbogen, B. S.; Katzenellenbogen, K. A. J. Med. Chem. 2005, 48, 1132.
(b) A Haddadin, M. J.; Conrad, W. E.; Kurth, M. J. Mini-Rev. Med. Chem. 2012, 12, 1293.
(c) Hu, J.-T.; Cheng, Y.-F.; Yang, Y.-Q.; Rao, Y. Chem. Commun. 2011, 47, 10133.
(d) Magano, J.; Waldo, M.; Greene, D.; Nord, E. Org. Process Res. Dev. 2008, 12, 877.
(a) De Lena, M.; Lorusso, V.; Latorre, A.; Fanizza, G.; Gargano, G.; Caporusso, L.; Guida, M.; Catino, A.; Crucitta, E.; Sambiasi, D.; Mazzei, A. Eur. J. Cancer 2001, 37, 364.
(b) Atta-ur-Rahman; Malik, S.; Cun-heng, H.; Clardy, J. Tetrahedron Lett. 1985, 26, 2759.
(c) Halim, R.; Harding, M.; Hufton, R.; Morton, C. J.; Jahangiri, S.; Pool, B. R.; Jeynes, T. P.; Draffan, A. G.; Lilly, M. J.; Frey, B. WO 2012051659, 2011.
(d) Steffan, R. J.; Matelan, E. M. WO 2006050006, 2006.
(a) Luo, G.; Chen, L.; Dubowchik, G. J. Org. Chem. 2006, 71, 5392.
(b) Unsinn, A.; Knochel, P. Chem. Commun. 2012, 48, 2680.
(c) Bunnell, A.; Yang, C.; Petrica, A.; Soth, M. J. Synth. Commun. 2006, 36, 285.
For selected reviews, see: (a) Ackermann, C. Acc. Chem. Res. 2014, 47, 281.
(b) Song, G.; Wang, F.; Li, X. Chem. Soc. Rev. 2012, 41, 3651.
(c) Huang, Z.; Lim, H, N.; Mo, F.; Young, M. C.; Dong, G. Chem. Soc. Rev. 2015, 44, 7764.
(d) Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T. S. A.; Liu, X. Chem. Soc. Rev. 2015, 44, 291.
(e) Chen, Z.; Wang, B.; Zhang, J.; Yu, W.; Liu, Z.; Zhang, Y. Org. Chem. Front. 2015, 2, 1107.
(f) Song, G.; Li, X. Acc. Chem. Res. 2015, 48, 1007.
(g) Zhang, F.; Spring, D. R. Chem. Soc. Rev. 2014, 43, 6906.
(h) Wencel-Delord, J.; Glorius, F. Nat. Chem. 2013, 5, 369.
(Ⅰ) Chen, X., Bai, L.; Zeng, W. Chin. J. Org. Chem. 2018, 38, 1859(in Chinese).
(陈训, 白丽丽, 曾伟, 有机化学, 2018, 38, 1859.)
(j) Zhang, Z.; Butt, N. A.; Zhou, M.; Liu, D.; Zhang, W. Chin. J. Chem. 2018, 36, 443.
(k) Jeong, T.; Han, S. H.; Han, S.; Sharma, S.; Park, J.; Lee, J.; Kwak, J. H.; Jung, Y. H.; Kim, I. S. Org. Lett. 2016, 18, 232.
(l) Chen, W.; Yang, W.; Wu, R.; Yang, D. Green Chem. 2018, 20, 2512.
(m) Yang, W.; Chen, Y.; Yao, Y.; Yang, X.; Lin, Q.; Yang, D. J. Org. Chem. 2019, 84, 11080.
(n) Yao, Y.; Yang, W.; Lin, Q.; Yang, W.; Li, H.; Wang, L.; Gu, F.; Yang D. Org. Chem. Front.. 2019, 6, 3360.
Lian, Y.; Bergman, R. G.; Lavis, L. D.; Ellman, J. A. J. Am. Chem. Soc. 2013, 135, 7122. doi: 10.1021/ja402761p
(a) Hummel, J. R.; Ellman, J. A. J. Am. Chem. Soc. 2015, 137, 490.
(b) Geng, X.; Wang, C. Org. Lett. 2015, 17, 2434.
(a) T. Gensch, M. N. Hopkinson, F. Glorius, J. Wencel-Delord, Chem. Soc. Rev. 2016, 45, 2900.
(b) Suzuki, C.; Morimoto, K.; Hirano, K.; Satoh, T.; Miura, M. Adv. Synth. Catal. 2014, 356, 1521.
(c) Itoh, M.; Hashimoto, Y.; Hirano, K.; Satoh, T.; Miura, M. J. Org. Chem. 2013, 78, 8098.
(d) Suzuki, C.; Hirano, K.; Satoh, T.; Miura, M. Org. Lett. 2013, 15, 3990.
(e) Kong, D.; Lu, G.; Wu, M.; Shi, Z.; Lin, Q. ACS Sustainable chem. Eng. 2017, 5, 3465.
(f) De Sarkar, S.; Liu, W.; Kozhushkov, S. I.; Ackermann, L. Adv. Synth. Catal. 2014, 356, 1461.
(g) Zhang, Y.; Yang, Z.; Yu, X.; Cheng, X.; Li, W.; Guo, L.; Hai, L.; Guo, L.; Wu, Y. Chin. J. Org. Chem. 2018, 38, 3211(in Chinese).
(张勇, 杨中振, 余昕玲, 程旭, 李伟剑, 郭令妹, 海俐, 郭丽, 吴勇, 有机化学, 2018, 38, 3211.)
(h) Xu, W.; Wang, N.; Zhang, M.; Shi, D. Chin. J. Org. Chem. 2019, 39, 1735(in Chinese).
(徐文韬, 王宁, 张梦烨, 史达清, 有机化学, 2019, 39, 1735.)
白丽丽, 王颖, 王烁今, 孔杜林, 付艳, 彭德乾, 文丽君, 陈训, 有机化学, 2018, 38, 3242.Bai, L.; Wang, Y.; Wang, S.; Kong, D.; Fu, Y.; Peng, D.; Wen, L.; Chen, X. Chin. J. Org. Chem. 2018, 38, 3242(in Chinese).
(a) Li, B.; Xu, H.; Wang, H.; Wang, B. ACS Catal. 2016, 6, 3856.
(b) Huang, X.; Huang, J.; Du, C.; Zhang, X.; Song, F.; You, J. Angew. Chem., Int. Ed. 2013, 52, 12970.
(c) Thirunavukkarasu, V. S.; Raghuvanshi, K.; Ackermann, L. Org. Lett. 2013, 15, 3286.
(a) Chen, X.; Hu, X.; Deng, Y.; Jiang, H.; Zeng, W. Org. Lett. 2016, 18, 4742.
(b) Jeon, B.; Yeon, U.; Son, J. Y.; Lee, P. H. Org. Lett. 2016, 18, 4610.
Table 1. Optimization of the reaction parametersa
|
|
||||
| Entry | Catalyst | Additive | Solvent | Yieldb/% |
| 1 | [Ru(p-cymene)Cl2] | None | Dioxane | 24 |
| 2 | [Ru(p-cymene)Cl2] | NaOAc | Dioxane | 51 |
| 3 | [Ru(p-cymene)Cl2] | KOAc | Dioxane | 46 |
| 4 | [Ru(p-cymene)Cl2] | LiOAc | Dioxane | 47 |
| 5 | [Ru(p-cymene)Cl2] | CsOAc | Dioxane | 40 |
| 6 | [Ru(p-cymene)Cl2] | AgOAc | Dioxane | 38 |
| 7 | [Ru(p-cymene)Cl2] | PivOH | Dioxane | 41 |
| 8 | [Ru(p-cymene)Cl2] | AcOH | CH3CN | 26 |
| 9 | [Ru(p-cymene)Cl2] | NaOAc | Toluene | Trace |
| 10 | [Ru(p-cymene)Cl2] | NaOAc | THF | 48 |
| 11 | [Ru(p-cymene)Cl2] | NaOAc | TFE | 21 |
| 12 | [Ru(p-cymene)Cl2] | NaOAc | MeOH | Trace |
| 13 | [Ru(p-cymene)Cl2] | NaOAc | DCE | 64 |
| 14 | [Ru(p-cymene)Cl2] | NaOAc | DCE | 76c |
| 15 | [Ru(p-cymene)Cl2] | NaOAc | DCE | 82d |
| 16 | RuCl2(PPh3)4 | NaOAc | DCE | 0 |
| 17 | Ru3(CO)12 | NaOAc | DCE | 0 |
| 18 | RuHCl(CO)(PPh3)3 | NaOAc | DCE | 0 |
| a Unless otherwise noted, all the reactions were carried out using azobenzenes (1a, 0.20 mmol) and ethyl glyoxalate (2a, 0.40 mmol) with catalysts (5 mol%) in the presence of AgSbF6 (20 mol%) and additive (50 mol%) in solvent (2.0 mL) at 80 ℃ for 12 h under Ar in a sealed reaction tube, followed by flash chromatography on SiO2. b Isolated yield. c The reaction temperature is 100 ℃. d The reaction time is 24 h. TFE=2, 2, 2-trifluoroethanol. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们