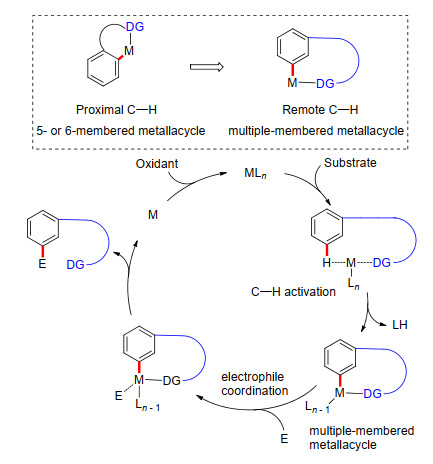
图式 1.
U型模板策略下芳烃远程meta-C—H键选择性官能化反应机理
Scheme 1.
Remote meta-C—H functionalization of arenes directed by a U-shaped template

基于过渡金属催化的C—H键活化策略的化学合成因无需对底物进行预官能化, 具有步骤经济性和原子经济性等独特的优势, 已发展成为有机合成化学中非常重要的工具[1].化学家通过引入导向基团, 利用导向基团与过渡金属催化剂配位, 以特定的空间构型拉近过渡金属中心与底物中目标C—H键的距离, 并形成五元或六元环金属过渡态, 以此诱导C—H键选择性活化, 实现芳烃邻位的各种官能化反应[2~8].而由于大环过渡态的环张力影响, 过渡金属催化的定向远程meta-C—H键活化变得十分困难.
配体是有机合成反应中常用的一类小分子化合物, 能够对催化中心的空间及电子效应进行调控, 不仅有助于C—H键断裂, 提高反应效率, 而且能够促进芳烃远程C—H键的选择性官能化反应[9].近年来, 发展配体参与的过渡金属催化芳烃远程C—H键选择性官能化反应成为相关领域的研究焦点, 已取得了突破性进展.根据反应机制, 系统总结了近几年来配体参与的过渡金属催化芳烃远程meta-C—H键选择性官能化反应的研究进展, 并就该领域亟待解决的关键问题和未来的发展前景进行了总结和展望.
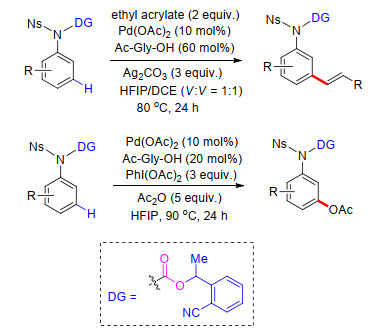
余金权课题组首先发展了一种在底物结构中引入U型模板, 再通过远端导向基团与金属配位, 从而实现芳烃远程C—H键的选择性活化.该策略可能的反应途径如下: (1)过渡金属催化剂与底物U型模板远端杂原子进行络合配位; (2)在外加配体协助作用下完成meta-C—H活化, 生成相应的多元金属大环过渡态; (3)亲电试剂在过渡金属催化中心进行配位并通过氧化加成/迁移插入和还原消除等过程实现了芳烃meta-C—H键选择性官能化; (4)过渡金属催化剂在氧化剂(如银离子)存在下被氧化到相应的高价态, 从而完成了催化循环(Scheme 1).这一策略突破了传统C—H键活化的局限性, 在底物中预修饰能够与过渡金属进行可逆性配位的氰基等基团作为远端导向基, 实现了一系列芳烃meta-C—H键选择性官能团化反应.
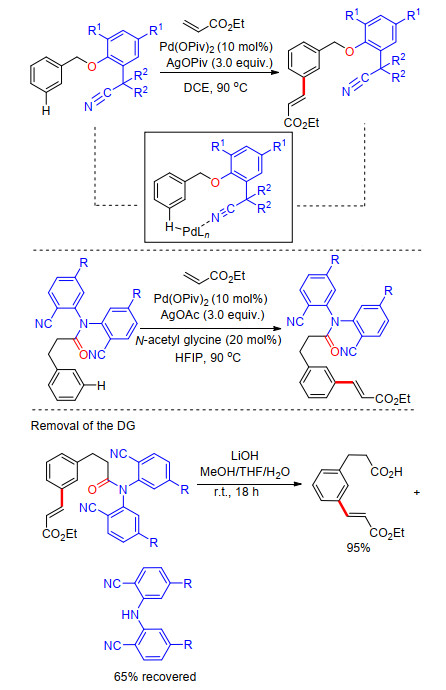
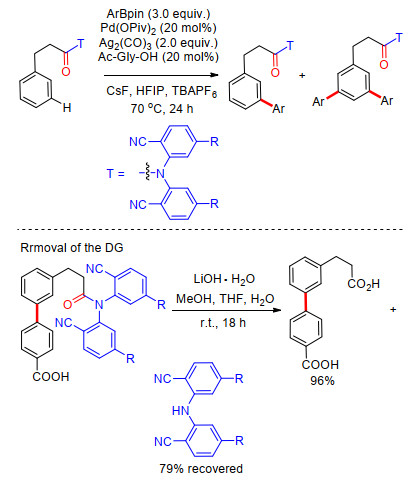
2012年, 余金权课题组[10]首次以醚基连接氰基芳基建立U型模板导向基, 成功完成了苄醇类衍生物meta-C—H键选择性烯基化反应.氰基作为远端导向基团及配体基团, 能够与过渡金属进行可逆配位, 从而在空间上拉近过渡金属与远端meta-C—H键的距离, 顺利完成meta-C—H活化, 最终成功得到一系列苄醇类化合物meta-C—H键烯基化产物.反应以Pd(OAc)2为催化剂, 二氯乙烷为溶剂, 在氧化剂存在条件下进行.反应具有非常高的区域选择性, 间位烯基化产率最高可达93% (Scheme 2).苯丙酸类化合物也能使用类似的模板进行meta-C—H键烯基化反应, 反应溶剂为六氟异丙醇(HFIP), 并添加乙酰甘氨酸为外加配体, 反应同样具有很好的区域选择性及底物适应性, 间位烯基化产率最高可达95% (Scheme 2).在完成meta-C—H键烯基化反应的基础上, 他们对U型模板导向基的脱除进行了研究, 最终在LiOH作碱, MeOH/THF/H2O混合溶剂中, 室温条件下以95%的产率得到间位烯基化苯丙酸(Scheme 2).
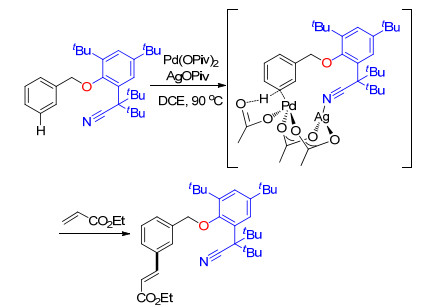
2014年, 余金权课题组[11]对U型模板导向的芳烃meta-C—H键烯基化反应机理进行了研究.研究表明, 反应经历了四个基本过程, 即C—H活化、烯烃插入、β-H消除和还原消除.根据理论计算, C—H活化过程是反应中活化能最高的一步, 即反应的决速步.此外, 通过计算, 他们提出了四种可能的催化过渡态, 其中异二聚体Pd-Ag过渡态具有最低的反应活化能, 理论上是更有利的中间过渡态.在这种过渡态中, 与氰基直接进行配位的是金属银而不是金属钯, Pd物种被选择性地带到远端meta-C—H键的近端, 形成唯一的间位区域选择性(Scheme 3).至此, 利用计算辅助机理研究方法, 余金权等给出了U型模板底物meta-C—H键选择性烯基化反应可能的反应机理.
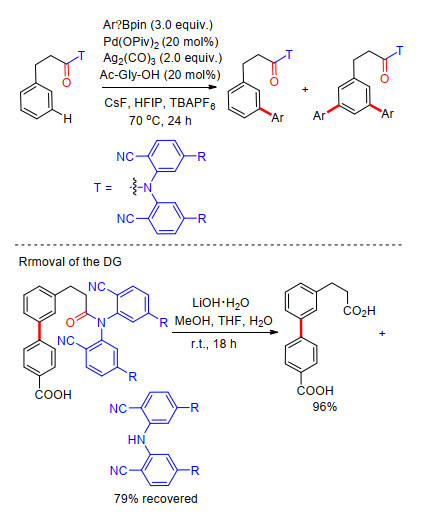
2013年, 余金权课题组[12]将苯丙酸类化合物meta-C—H键选择性官能化反应拓展到了芳基化过程.反应中同样选择羰基共价连接氰基芳基构建U型模板, 以芳基硼试剂为芳基化试剂, Pd(OPiv)2为催化剂, Ag2(CO)3为氧化剂, 能够促进C—H键与有机硼试剂偶联的甘氨酸类为外加配体, 四丁基六氟硼酸铵(TBAPF6)为添加剂, CsF为碱, HFIP为溶剂, 得到meta-C—H键芳基化和少量meta-C—H键双芳基化产物(Scheme 4).反应具有良好的底物适应性, 在以温和的LiOH·H2O为碱, MeOH/THF/H2O为混合溶剂条件下, 能够顺利脱除U型模板, 以96%的产率得到相应的meta-C—H键芳基化苯丙酸类化合物(Scheme 4).该反应进一步拓展了U型模板策略, 实现了苯丙酸类化合物meta-C—H键选择性芳基化.
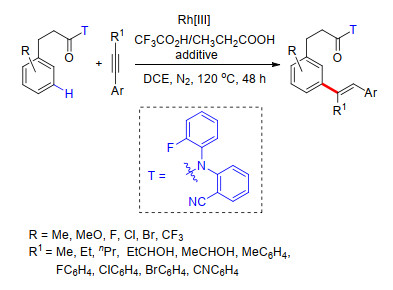
2017年, 余金权课题组[13]利用铑作为催化剂, 同样选择羰基共价连接氰基芳基构建U型模板, 在模板远端导向基团与金属配体共同作用下, 完成了苯丙酸类化合物的meta-C—H键烯基化反应(Scheme 5).他们给出了可能的反应机理, 反应经历了在模板远端导向基团与配体共同作用下苯丙酸衍生物meta-C—H键的选择性活化, 紧接着发生烯基插入和β-H消除过程, 得到最终产物.反应具有良好的底物适应性, 多种取代芳烃都能够很好地适应反应条件, 烯基化试剂同样具有很好的底物适应性, 多种吸电取代烯烃, 如丙烯酸酯、烯丙基酰胺、烯丙基醛、丙烯酸硫酸酯和丙烯酸磷酸酯都能够以很好的产率得到相应的产物(Scheme 5).
近期, 余金权课题组[14]同样以铑作催化剂, 以羰基共价连接氰基芳基构建U型模板, 以二苯乙炔为官能化试剂, 得到一系列苯丙酸衍生物meta-C—H键烯基化产物.反应中U型模板远端导向基团作为配体导向基团, 对反应间位选择性起到至关重要的作用.他们对两种反应底物适应性分别进行了考察, 发现两种反应底物都具有较好的适应性, 多种苯乙炔均以较高的产率与苯丙酸类化合物发生偶联, 生成相应的产物(Scheme 6).
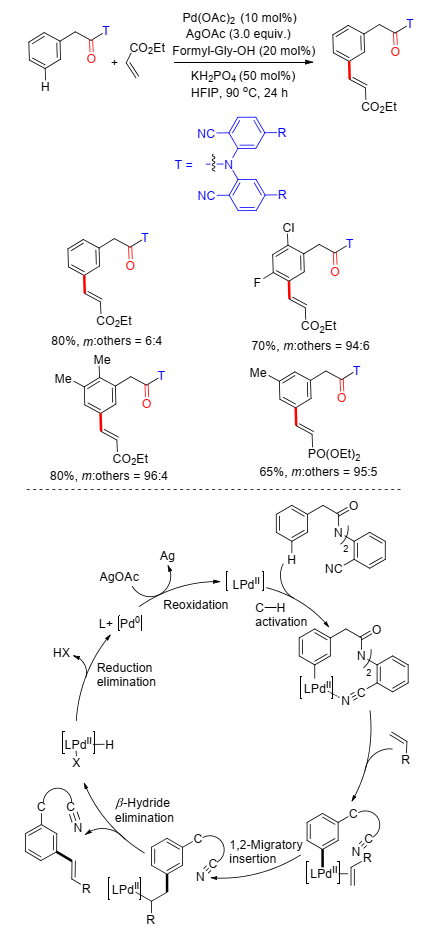
2015年, 余金权课题组[15]将底物拓展到苯乙酸类化合物, 同样利用氰基芳基作为U型模板导向基, 以96:4的间/邻比率选择性地得到了间位烯基化苯乙酸化合物. N-甲酰甘氨酸作为配体与KH2PO4共同协助稳定11元钯环中间过渡态(Scheme 7), 在反应中起到重要作用.脱U型导向基实验中, 发现在碱与溶剂组成的基本水解条件下得到水解混合物, 而在加入双氧水时, 能够保留烯基酯, 得到单一的间位烯基化苯乙酸化合物.这项工作中, 作者给出了可能的反应机理:在C—H活化过程中, 碱与配体共同作用形成了较稳定的11元钯环中间体, 烯烃配位插入钯催化中心后经β-H消除过程得到最终产物, AgOAc将Pd(0)氧化到Pd(Ⅱ)从而完成了催化循环(Scheme 7)
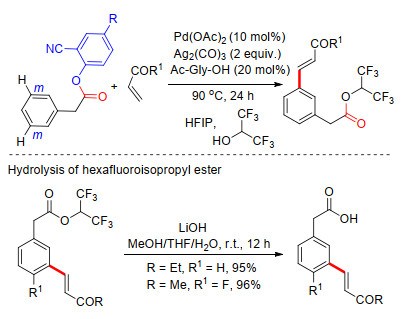
2014年, Maiti课题组[16]以苯乙酸连接2-羟基氰基芳基构建U型模板骨架, 完成了苯乙酸类化合物meta-C—H键选择性烯基化反应.他们巧妙地使用六氟异丙醇作为溶剂, 可使羧基连接桥发生酯交换反应, 一锅法实现了脱U型模板导向基过程, 得到间位烯基化的苯乙酸三氟异丙酯.该反应具有很高的立体选择性及良好的底物适应性, 一系列苯乙酸通过U型模板途径实现了meta-C—H键烯基化反应.酯类化合物在碱性条件下更易水解得到间位烯基化苯乙酸类化合物(Scheme 8).
2015年, 李纲课题组[17]利用酰胺结构共价连接氰基芳基构建U型模板, 完成了芳烃meta-C—H键烯基化反应.通过优化反应条件, 发现在钯催化剂和氨基酸配体作用下, 在HFIP/1, 2-二氯乙烷(DCE)混合溶剂中, 反应收率最高, 且具有良好的选择性及底物适应性(Scheme 9).该方法中需对酰胺结构氮原子进行甲基化后才能够得到间位烯基化产物, 而非甲基化酰胺在U型模板条件下只得到单一的邻位烯基化产物, 他们利用这一特性对该酰胺类化合物进行了邻位-间位顺序烯基化反应实验, 成功合成了多取代芳烃化合物(Scheme 9).最后在酸水解条件下去除U型模板导向基, 得到间烯基化苯乙胺.
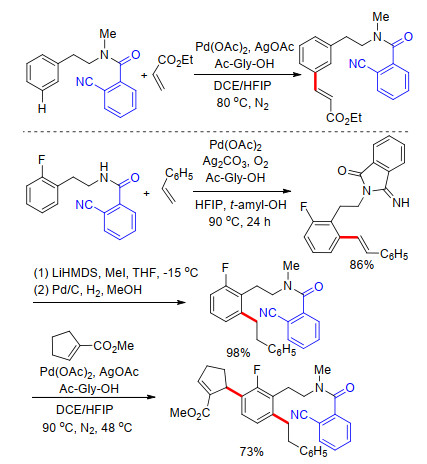
紧接着, 李纲课题组[18]选用酰胺基连接氰基苯乙基结构构建U型模板, 实现了苯甲酸类化合物间位官能化.在该U型模板条件下, 多种取代苯甲酸能够顺利完成间位烯基化及乙酰基化反应, 反应具有良好的底物适应性.与之前报道的利用银作氧化剂不同, 李纲课题组利用催化量的醋酸铜与氧气为共氧化剂, 完成了苯甲酸类化合物间位烯基化反应, 利用廉价易得的醋酸碘苯作为氧化剂, 实现了苯甲酸间位乙酰基化(Scheme 10).在此基础上, 通过筛选反应条件, 以更温和更高效的脱U型模板导向基方法得到间位烯基化苯甲酸.该合成方法的建立为几类常见苯乙基酰胺类药物的合成提供了新方法.
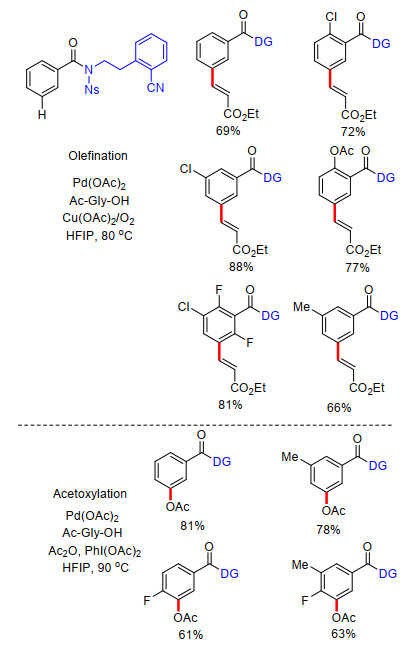
2017年, 余金权课题组[19]对上述苯甲酸类化合物的间位烯基化反应进行了改进, 以2-(2-(甲氨基)乙氧基)氰基苯为导向基团与苯甲酸连接建立U型模板, 顺利完成了苯甲酸类化合物间位烯基化反应.与他们组之前的工作相比, 该工作以具有长侧链的氰基苯为模板导向基及配体基团, 这样目标底物中连接模板导向基的侧链原子数可相应减少, 反应中在形成金属过渡态时, 同样能够生成相对稳定的大环金属中间体, 从而促进反应进行.与之前工作相比, 该反应有更高的反应效率及良好的底物适应性.通过理论计算, 作者提出反应中可能形成具有最低反应活化能的Pd-Ag双金属中间过渡态机制(Scheme 11).此外, 动力学实验表明, 单保护氨基酸的加入大大提高了反应速率, 与U型模板中氰基共同作为配体在反应中起到至关重要的作用.该工作增加了模板导向基侧链长度, 减少了底物侧链长度, 对新模板的提出具有重要的指导性意义, 为更多简单芳烃的间位官能化反应指出新思路.最后反应在酸性介质中完成了水解酯化过程, 顺利脱除了U型模板导向基, 得到间位烯基化苯甲酸酯类化合物.
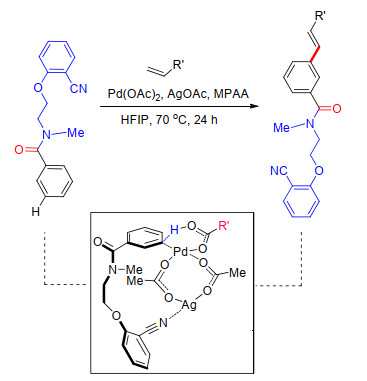
2017年, 李纲课题组[20]继续以酰胺键桥接氰基苯基构建U型模板, 实现了苯胺类化合物meta-C—H键官能化.他们将廉价易得的无毒气体CO2引入体系中, 依次与苯胺成酯并连接氰基苯基构建U型模板, 在Pd(OAc)2催化的温和条件下, 顺利发生苯环meta-C—H键烯基化及乙酰基化反应(Scheme 12).在碱性条件下, 能够顺利脱去U型模板, 得到相应的间位官能化苯胺类化合物.
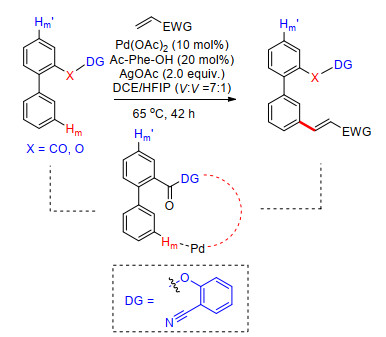
2016年, Maiti课题组[21]利用苯氧基及苯羰基连接氰基芳基构建U型模板, 完成了联二苯类化合物远端苯基meta-C—H键烯基化反应.反应在Pd(OAc)2催化下, 以苯基氨基酸为配体, 醋酸银为氧化剂, 在二氯乙烷及六氟异丙醇混合溶剂中, 高效完成了多种取代联二苯类化合物的间位烯基化, 烯基化试剂在反应体系中具有很好的适应性(Scheme 13).最后在对苯甲磺酸及甲醇中加热至105 ℃条件下完成了模板导向基的脱除.
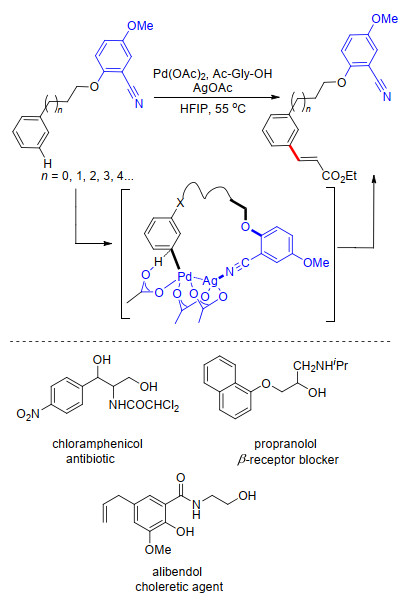
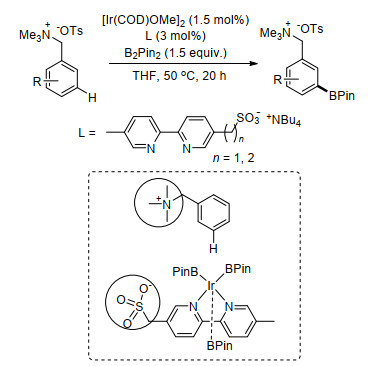
2017年, 金钟课题组[22]利用长链芳基烷醇类连接氰基苯基构建U型模板, 完成了一系列芳醇类化合物间位烯基化.在醋酸钯催化下, 芳基烷醇如3-苯丙醇、2-苯氧基乙醇和2-(苯拉米诺)乙醇等以高达91%的产率获得相应的meta-C—H键烯基化产物, 区域选择性良好, 最优选择比率达到20:1 (m:others=20:1).在反应中, AgOAc作为氧化剂起到重要作用, 在不添加AgOAc条件下, 无法得到相应产物.机理实验和密度泛函理论(DFT)计算结果证实了反应中可能的催化剂为杂双金属聚合物PdAg(OAc)3, 反应过程中与氰基直接配位的为金属Ag, 形成了C-N-Ag-Pd催化过渡态(Scheme 14).该反应可用于一些药物的合成.
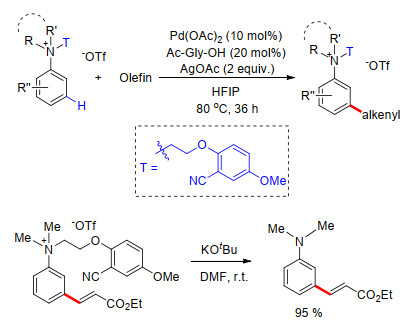
近期, 金钟课题组[23]继续以芳叔胺基桥接苯基氰基构建U型模板, 巧妙完成了一系列芳叔胺类化合物的间位烯基化.反应具有良好的底物适应性和高效的区域选择性, 该方法利用了叔胺经历季铵盐过程完成间位烯基化反应.最后, 在叔丁醇钾的N, N-二甲基甲酰胺溶液中能够顺利脱除U型模板(Scheme 15).
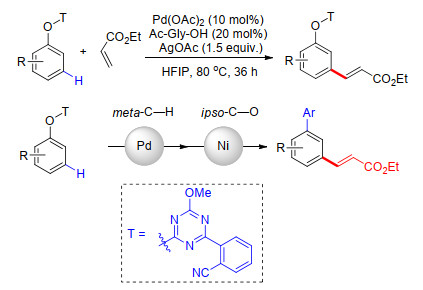
以往的工作多以羰基或醚基桥接饱和烷烃连接氰基苯基构建U型模板.近期, 余金权课题组[24]设计合成了氰基苯基取代的1, 3, 5-三嗪结构连接氰基苯基构建U型模板, 完成了Pd催化苯酚类化合物间位烯基化和Ni催化C—O键芳基化的有序串联反应.利用这种合成策略可以方便地修饰联芳基和氨基酸残基, 以便进行这类天然产物结构与活性关系的研究(Scheme 16).
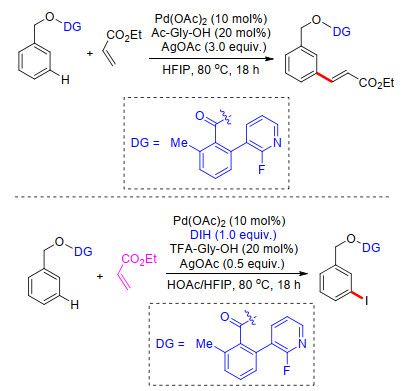
在羰基或醚基桥接氰基芳基构建的U型模板策略中, 充分利用了末端氰基的弱配位能力, 化学家们在模板设计中做了各种各样的改变.然而这些工作都是以氰基作为中心弱配位基团与过渡金属催化剂进行可逆配位.若体系中存在其他强配位能力的基团或者溶剂, 氰基与催化剂的配位作用将被强配位基团取代并使催化体系失活, 这严重限制了芳烃C—H键选择性官能化反应的适用性. 2015年, 余金权课题组[25]在氰基U型模板作用机制的基础上, 利用具有强配位作用的吡啶作为远端导向基团, 吡啶N原子作为远端配位中心, 利用醚基桥接苯基吡啶基团构建U型模板, 在Pd(OAc)2催化下, 以AgOAc为氧化剂, N-乙酰甘氨酸(Ac-Gly-OH)为外加配体, 在HFIP溶剂中, 顺利完成了苄醇类化合物meta-C—H键选择性烯基化(Scheme 17).在标准反应条件基础上, 通过添加1, 3-二碘-5, 5-二甲基乙内酰脲(DIH)为碘化剂, 完成了系列苄醇类化合物的间位碘化(Scheme 17).最后, 在温和的碱性条件下, 水解去除U型模板导向基, 得到间位碘化苄醇化合物, 产率高达94%.
2018年, 余金权课题组[26]将远端导向基团由氰基拓展到吡啶基, 他们以羰基连接苯基吡啶基构建U型模板, 反应中吡啶氮原子作为远端配位中心, 完成了苯乙酸类化合物的间位烯基化、芳基化及碘化(Scheme 18).至此, 余金权课题组通过设计和调整吡啶基位置, 完成了芳烃远端meta-C—H键多种选择性官能化反应.
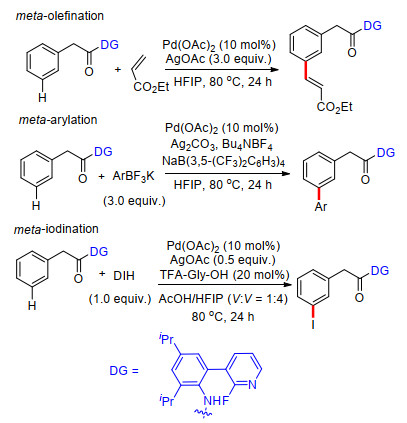
除此之外, 孔令仪课题组[27]近期利用U型模板法完成了多种苯基烷基二醇类天然产物的合成.他们利用1, 3-二氧戊环桥接苯基嘧啶, 以嘧啶氮原子为远端导向基团, 完成了苯环meta-C—H键烯基化反应.该方法可用于香豆素、苯丙氨酸、二苯乙烯和查尔酮类天然产物的合成.该方法能够顺利脱去U型模板, 并能以较高效率发生克级反应, 具有重要的应用价值.此外, 他们通过1H NMR, ESI-MS和IR数据, 结合理论计算对反应机理进行了研究, 给出了可能的反应中间过渡态及反应机理(Scheme 19).
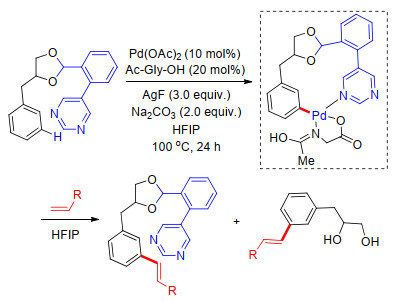
在U型模板策略中, 大部分反应主要生成单间位官能化产物, 而生成双间位官能化产物的报道很少, 这是由于在反应过程中, 生成单官能化产物后, 要继续进行间位官能化时, 底物立体效应和电子效应受先进入的间位官能团影响而发生改变, 使进一步的间位官能化反应变得困难. 2015年, Maiti课题组[28]利用磺酰基桥接U型模板, 在Pd(OAc)2催化下, 以氨基酸为外加共配体, Ag2CO3为氧化剂, 六氟异丙醇(conditions B, C)或二氯乙烷/六氟异丙醇(conditions A)为溶剂, 通过优化反应中配体及氧化剂用量, 完成了一系列苯甲磺酸类化合物的单间位烯基化或双间位烯基化反应.反应中, 不同底物在各个反应条件下主要生成单烯基化产物或双烯基化产物.最后在碱性条件下, 脱除模板导向基, 以高达95%的回收率得到U型模板氰基芳烃底物(Scheme 20).
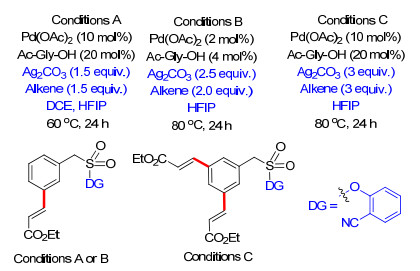
2016年, Maiti课题组[29]利用相同的磺酰基桥接的U型模板, 以不同的氨基酸为外加共配体, 在PhI(OAc)2为氧化剂条件下完成了芳烃间位乙酰基化, 在三氟醋酸碘苯为氧化剂时得到羟基化产物(Scheme 21).反应具有良好的底物适应性和高效的选择性, 该反应进一步拓展了芳烃间位官能化反应.
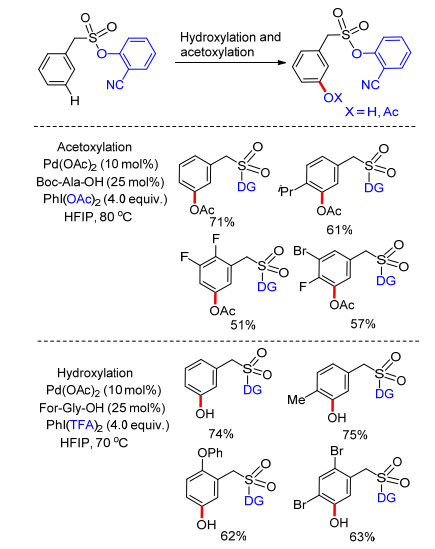
2017年, Maiti课题组[30]报道了以磺酰基桥接2-氰基苯酚为导向基团构建U型模板, 实现了芳基烷磺酸类化合物meta-C—H键选择性硅烷化及锗烷化.反应以六甲基二硅烷为硅化剂, 六甲基二锗烷为锗化剂.反应在钯催化剂体系中进行, 以银盐作为氧化剂, 六氟异丙醇为溶剂, 添加氨基酸为配体, 反应条件温和, 具有很高的间位单一选择性和良好的底物适应性.锗烷化反应最高间位单取代产物产率可达到85% (Scheme 22).最后, 在碱性条件下, 以高达92%的收率实现了脱U型模板导向基过程.这项工作将U型模板策略中芳烃间位选择性官能化反应由简单的烯基化和乙酰基化扩展到硅烷化及锗烷化反应, 顺利构建了芳烃meta-C—Si键及C—Ge键.含硅化合物常存在于一些药物、农药及高分子化合物中, 因此该反应的发现对药物、农药及高分子产业具有重要意义.
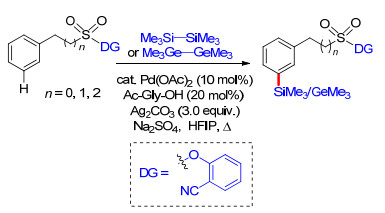
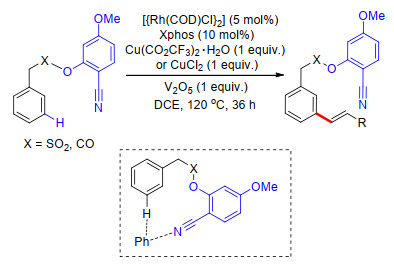
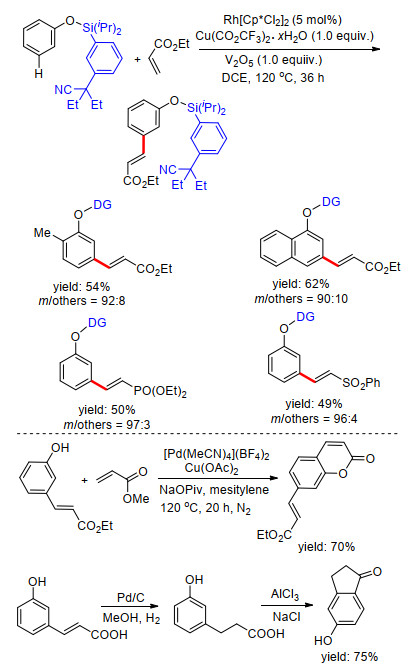
同年, Maiti课题组[31]以磺酰酯基桥接氰基苯基构建U型模板, 在Rh催化体系中完成了苯磺酸及苯乙酸类化合物meta-C—H键烯基化.在这项工作中, 他们对反应中外加的共配体进行了大量筛选工作, 发现磷配体对间位选择性具有重要作用, 最终确定了最高效的Xphos为标准条件配体, 反应中还需加入铜盐作为氧化剂, V2O5为共氧化剂才能够保证反应顺利发生.此外, 具有给电子效应的甲氧基氰基苯基导向基具有更好的导向活性.反应具有很好的底物适应性, 多种取代芳烃及烯基化试剂能够很好地适应反应条件.在温和的碱性条件下, 能够脱去U型模板, 得到相应的间位烯基化产物.最后, 通过机理研究, 给出了可能的反应机理(Scheme 23).
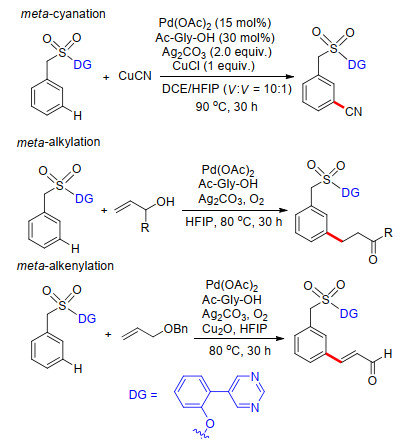
2017年, Maiti课题组[32]以嘧啶基作为远端导向基团及配体基团, 以磺酰基桥接苯基嘧啶基构建U型模板, 完成了苯甲磺酸类化合物meta-C—H键氰基化反应.反应中以Pd(OAc)2为催化剂, N-乙酰氨基酸为外加共配体, 碳酸银及氯化亚铜共同作为氧化剂, 氰化亚铜为氰基化试剂, 一系列取代苯甲磺酸化合物顺利发生间位氰基化反应, 以非常高的产率得到相应的氰基化产物(Scheme 24).同样利用嘧啶基作为远端导向基, Maiti课题组[33]完成了苯甲磺酸meta-C—H键烷基化及烯基化反应(Scheme 24).
除上述简单芳烃外, 2014年, 余金权课题组[34]利用磺酰基桥接苯基氰基构建了U型模板, 完成了吲哚类化合物C(6)—H键的选择性烯基化、芳基化及乙酰基化.反应中以过渡金属钯为催化剂, 远端氰基配位基团与N-乙酰基氨基酸共同作为配体对反应选择性起到重要的作用.转化过程均具有良好的底物适应性和反应选择性.此外, 作者利用该方法成功合成了多种具有生物活性的吲哚骨架化合物(Scheme 25).
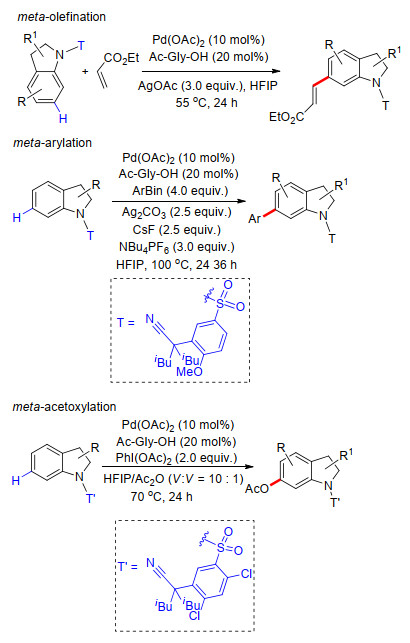
2018年, 余金权组[35]在上述工作的基础上, 继续以磺酰基桥接苯甲氰基构建U型模板, 完成了多种苯并N-杂环类化合物远程meta-C—H键烯基化反应.在这项工作种, 多种喹啉、异喹啉、吲哚、2-苯基吡咯和苯并吡咯结构都能够很好地适应反应条件, 经不同原子数目的大环中间过渡态完成远程苯环上meta-C—H键烯基化反应.烯基化试剂同样具有很好的底物适应性.最后, 在甲醇溶液中, 零价镁存在条件下能够顺利发生脱U型模板过程, 得到相应的烯基化产物(Scheme 26).
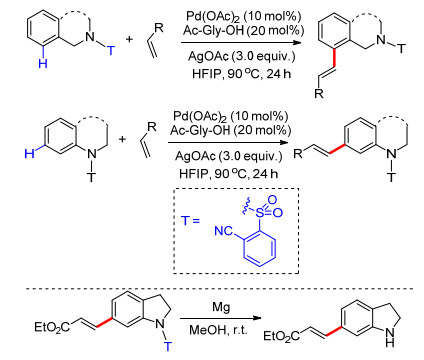
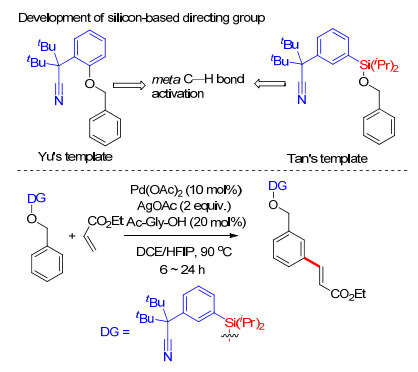
2013年, Tan课题组[36]在余金权工作的基础上, 利用硅醚基桥接丁氰基芳基构建U型模板.这种硅醚基桥接建立的U型模板具有更易构建和更易脱除的优点.硅原子的加入延长了丁氰基与芳香环的距离, 在反应中氰基更易与芳香环分离, 从而提高了反应效率, 该方法具有更高的应用价值.在钯(Ⅱ)催化剂、N-乙酰基甘氨酸(N-Ac-Gly-OH)配体和六氟异丙醇作为溶剂的条件下, 克服了苄基底物固有的电子性质, 获得了98:2的超高间/邻位选择性.反应具有良好的底物适应性(Scheme 27).
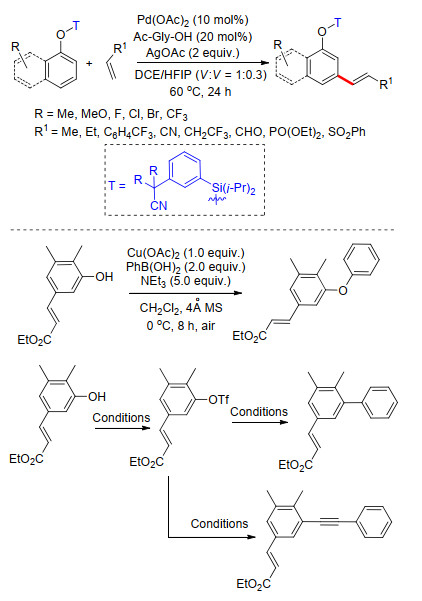
2017年, 许兆青课题组[37]以苯酚类化合物为底物, 以硅醚基桥接苯基氰基构建U型模板, 完成了苯酚类化合物间位烯基化反应.反应具有良好的底物适应性, 多种取代苯酚及萘酚能够以较高产率得到相应产物, 多种取代烯烃能够适应反应条件(Scheme 28).在温和反应条件下能够顺利脱除U型模板, 得到间位烯基化酚类化合物.由于酚羟基具有较好的反应活性, 可直接进行官能团转化, 他们对间位烯基化的苯酚衍生物进行了一系列官能团转化反应实验, 利用这种策略完成了间位烯烃化苯酚的醚化、芳基化及芳炔化(Scheme 28).该策略为一系列间烯烃化芳香族化合物的制备提供了简便易行的方法.
2018年, 周明东课题组[38]在铑催化下, 利用硅醚基桥接丁氰基芳基构建U型模板, 完成了苯酚类化合物meta-C—H键选择性烯基化反应.反应以铜盐作为氧化剂, 五氧化二钒为共氧化剂, 二氯乙烷为溶剂, 反应条件温和, 具有高效的选择性, 底物适应性良好, 烯基化试剂同样具有很好的底物适应性, 多种吸电子基团取代的烯烃都能够得到较好的产率(Scheme 29).在完成苯酚间位烯基化反应的基础上, 他们利用所得产物进一步合成了几种常见的药物分子结构(Scheme 29), 该反应具有很好的应用价值.
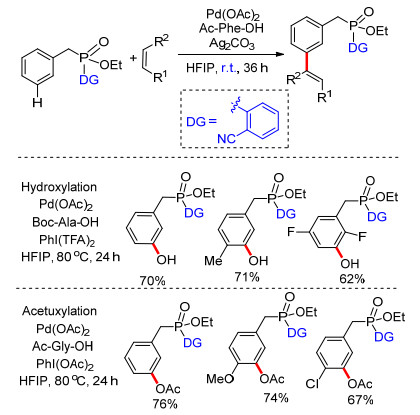
2012年以来, 芳烃meta-C—H活化得到了快速发展, 化学家们利用U型模板策略完成了一系列芳烃间位选择性官能化反应, 然而为了得到较高的转化产率, 大部分反应需要在加热或者较高的温度条件下进行.直至2016年, Maiti课题组[39]利用磷酸酯基桥接简单氰基芳基构建U型模板, 在室温条件下实现了芳烃meta-C—H键选择性烯基化.反应在醋酸钯催化下, 以N-乙酰基苯丙氨酸为外加共配体(Ac-Phe-OH), 银盐作为氧化剂, 六氟异丙醇为溶剂, 以高达84%的产率得到间位单一烯基化产物.反应具有良好的底物适应性.通过调整反应条件, 以二(三氟乙酸)碘苯为氧化剂时得到间位羟基化产物, 以醋酸碘苯为氧化剂时得到间位乙酰基化产物(Scheme 30).首次在室温条件下完成了芳烃meta-C—H键选择性官能化, 对该类反应的发展具有重要意义.
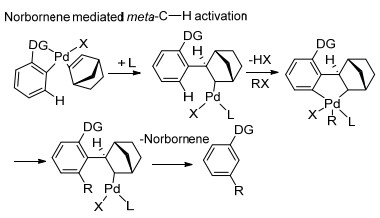
除以上U型模板策略能够实现芳烃meta-C—H键选择性官能化反应外, 余金权等课题组采用降冰片烯(NBE)作为瞬态介质, 使用常见的邻位定位基实现了间位选择性C—H键活化.反应首先经历了芳基邻位C—H键活化, 成功地将降冰片烯引入邻位作为新的瞬态导向基团, 进而发生降冰片烯导向的邻位C—H键活化, 随后降冰片烯通过β-消除过程从底物中脱离, 这样一个接力转移的过程最终完成了芳烃间位选择性官能化反应(Scheme 31).在这种机制中, 降冰片烯化合物作为过渡导向介质对促进芳烃meta-C—H键选择性官能化起到关键性作用, 反应中选用合适配体使降冰片烯参与导向的中间过渡态更稳定同样具有至关重要的作用, 化学家们[40]对该策略进行了一系列研究工作, 以下将对不同条件下瞬态媒介法进行介绍.
2015年, 余金权课题组[40a]首次报道了以降冰片烯作为瞬态导向基, 实现芳烃meta-C—H键选择性烷基化及芳基化反应的新方法.反应中以酰胺为导向基, 在Pd(OAc)2催化下, 以AgOAc为氧化剂, 通过筛选一系列吡啶类配体, 发现添加一种吡啶并呋喃环类配体, 能够以高达91%的产率得到间位烷基化产物.同样的反应条件也可用于芳基化反应中.以碘代苯为芳基化试剂, 以高达82%的产率得到间位芳基化产物.反应经历了导向基邻位C—H活化过程, 将瞬态过渡介质降冰片烯引入邻位, 接着以降冰片烯作为临时导向基团, 发生降冰片烯导向的邻位C—H活化, 进而发生官能化反应, 最后降冰片烯发生β-消除过程从体系中脱离, 得到间位官能化产物(Scheme 32).
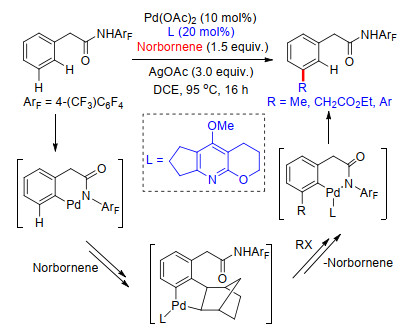
同年, 余金权课题组[41]进一步探究了该反应, 在2-甲酯降冰片烯调控下, 完成了酰胺基导向的芳烃间位烷基化及芳基化反应.反应中吡啶并呋喃结构配体对选择性C—H活化起到重要作用, 反应以碘代烃或碘代芳烃为烷基化试剂或芳基化试剂, 碘代物具有很好的底物适应性, 一系列碘代烃或碘代芳烃能够以较好的收率得到相应的产物(Scheme 33).
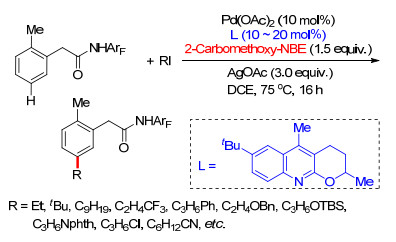
2015年, 董广彬课题组[42]利用降冰片烯作为瞬态导向基, 完成了简单芳烃胺基苯meta-C—H键选择性芳基化反应.其中, 选用能够在弱酸性条件下发生可逆邻位金属化的二甲胺基作为导向基[43], 对完成这项工作起到重要作用.通过对反应条件进行优化, 在Pd(OAc)2催化下, 以三苯基胂(AsPh3)为配体, AgOAc为氧化剂, 碘代苯为芳基化试剂, 添加降冰片烯为瞬态导向介质, 能够顺利完成苄胺类化合物meta-C—H键选择性芳基化反应(Scheme 34).反应具有良好的底物适应性, 一系列吸电基或供电基取代的苄胺都能够以较好的产率得到间位芳基化产物, 取代碘代苯同样具有良好的底物适应性, 吸电子基取代碘代苯具有非常好的反应活性.该工作进一步发展了芳烃间位选择性官能化反应, 将Pd/ NBE体系拓展到了更多芳烃间位官能化反应.
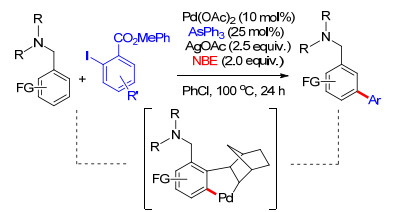
2016年, 余金权课题组[44]继续拓展底物适应性及官能化试剂, 在Pd/NBE体系中, 完成了芳烃化合物的间位选择性氯化反应.该体系表现出非常好的官能团耐受性, 利用该体系能够顺利合成一系列含间氯的苯酚及苯胺类药物分子(Scheme 35).反应中以吡啶酮类化合物为配体, 在反应过程中, 取代吡啶酮氮原子与氧原子能够首先与过渡金属钯进行配位, 接着钯转移到取代基酰基氮原子, 形成较稳定的六元环结构(Scheme 35), 因此在该配体作用下能够在反应过程中生成较稳定的中间过渡态, 从而使反应顺利发生, 取代吡啶酮类配体在反应中起到至关重要的作用.
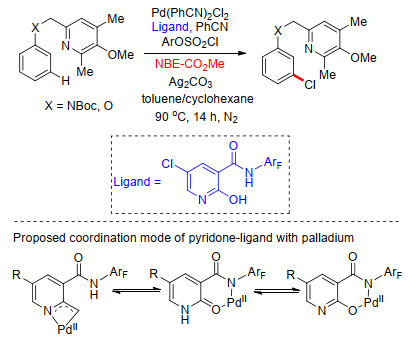
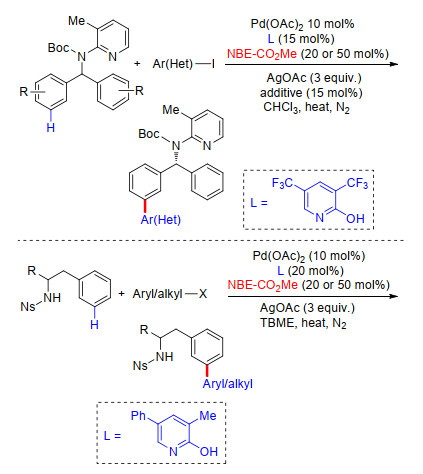
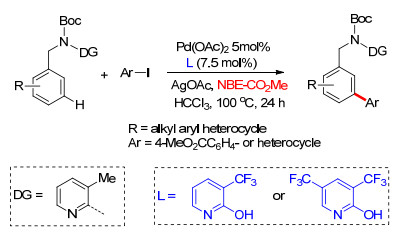
2017年, 余金权课题组[45]接着通过筛选不同配体, 在Pd/NBE体系中完成了N-取代苯乙胺类、苄胺类及2-芳基苯胺类化合物间位芳基化反应.他们以对硝基苯磺酰基(Ns)对底物氨基进行保护, Ns基团同时作为导向基团及配体基团, 与吡啶类配体共同作用, 并在降冰片烯作为瞬态介质机制中顺利发生meta-C—H键选择性活化.通过筛选大量吡啶类配体, 发现在4-乙酰基吡啶作为配体条件下, 能够以最高产率得到间位双芳基化产物.通过对照实验发现, 在不添加吡啶配体时, 反应不能发生.多种取代碘化芳烃及杂环碘代芳烃能够较好地适应反应条件, 得到相应的芳基胺类间位芳基化产物, 其中2-芳基苯胺类能够得到远端芳基的间位芳基化产物(Scheme 36), 该反应体系同样适用于克级反应的发生, 产率可达86%, 在碱性条件下, 保护基团(Ns)可顺利脱除, 表现出潜在的应用价值.
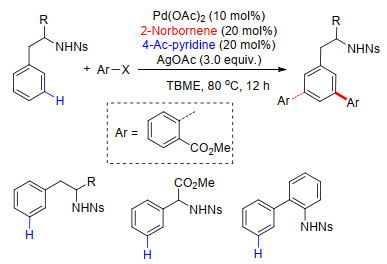
2017年, 在上述工作的基础上, 余金权课题组[46]继续对该类反应进行了拓展研究, 在Pd/NBE体系中完成了苄叔胺类化合物间位官能化反应.反应中以取代2-吡啶酮类为配体, 在不同取代2-吡啶酮配体作用下分别实现了吡啶基导向苄叔胺类化合物meta-C—H键芳基化、胺化及氯化反应.反应中以2-酯基降冰片烯为瞬态介质诱导反应进行, 反应底物具有良好的普适性, 杂环类卤代芳烃同样能够作为芳基化试剂, 得到相应的偶联产物(Scheme 37).在最优的取代吡啶酮类配体条件下, 催化剂Pd用量可以降低到2.5 mol%.此外, 吡啶导向基团易于离去, 且该体系适用于克级反应, 为芳基苄叔胺类化合物的应用研究提供了基础.
接着, 余金权课题组[47]利用2-酯基降冰片烯为瞬态介质, 诱导完成了苯乙酸类化合物meta-C—H键芳基化反应.反应中以金刚烷酸保护3-氨基-2-羟基吡啶型化合物为外加配体, 不仅能够促进羧酸导向的邻位C—H键活化过程, 利于将降冰片烯进入邻位作为瞬态导向介质, 而且能够在降冰片烯导向的C—H活化过程中与过渡金属催化剂配位, 稳定反应中间过渡态, 对间位选择性芳基化起到至关重要的作用.一系列芳基烷酸包括苯乙酸和苯甘氨酸都能够很好地适应反应条件, 以较高的产率得到相应的meta-C—H键芳基化产物(Scheme 38).同样在Pd/NBE-CO2Me体系中, 通过筛选配体, 在异喹啉作为外加配体条件下, 余金权课题组[48]完成了苄基磺酰胺类化合物间位芳基化及烷基化反应.反应中利用了一个新型吸电子基团作为导向基, 异喹啉配体的加入使导向基与催化体系发生配位作用而有利于间位官能化反应的顺利发生(Scheme 38).间位取代的苯基磺酰胺可以很容易地转化为磺酸钠、磺酸酯和磺酰胺以及通过烯化反应转化为苯乙烯, 为更多化合物的合成提供了条件.
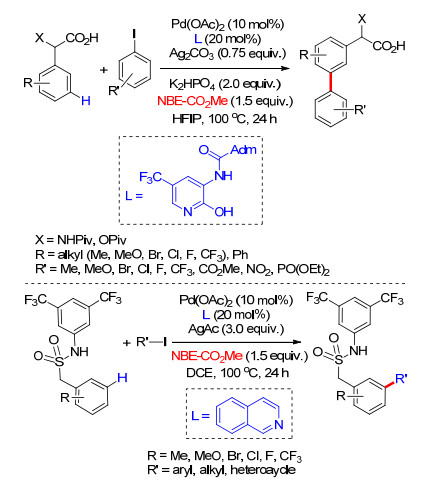
2018年, 余金权课题组[49]进一步对降冰片烯介导的远程meta-C—H键官能化反应进行了研究.他们以吡啶导向基保护的对称二芳基苄胺类化合物为模板底物, 研究了不同取代基的降冰片烯对meta-C—H活化反应的影响, 发现2-酯基降冰片烯具有最高的反应活性.筛选了磷配体及吡啶酮配体, 发现2-吡啶酮类作为配体具有更高的反应活性(Scheme 39).最后, 以对硝基苯磺酰基(Ns)保护的氨基为导向基完成了对芳基苄胺类化合物单meta-C—H键芳基化及烷基化反应, 该方法在合成中具有显著的应用价值.此外, 该反应体系具有良好的底物适应性, 多种芳基苄胺类化合物能够得到单间位官能化产物, 官能化试剂同样具有良好的适应性(Scheme 39).
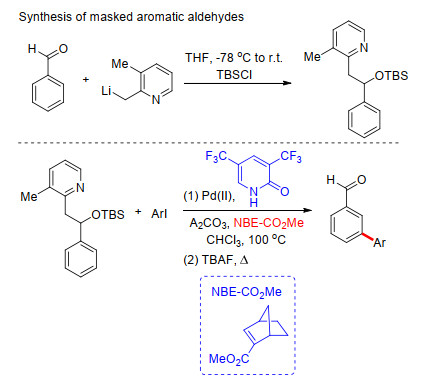
2018年, 余金权课题组[50]进一步拓展上述反应, 在吡啶酮为配体的Pd/NBE体系中完成了芳香醛类化合物的间位芳基化反应.他们首先对芳基醛类化合物进行了修饰(Scheme 40), 经修饰后吡啶基起到导向基作用.在取代2-吡啶酮为配体, Ag2CO3为氧化剂条件下, 顺利完成了吡啶基导向降冰片烯调控的芳香醛类化合物meta-C—H键芳基化反应(Scheme 40).最后, 在四丁基氟化铵(TBAF)存在条件下加热可以脱去吡啶导向基, 得到间位芳香醛类化合物.
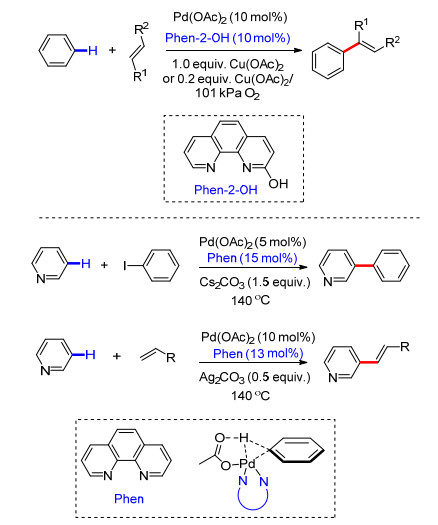
过渡金属与底物中导向基配位可以改变底物中碳原子的电性, 同时在配体的作用下, 通过空间立体效应可以实现芳烃间位选择性C—H键活化.反应中C—H键活化一般认为经历了协同金属化-去质子化过程, 再经氧化加成/迁移插入和还原消除等过程完成远程C—H键选择性官能化.段伟良课题组[51]利用2-羟基邻菲罗啉为配体, 完成了取代芳烃远程meta-C—H键选择性烯基化反应.反应中, 多种取代芳烃能够以高产率及较高的区域选择性得到间位烯基化产物, 烯基化试剂具有良好的底物适应性(Scheme 41).余金权课题组[52]利用邻菲罗啉(Phen)为配体, 在醋酸钯催化下, 完成了吡啶C(3)—H选择性烯基化及芳基化反应(Scheme 41).
2016年, Chattopadhyay课题组[53]对苯甲醛类化合物邻位和间位选择性硼化反应进行了研究, 他们将苯甲醛转化成能够与催化剂发生配位作用的亚胺基团.通过配体的筛选, 他们发现在8-氨基喹啉作为配体时, 反应以邻位C—H键活化为主, 主要生成邻位硼化产物, 选择性o/m比值最高可达97:3;以四甲基邻非罗琳为配体时, 主要生成亚胺指向的meta-C—H键硼化反应, 得到meta-C—H键硼化产物.他们对反应中电子效应及立体效应进行了验证性实验, 发现甲基亚铵反应活性最高, 并给出了可能的反应中间过渡态(Scheme 42).
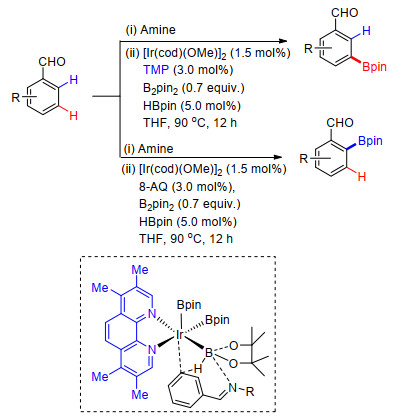
除此之外, 曾小明课题组[54]利用氨基酸类配体, 完成了吸电取代芳烃远程meta-C—H键及吡啶化合物远程C(3)—H键的选择性烯基化反应.他们对多种氨基酸类配体进行了筛选, 发现在Ac-Val-OH作为配体时, 能够以高达14:1 [C(3):C(2):C(4)=14:1:0]的比率高选择性地得到间位或C-3位烯基化产物.反应中以Pd(OAc)2为催化剂, 氧气作为氧化剂, 反应条件温和.各种吸电及供电取代吡啶能够很好地适应反应条件, 烯基化产物产率最高达96% (Scheme 43).他们通过理论计算及动力学同位素效应(KIE)实验对反应机理进行了研究, 给出了可能的反应机理.
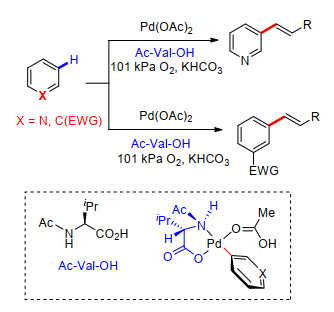
2016年, 我们课题组[55]在以上工作基础上, 对配体进行了筛选与优化, 在金刚烷甲酸作为配体及添加剂条件下, 经过双分子C—H活化过程, 完成了喹啉类化合物C(3)—H键选择性芳基化反应.反应中以Pd(OAc)2为催化剂, Ag2CO3为氧化剂, 卤代苯为芳基化试剂.多种取代喹啉能够很好的适应反应条件, 以较高的产率得到C(3)—H键芳基化产物, 多种取代卤代苯也能够顺利发生C—H活化过程, 通过双分子C—H键活化过程, 与喹啉化合物进行偶联(Scheme 44).
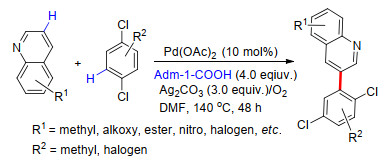
2017年, 余金权课题组[56]报道了无导向基参与的芳烃C—H键选择性烯基化及羧化反应, 利用3, 5-二(三氟甲基)-2-羟基吡啶配体与过渡金属中心配位, 并辅助参与C—H键断裂过程, 实现了一系列富电子及缺电子芳烃meta-C—H键的选择性烯基化反应.此外, 该反应还可用于杂芳香烃、氨基酸衍生物及其他复杂天然产物分子的选择性C—H键官能化.他们通过X单晶衍射发现, 2-吡啶酮配体与催化中心的配位方式与羧酸类似(Scheme 45), 该结果与DFT理论计算所得结果一致.
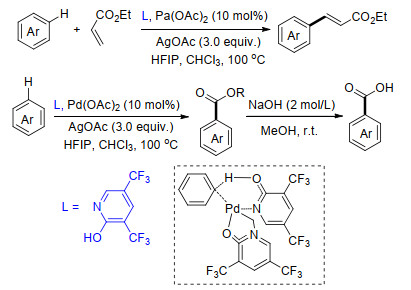
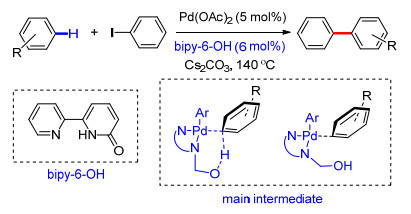
2018年, Albeniz课题组[57]利用联二吡啶酮(bipy-6-OH)为配体, 在Pd(OAc)2催化下, 以卤代烃为芳基化试剂, 顺利完成了吡啶类化合物C(3)—H键及简单取代芳烃meta-C—H键选择性芳基化反应.他们通过分离反应中间体对反应机理进行研究, 发现联二吡啶酮不仅能够作为配体, 同时在反应体系中起到碱的作用, 能够促进C—H键的断裂[58].反应具有很好的区域选择性, 其中选择性比率最高达到100:5:1 [C(3):C(2):C(4)=100:5:1].反应中多种卤代烃能够很好地适应反应条件, 以最高达98%的产率得到相应的芳基化产物(Scheme 46).
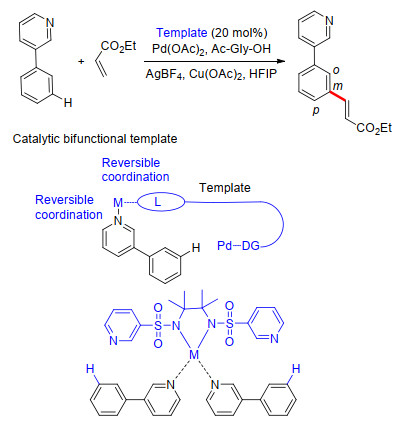
2017年, 余金权课题组[59]报道了一种催化双功能配体模板, 一方面通过模板骨架(配体)-金属-底物的可逆配位作用结合底物, 一方面通过模板(配体)另一端的导向基团引导钯催化剂至底物远端特定位点的C—H键, 实现杂环类底物远程位点C—H键选择性活化.作者选择了双磺酰胺类结构作为模板实现了该过程, 并对3-苯基吡啶类衍生物和烯烃类衍生物的普适性进行了考察, 底物间位选择性及单烯基化选择性都非常好.该反应具有反应产率高和区域选择性好的特点.这是目前C—H键活化概念的又一新突破(Scheme 47).
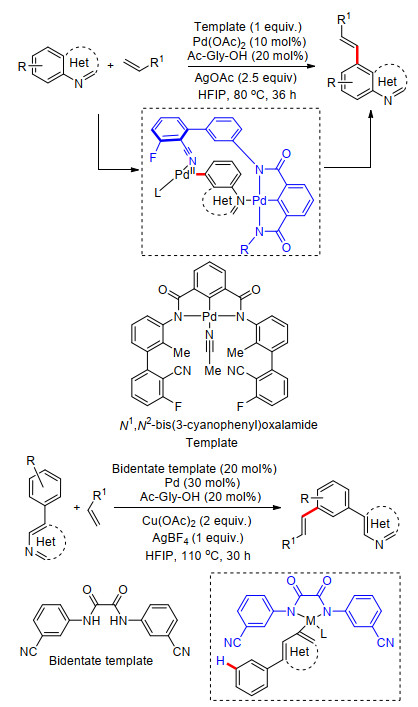
2018年, Maiti课题组[60]设计了螯合钯配合物作为双功能模板, 完成了一系列喹啉类化合物远端C(5)—H键的烯基化反应.他们给出了可能的反应机理, 反应中喹啉氮原子能够与模板中金属钯进行配位, 模板远端配体基团与催化体系中金属钯配位后, 选择性地活化喹啉C(5)—H键, 接着发生C(5)—H键烯基化反应, 最后脱除模板, 得到最终产物.该反应具有良好的底物适应性及反应活性, 多种取代喹啉及苯并噻唑能够以较高产率得到相应的产物(Scheme 48).除此之外, 他们还设计了N1, N2-双(3-氰基苯基)乙二酰胺与铜盐组合的双功能模板, 完成了一系列2-苯基吡啶及2-苯基杂芳烃化合物远端苯基meta-C—H键的活化.反应中, 在远端导向基苯基氰基与配体共同作用下, 完成了多种苯基取代杂环芳烃的远端苯基meta-C—H键烯基化反应(Scheme 48).
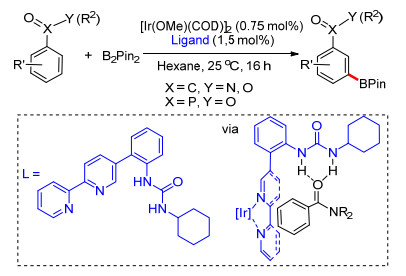
2015年, Kuninobu课题组[61]设计了铱催化芳烃化合物meta-C—H键选择性硼化反应的催化体系.在该体系中, 具有一个侧链脲部分的联吡啶衍生物作为配体能够与铱络合配位, 配体中脲结构与底物中的氢键受体之间形成相互作用, 使铱接近meta-C—H键, 从而控制区域选择性.反应具有广泛的底物适应性, 多种酰胺类、羧酸酯及磷酸酯均能适应反应条件, 得到相应的meta-C—H键硼化产物(Scheme 49).此外, 他们通过核磁数据和控制实验证明了这种分子间氢键参与诱导区域选择性的可行性.
2016年, Phipps课题组[62]利用弱配位基团离子对定向法控制铱催化的芳香季铵盐的硼化反应区域选择性, 得到多用途的meta-C—H键硼化产物(Scheme 50).反应中联吡啶配体与弱配位基团离子对共同作用, 对形成铱金属过渡态具有重要作用.该体系具有良好的底物适应性, 适用于多种简单芳基季铵盐及杂芳基季铵盐, 以较高的产率及良好的区域选择性得到相应的间位硼化产物, 为进一步官能化做好准备.该方法证明了利用非共价相互作用控制过渡金属催化区域选择性的可行性, 为离子对控制过渡金属催化区域选择性官能化反应提供了新方法.
配体参与的过渡金属催化芳烃远程C—H键官能化反应能够专一地将官能团引入芳烃的特定位置, 有效控制反应的区域选择性, 实现多种芳烃meta-C—H键及含氮杂芳烃C(3)—H键的选择性官能化反应, 包括烯基化反应、芳基化反应、烷基化反应、卤化反应、羟基化反应及乙酰氧基化反应等.这些反应为配体促进过渡金属催化芳烃间位选择性C—H键活化提供了新方法, 表现出无可替代的优越性.但是, U型模板法底物需预修饰几种特定的导向基, 瞬态导向法和双功能模板法对导向基要求较高, 底物普适性不足.特定配体调控制法是芳烃meta-C—H选择性官能化普适性最好的方法, 但是这种策略仍存在以下几个问题: (1)相关反应机理尚不明确; (2)能够利用该策略完成芳烃远程C—H键选择性官能化反应的配体种类非常有限; (3)底物适用范围有待拓展.深入研究配体在芳烃meta-C—H键选择性官能化反应中的作用机理, 探索新型多功能配体将成为芳烃远程C—H键选择性官能化反应未来的主要研究方向.
For selected reviews on transition metal catalyzed C—H activation, see: (a) Chen, X.; Engle, K. M.; Wang, D.-H.; Yu, J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094.
(b) Lyons, T. W.; Sanford, M. S. Chem. Rev. 2010, 110, 1147.
(c) Daugulis, O.; Do, H.-Q.; Shabashov, D. Acc. Chem. Res. 2009, 42, 1074.
(d) Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624.
(e) Arockiam, P. B.; Bruneau, C.; Dixneuf, P. H. Chem. Rev. 2012, 112, 5879.
(f) Kuhl, N.; Hopkinson, M. N.; Wencel-Delord, J.; Glorius, F. Angew. Chem., Int. Ed. 2012, 51, 10236.
(g) Li, B. J.; Shi, Z. J. Chem. Sci. 2011, 2, 488.
(h) Fischer, D. F.; Sarpong, R. J. Am. Chem. Soc. 2010, 132, 5926.
(i) Wang, Y.-C.; Ye, Q.-X.; Qiu, G.-S.; Liu, J.-B. Chin. J. Org. Chem. 2018, 38, 1650 (in Chinese).
(王玉超, 叶秋香, 邱观音生, 刘晋彪, 有机化学, 2018, 38, 1650.)
(a) Engle, M. K.; Mei, T.-S.; Wasa, M.Y.; Yu, J.-Q. Acc. Chem. Res. 2012, 45, 788.
(b) David, L. D.; Stuart, A. M.; Claire, L. M. Chem. Rev. 2017, 117, 8649.
Ozdemir, I.; Demir, S.; Çetinkaya, B.; Gourlaouen, C.; Maseras, F.; Bruneau, C.; Dixneuf, P. H. J. Am. Chem. Soc. 2008, 130, 1156. doi: 10.1021/ja710276x
(a) Berman, A. M.; Lewis, J. C.; Bergmann, R. G.; Ellman, J. A. J. Am. Chem, Soc. 2008, 130, 14926.
(b) Giri, R.; Yu, J.-Q. J. Am. Chem. Soc. 2008, 130, 14082.
(a) Ren, X. Y.; Wen, P.; Shi, X. K.; Wang, Y. L.; Li, J.; Yang, S. Z.; Yan, H.; Huang, G. S. Org. Lett. 2013, 15, 5194.
(b) Wen, P.; Li, Y. M.; Zhou, K.; Ma, C.; Lan, X. B.; Ma, C. W.; Huang, G. S. Adv. Synth. Catal. 2012, 354, 2135.
程辉成, 林锦龙, 张耀丰, 陈冰, 王敏, 程丽华, 马姣丽, 有机化学, 2019, 39, 318. doi: 10.6023/cjoc201807002Cheng, H.-C.; Lin, J.-L.; Zhang, Y.-F.; Chen, B.; Wang, M. C.; Li, H.; Ma, J.-L. Chin. J. Org. Chem. 2019, 39, 318 (in Chinese). doi: 10.6023/cjoc201807002
黄鸿泰, 李涛, 王家状, 秦贵平, 肖铁波, 有机化学, 2019, 39, 1511. doi: 10.6023/cjoc201903078Huang, H.-T.; Li, T.; Wang, J.-Z.; Qin, G.-P.; Xiao, T.-B. Chin. J. Org. Chem. 2019, 39, 1511 (in Chinese). doi: 10.6023/cjoc201903078
Jiang, Z.-T.; Wang, B.-Q.; Shi, Z.-J. Chin. J. Chem. 2018, 36, 950. doi: 10.1002/cjoc.201800223
(a) Yang, Y.-F.; Hong, X.; Yu, J.-Q.; Houk, K. N. Acc. Chem. Res. 2017, 50, 2853.
(b) Dey, A.; Sinha, S. K.; Achar, T. K.; Maiti, D. Angew. Chem., Int. Ed. 2019, 131, 10934.
Leow, D.; Li, G.; Mei, T. S.; Yu, J.-Q. Nature 2012, 486, 518. doi: 10.1038/nature11158
Yang, Y.-F.; Cheng, G.-J.; Liu, P.; Leow, D.; Sun, T.-Y.; Chen, P.; Zhang, X.; Yu, J.-Q.; Wu, Y.-D.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 344. doi: 10.1021/ja410485g
Wan, L.; Dastbaravardeh, N.; Li, G.; Yu, J.-Q. J. Am. Chem. Soc. 2013, 135, 18056. doi: 10.1021/ja410760f
Xu, H.-J.; Lu, Y.; Farmer, M. E.; Wang, H.-W.; Zhao, D.; Kang, Y.-S.; Sun, W.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 2200. doi: 10.1021/jacs.6b13269
Xu, H.-J.; Kang, Y.-S.; Shi, H.; Zhang, P.; Chen, Y.-K.; Zhang, B.; Liu, Z.-Q.; Zhao, J. Sun, W.-Y.; Yu, J.-Q.; Lu, Y. J. Am. Chem. Soc. 2019, 141, 76. doi: 10.1021/jacs.8b11038
Deng, Y.; Yu, J. Q. Angew. Chem., Int. Ed. 2015, 54, 888. doi: 10.1002/anie.201409860
Bera, M.; Modak, A.; Patra, T.; Maji, A.; Maiti, D. Org. Lett. 2014, 16, 5760. https://www.ncbi.nlm.nih.gov/pubmed/25310718
Li, S.; Ji, H.; Cai, L.; Li, G. Chem. Sci. 2015, 6, 5595. doi: 10.1039/C5SC01737H
Li, S.; Cai, L.; Ji, H.; Yang, L.; Li, G. Nat. Commun. 2016, 7, 10443. doi: 10.1038/ncomms10443
Fang, L.; Saint-Denis, T. G.; Taylor, B. L. H.; Ahlquist, S.; Hong, K.; Liu, S.; Han, L.; Houk, K. N.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 10702. doi: 10.1021/jacs.7b03296
Long, Y.; Lei, F.; Li, G. Adv. Synth. Catal. 2017, 359, 2235. doi: 10.1002/adsc.201700261
Maity, S.; Hoque, E.; Dhawa, U.; Maiti, D. Chem. Commun. 2016, 52, 14003. doi: 10.1039/C6CC07861C
Zhang, L.; Zhao, C.; Liu, Y.; Xu, J.; Xu, X.; Jin, Z. Angew. Chem., Int. Ed. 2017, 56, 12245. doi: 10.1002/anie.201705495
Wang, B.; Zhou, Y.; Xu, N.-N.; Xu, X.-F.; Xu, X.-H.; Jin, Z. Org. Lett. 2019, 21, 1885. doi: 10.1021/acs.orglett.9b00499
Xu, J.-C.; Chen, J.-J.; Gao, F.; Xie, S.-G.; Xu, X.-H.; Jin, Z.; Yu, J.-Q. J. Am. Chem. Soc. 2019, 141, 1903. doi: 10.1021/jacs.8b13403
Chu, L.; Shang, M.; Tanaka, K.; Chen, Q.; Pissarnitski, N.; Streckfuss, E.; Yu, J.-Q. ACS Cent. Sci. 2015, 1, 394.
Jin, Z.; Chu, L.; Chen, Y.-Q.; Yu, J.-Q. Org. Lett. 2018, 20, 425.
Fang, S.-Q; Wang, X.-B.; Yin, F.-C.; Cai, P.; Yang, H.-L.; Kong, L.-Y. Org. Lett. 2019, 21, 1841. doi: 10.1021/acs.orglett.9b00433
Bera, M.; Maji, A.; Sahoo, S. K.; Maiti, D. Angew. Chem., Int. Ed. 2015, 54, 8515. doi: 10.1002/anie.201503112
Maji, A.; Bhaskararao, B.; Singha, S.; Sunoj, R. B.; Maiti, D. Chem. Sci. 2016, 7, 3147.
Modak, A.; Patra, T.; Chowdhury, R.; Raul, S.; Maiti, D. Organometallics 2017, 36, 2418. doi: 10.1021/acs.organomet.7b00309
Bera, M.; Agasti, S.; Chowdhury, R.; Mondal, R.; Pal, D.; Maiti, D. Angew. Chem., Int. Ed. 2017, 56, 5272. doi: 10.1002/anie.201701579
Bag, S.; Jayarajan, R.; Dutta, U.; Chowdhury, R.; Mondal, R.; Maiti, D. Angew. Chem., Int. Ed. 2017, 56, 12538. doi: 10.1002/anie.201706360
Bag, S.; Jayarajan, R.; Mondal, R.; Maiti, D. Angew. Chem., Int. Ed. 2017, 56, 3182. doi: 10.1002/anie.201611360
Yang, G.-Q.; Lindovska, P.; Zhu, D.-J.; Kim, J.; Wang, P.; Tang, R.-Y.; Movassaghi, M.; Yu, J.-Q. J. Am. Chem. Soc. 2014, 136, 10807. doi: 10.1021/ja505737x
Yang, G.-Q.; Zhu, D.-J.; Wang, P.; Tang, R.-Y.; Yu, J.-Q. Chem.-Eur. J. 2018, 24, 3434. doi: 10.1002/chem.201800105
Lee, S.; Lee, H.; Tan, K. L. J. Am. Chem. Soc. 2013, 135, 18778. doi: 10.1021/ja4107034
Mi, R.-J.; Sun, J.; Kuhn, F. E.; Zhou, M.-D.; Xu, Z.-Q. Chem. Commun. 2017, 53, 13209. doi: 10.1039/C7CC07093D
Mi, R.-J.; Sun, Y.-Z.; Wang, J.-Y.; Sun, J.; Xu, Z.; Zhou, M.-D. Org. Lett. 2018, 20, 5126. doi: 10.1021/acs.orglett.8b01995
Bera, M.; Sahoo, S. K.; Maiti, D. ACS Catal. 2016, 6, 3575. doi: 10.1021/acscatal.6b00675
(a) Wang, X.-C.; Gong, W.; Fang, L. Z.; Zhu, R. Y.; Li, S.; Engle, K. M.; Yu, J.-Q. Nature 2015, 519, 334.
(b) Ye, J.; Lautens, M. Nat. Chem. 2015, 7, 863.
(c) Della C. N.; Fontana, M.; Motti, E.; Catellani, M. Acc. Chem. Res. 2016, 49, 1389.
Shen, P.-X.; Wang, X.-C.; Wang, P.; Zhu, R.-Y.; Yu, J.-Q. J. Am. Chem. Soc. 2015, 137, 11574. doi: 10.1021/jacs.5b08914
Dong, Z.; Wang, J. C.; Dong, G. B. J. Am. Chem. Soc. 2015, 137, 5887. doi: 10.1021/jacs.5b02809
(a) Ryabov, A. D.; Yatsimirskii, A. K. Inorg. Chem. 1984, 23, 789.
(b) Ryabov, A. D. Inorg. Chem. 1987, 26, 1252.
Shi, H.; Wang, P.; Suzuki, S.; Farmer, M. E.; Yu, J.-Q. J. Am. Chem. Soc. 2016, 138, 14876. doi: 10.1021/jacs.6b11055
Ding, Q.; Ye, S.; Cheng, G.; Wang, P.; Farmer, M. E.; Yu, J.-Q. J. Am. Chem. Soc. 2017, 139, 417. doi: 10.1021/jacs.6b11097
Wang, P.; Farmer, M. E.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 5125. doi: 10.1002/anie.201701803
Li, G.-C.; Wang, P.; Farmer, M. E.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 6874. doi: 10.1002/anie.201702686
Cheng, G.-L.; Wang, P.; Yu, J.-Q. Angew. Chem., Int. Ed. 2017, 56, 8183. doi: 10.1002/anie.201704411
Shi, H.; Herron, A. N.; Shao, Y.; Shao, Q.; Yu, J.-Q. Nature 2018, 558, 581. doi: 10.1038/s41586-018-0220-1
Farmer, M. E.; Wang, P.; Shi, H.; Yu, J.-Q. ACS Catal. 2018, 8, 7362. doi: 10.1021/acscatal.8b01599
Ying, C. H.; Yan, S.-B.; Duan, W.-L. Org. Lett. 2014, 16, 500. doi: 10.1021/ol4033804
(a) Ye, M. C.; Gao, G. L.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 6964.
(b) Ye, M.; Gao, G. L.; Edmunds, A. J. F.; Worthington, P. A.; Morris, J. A.; Yu, J.-Q. J. Am. Chem. Soc. 2011, 133, 19090.
Bisht, R.; Chattopadhyay, B. J. Am. Chem. Soc. 2016, 138, 84. doi: 10.1021/jacs.5b11683
Cong, X. F.; Tang, H. R.; Wu, C.; Zeng, X. M. Organometallics 2013, 32, 6565. doi: 10.1021/om400890p
He, Y. Q.; Wu, Z. Y.; Ma, C. W.; Zhou, X. Q.; Liu, X. X.; Wang, X. J.; Huang, G. S. Adv. Synth. Catal. 2016, 358, 375. doi: 10.1002/adsc.201500763
Wang, P.; Verma, P.; Xia, G.-Q.; Shi, J.; Qiao, J. X.; Tao, S.-W.; Cheng, P. T. W.; Poss, M. A.; Farmer, M. E.; Yeung, K.-S.; Yu, J.-Q. Nature 2017, 551, 489. doi: 10.1038/nature24632
Salamanca, V.; Toledo, A.; Albeniz, A. C. J. Am. Chem. Soc. 2018, 140, 17851. doi: 10.1021/jacs.8b10680
Haines, B. E.; Musaev, D. G. ACS Catal. 2015, 5, 830. doi: 10.1021/cs5014706
Zhang, Z. P.; Tanaka, K.; Yu, J.-Q. Nature 2017, 543, 538. doi: 10.1038/nature21418
Achar, T. K.; Ramakrishna, K.; Pal, T.; Porey, S.; Dolui, P.; Biswas, J. P.; Maiti, D. Chem.-Eur. J. 2018, 24, 17906. doi: 10.1002/chem.201804351
Kuninobu, Y.; Ida, H.; Nishi, M.; Kanai, M. Nat. Chem. 2015, 7, 712. doi: 10.1038/nchem.2322
Davis, H. J.; Mihai, M.; Phipps, R. J. J. Am. Chem. Soc. 2016, 138, 12759. doi: 10.1021/jacs.6b08164

图式 1 U型模板策略下芳烃远程meta-C—H键选择性官能化反应机理
Scheme 1 Remote meta-C—H functionalization of arenes directed by a U-shaped template

图式 2 苄醇及苯丙酸类化合物远程meta-C—H键选择性烯基化反应
Scheme 2 meta-Selective C—H bond olefination of benzyl alcohol derivatives and hydrocinnamic acid derivatives

图式 3 苄醇类化合物meta-C—H键选择性烯基化反应
Scheme 3 meta-Selective C—H bond olefination of benzyl alcohol derivatives

图式 4 苯丙酸类化合物meta-C—H键选择性芳基化反应
Scheme 4 meta-Selective C—H bond arylation of 3-phenyl-propanoic acid derivatives

图式 5 苯丙酸类化合物meta-C—H键选择性芳基化反应
Scheme 5 meta-Selective C—H bond arylation of 3-phenyl-propanoic acid derivatives

图式 6 苯丙酸类化合物meta-C—H键选择性芳基化反应
Scheme 6 meta-Selective C—H bond arylation of 3-phenyl-propanoic acid derivatives

图式 7 苯乙酸类化合物meta-C—H键选择性烯基化反应
Scheme 7 meta-Selective C—H bond olefination of phenylacetic acid derivatives

图式 8 苯乙酸类化合物meta-C—H键选择性烯基化反应
Scheme 8 meta-Selective C—H bond olefination of phenylacetic acid derivatives

图式 10 苯甲酸类化合物间位烯基化及乙酰基化反应
Scheme 10 Directed meta-olefination and meta-acetoxylation of benzoic acid derivatives

图式 12 苯胺类化合物间位烯基化及乙酰基化反应
Scheme 12 Directed meta-olefination and acetoxylation of aniline derivatives

图式 13 联二苯甲酸类化合物远端meta-C—H键烯基化反应
Scheme 13 Distal C—H olefination at the meta-position of biphenyl carboxylic acid

图式 14 苯基烷醇类化合物meta-C—H键烯基化反应
Scheme 14 Directed meta-olefination of arenes with tethered alcohols derivatives

图式 17 苄醇类化合物间位烯基化及碘化反应
Scheme 17 Directed meta-olefination of benzyl alcohol derivatives

图式 18 苯乙酸类化合物meta-C—H键烯基化、芳基化及碘化反应
Scheme 18 Directed meta-olefination, meta-arylation and meta-iodination of phenylacetic acid derivatives

图式 19 芳烷基二醇类化合物meta-C—H键烯基化反应
Scheme 19 Directed meta-olefination of arene-tethered diols

图式 20 苯甲磺酸类化合物间位烯基化反应
Scheme 20 Directed meta-olefination of benzylsulfonate derivatives

图式 21 苯甲磺酸类化合物间位乙酰基化及羟基化反应
Scheme 21 Directed meta-acetoxylation and meta-hydroxy-lation of benzylsulfonate derivatives

图式 22 芳基烷磺酸类化合物间位硅烷化及锗烷化反应
Scheme 22 Directed meta-silylation and germanylation of phenylalkanesulfonic acids

图式 23 芳基磺酸类及苯乙酸类化合物间位烯基化反应
Scheme 23 Directed meta-olefination of phenylalkanesulfonic acids and phenyl acetic acids

图式 24 苯甲磺酸类化合物meta-C—H键氰基化反应
Scheme 24 Directed meta-cyanation, meta-alkyation and alkenylation of arylsulfonate derivatives

图式 25 吲哚类化合物meta-C—H键烯基化、芳基化及乙酰基化反应
Scheme 25 Directed meta-olefination, meta-arylation and meta-acetoxylation of Indolines

图式 26 N-杂环类化合物远程meta-C—H键烯基化反应
Scheme 26 Directed remote meta-olefination of N-heterocycles

图式 30 芳基烷磷酸酯类化合物间位烯基化、羟基化及乙酰基化反应
Scheme 30 Directed meta-olefination, meta-hydroxylation and meta-acetoxylation of benzylphosphonate derivatives

图式 31 降冰片烯参与下芳烃远程meta-C—H键选择性官能化反应机理
Scheme 31 Norbornene mediated remote meta-C—H selective functionalization of arenes

图式 32 苯基酰胺类化合物meta-C—H键烷基化及芳基化反应
Scheme 32 Norbornene mediated meta-alkylation and meta-arylation of phenylacetic amides derivatives

图式 33 苯基酰胺类化合物meta-C—H键烷基化及芳基化反应
Scheme 33 Norbornene mediated meta-alkylation and meta-arylation of phenylacetic amides derivatives

图式 34 苄胺类化合物meta-C—H键芳基化反应
Scheme 34 Directed meta-arylation of benzylamine derivatives

图式 35 苯胺及苯酚类化合物间位氯化反应
Scheme 35 Directed meta-chlorination of anilines and phenols derivatives

图式 36 苯乙胺、苄胺及2-芳基苯胺类化合物间位芳基化反应
Scheme 36 Directed meta-arylation of Nosyl-protected phenethylamines, benzylamines and 2-aryl anilines

图式 38 苯乙酸类化合物间位芳基化反应及苄基磺酰胺类化合物间位芳基化/烷基化反应
Scheme 38 Directed meta-arylation of phenylacetic acids and meta-arylation/meta-alkylation of benzylsulfonamides

图式 39 取代二芳基甲胺及苄胺类化合物间位芳基化/烷基化反应
Scheme 39 Directed meta-arylation arylation and alkylation of diarylmethylamines and homobenzylamines

图式 40 芳香醛类化合物间位芳基化反应
Scheme 40 Directed meta-arylation of masked aromatic aldehydes derivatives

图式 41 配体促进芳烃远程meta-C—H烯基化反应及吡啶类化合物C(3)—H键选择性烯基化/芳基化反应
Scheme 41 Ligand-promoted remote meta-C—H olefination of arenes and C(3)-selective C—H olefination/arylation of pyridines

图式 42 配体促进苯甲醛类化合物远程meta-C—H硼化反应
Scheme 42 Ligand-promoted remote meta-C—H borylation of aromatic aldehydes

图式 43 氨基酸配体促进的芳烃远程meta-C—H键烯基化反应及吡啶类化合物C(3)—H键选择性烯基化反应
Scheme 43 Amino acid ligand-promoted remote meta-C—H olefination of arenes and C(3)-selective C—H olefination of pyridines

图式 44 配体促进喹啉C(3)—H键选择性芳基化反应
Scheme 44 Ligand-promoted C(3)-selective C—H arylation of quinolines

图式 45 配体促进芳烃meta-C—H键选择性芳基化反应
Scheme 45 Ligand-promoted selective meta-C—H arylation of arenes

图式 46 吡啶酮配体促进的芳烃远程meta-C—H键烯基化反应及吡啶类化合物C(3)—H键选择性烯基化反应
Scheme 46 Bipy-6-OH ligand-promoted remote meta-C—H olefination of arenes and C(3)-selective C—H olefination of pyridines

图式 47 双功能配体模板下2苯基吡啶远程C—H键选择性烯基化反应
Scheme 47 Remote C—H olefination of 2-phenyl pyridine directed by a catalytic bifunctional template

图式 48 双功能配体模板下杂环芳烃及苯基杂环芳烃远程C—H键选择性烯基化反应
Scheme 48 Remote C—H olefination of benzoheterocycle and phenyl heterocyclic directed by a catalytic bifunctional template

图式 49 配体导向的芳烃化合物meta-C—H键硼化反应
Scheme 49 Ligand directed meta-selective C—H borylation of arenes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:








