Scheme 1.
Methods for the synthesis of internal gem-diboro- nates from ketones
Iron-Catalyzed Deoxygenative Diborylation of Ketones to Internal gem-Diboronates
Zeyu He , Min Fan , Jia'neng Xu , Yue Hu , Lu Wang , Xudong Wu , Chungu Xia , Chao Liu

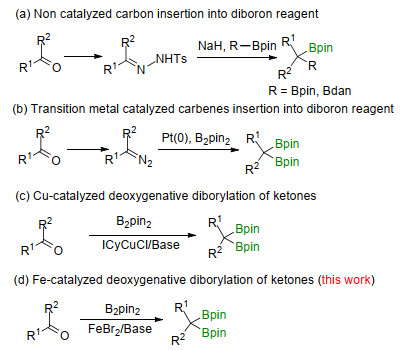
gem-Diboronates as novel dimetalated compounds are highly valuable building blocks in organic synthesis due to their selective mono- or di-functionalization.[1] Among them, the internal gem-diboronates are particular attractive for the construction of quaternary carbon centres.[1a, 1b, 2] The non-catalytic cross-coupling of internal gem-diborona- tes with electrophiles (eg. halides and N-heteroaromatic N-oxides) and the addition reaction of internal gem-di- boronates with vinyl, carbonyl and carboxylic derivatives have been proven to be an ideal methodology for C—C bond formation. Most of these reactions proceed through the α-monoboryl carbanion which was generated by base promoted monodeborylation of gem-diboronates.[1e, 2b, 3] Furthermore, the transition metal catalyzed cross coupling of one boron group of internal gem-diboro- nates with halides was an efficient method to construct mono boronic ester compounds, which could be further functionalized under appropriate reaction conditions.[1c, 4] Meanwhile, the C—B bond of internal gem-diboronates could be transformed to C—F bond to achieve gem- difluorides through the deboronofluorination.[5] In addition to the C—B bond transformation, the typical 1, 2-metalate rearrangement of boron was the other reaction mode of the internal gem-diboronates.[6] Although internal gem-diboronates have been deemed as attractive bifunctional reagents, the methods to access this kind of compounds are still limited. Early synthetic methods of deprotonation-alkylation of 1, 1-dibornates[1c, 1p, 1q, 1ag, 3b, 7] and alkylation of 1, 1, 1-tri- (boronates)[8] could lead to internal gem-diboronates. However, the preparation of such start materials are complicated. Moreover, the alkylarenes, [8] gem-dihaloal- kanes, [6] alkynes, [9] vinylboronates[10] and fully substituted benzylic lithiated carbamates[3a] have been also successfully applied to afford internal gem-diboronates. Furthermore, the strategies of direct diborylation of N-tosylhydro- drazones[11] (Scheme 1a) or diazoalkanes[12] (Scheme 1b) which pre-prepared form their corresponding ketones were also used to achieve the gem-diboronates (Scheme 1b). Compared to these reported methods, direct deoxygenative gem-diborylation of widely available ketones to access internal gem-diboronates would be more attractive. In 2017, our group reported an ICyCuCl (ICy=1, 3-dicyclohexylimidazol-2-ylidene) catalyzed deoxygenative diborylation of ketones to prepare these internal gem-diboronates (Scheme 1c).[13] Although various internal gem-diboronates could be well synthesized, the use of expensive NHC ligand ICy and the pre-preparation of ICyCuCl catalyst promoted us to further optimize this catalytic system with more practical and economical catalyst for this transformation.
Recently, our group have developed an iron catalyzed deoxygenative diborylation of aldehydes to synthesize corresponding gem-diboronates with B2pin2 as boron source, in which the addition of Fe-Bpin species to carbonyl group of aldehyde was assumed to be the crucial step.[14] Inspired by our previous studies, the deoxygenative diborylation reaction of ketones with the iron catalytic system was examined. 4-Phenylbutan-2-one (1a) was first selected as the model substrate and toluene was used as solvent. After considerable efforts, the reaction was achieved by using the following optimal conditions: B2pin2 (2.8 equiv.), FeBr2 (10 mol%), NaOtBu (1.3 equiv.) in toluene at 100 ℃ (Table 1, Entry 1). As shown in Table 1, the pre-catalyst has the crucial effects on the formation of gem-diboronates. Among all the catalysts tested, FeBr2 showed the best reactivity (Entries 2~4). And it should be emphasized that the necessity for iron has been confirmed by the result that no desired internal gem-diborylation product has been achieved in the absence of FeBr2 (Entry 5). In these reaction conditions, the amount of NaOtBu also played a key role. Catalytic amounts of NaOtBu (40 mol%) could only resulted in trace amount of 2a formation (Entry 6). However, the α-OH Boronate species which was generated by the borylation of ketone was observed. And the excess of NaOtBu (2.0 and 3.0 equiv.) would supress the formation of the desired compound 2a (Entries 7 and 8).
 下载:
导出CSV
下载:
导出CSV
 |
||
| Entry | Variation from "standard conditions" | Yieldb/% |
| 1 | None | 73 |
| 2 | FeF2 instead of FeBr2 | 14 |
| 3 | FeCl2 instead of FeBr2 | 39 |
| 4 | Fe(OAc)2 instead of FeBr2 | 59 |
| 5 | No FeBr2 | n.d. |
| 6 | NaOtBu (0.4 equiv.) | Trace |
| 7 | NaOtBu (2.0 equiv.) | 59 |
| 8 | NaOtBu (3.0 equiv.) | 32 |
| a Conditions: 1a (0.5 mmol), B2pin2 (2.8 equiv. 1.4 mmol), NaOtBu (1.3 equiv., 0.65 mmol), catalyst (10 mol%, 0.05 mmol), toluene (3.0 mL), 100 ℃, 5 h; b Determined by gas chromatography (GC) analysis using naphthalene as the internal standard. | ||
With the optimized conditions in hand, the substrates scope of this protocol have been explored (Table 2). Various of simple aliphatic ketones such as 4-phenylbutan-2- one, 2-pentanone and 3-pentanone proceeded smoothly to afford the corresponding internal gem-diboronates in moderate to good yields under this condition (2a, 2u and 2v). Notably, substrates bearing ketal (2r, 2s) and alkene (2t) group proceeded well to give the desired products with sufficient yields. Substrates such as piperonyl acetone, 2-tetralone, 7-methoxy-2-tetralone and indan-2-one were well gem-diborylated to afford their desired products (2b, 2o, 2p and 2q). Cyclic ketones such as five-membered ring, six-membered ring and seven-membered ring were all compatible in this reaction (2h~2n). Sterically demanding substrates containing an ortho substituent, proceeded smoothly to give the corresponding deoxygenative gem- diboronates 2i in yield of 40%. Aryl C-Cl group was also suitable with this protocol (2d), and provided a potential for further transformation. Aryl C-CF3, and aromatic methoxyl group were also tolerant to afford the target com-pounds (2e~2g). Acetone-d6 was also used to prepare corresponding deuteration internal gem-diboronates (2w) in 47% yield.
下载:
导出CSV
 |
 |
| a Conditions: 1 (1.0 mmol), B2pin2 (2.8 equiv. 2.8 mmol), NaOtBu (1.3 equiv. 1.3 mmol), FeBr2 (10 mol%, 0.1 mmol), toluene (5.0 mL), 100 ℃ for 5 h in N2 atmosphere, yields are based on isolated products; b 0.5 mmol scale, toluene (3.0 mL); c B2pin2 (2.0 mmol), NaOtBu (1.5 mmol). |
To demonstrate the practical applicability of this transformation, the 10.0 mmol scale reactions of 1s were carried out under standard conditions. As a result, 2.60 g (60% yield) of 2s were isolated, indicating the possibility to scale up this procedure (Scheme 2).
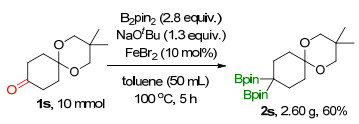
Conditions and regents: 1s (10.0 mmol), B2pin2 (2.8 equiv. 28.0 mmol), NaOtBu (1.3 equiv. 13.0 mmol), FeBr2 (10 mol%, 1.0 mmol), toluene (50.0 mL), 100 ℃ for 5 h in N2 atmosphere. Yields are based on isolated products
As above mentioned, internal gem-diboronates could be used as a building block to construct quaternary carbon centres through the transformation of C—B bond to C—C bond. Herein, the C—C bond formation reactions were explored by using this compounds. Initially, acetone, the common organic solvent, was gem-diborylated to afford internal gem-diboronate 2wa under the standard conditions (Scheme 3). Then, some modes of transformation of 2wa has been applied. Tertiary boronic ester 3a has been prepared in yield of 83% in the presence of internal gem-diboronates 2wa and 1-bromooctane via deborylative borylalkylation process.[11] Compound 4a, which containing a quandary carbon centres could also be achieved by the coupling reaction of 3a with 2-bromothiophene.[15] Furthermore, 1, 4-dione 3b has also been synthesized by dual-functionalization of 2wa through the deoxygenative enolization with carboxylic acid and methyl bromoacetate.[1e]
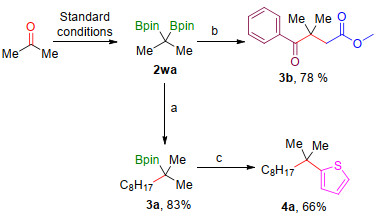
Reaction conditions: (a) 2wa (0.39 mmol), 1-bromoocatane (0.3 mmol), NaOtBu (0.9 mmol), THF, r.t., 12 h; (b) 2wa (0.375 mmol), MeLi (0.625 mmol, 1.6 mol/L in diethyl ether), benzoic acid (0.25 mmol), methyl bromoacetate (0.5 mmol), THF; (c) 2-bromothiophene (0.36 mmol), nBuLi (0.36 mmol, 1.6 mol/L in hexanes), 3a (0.3 mmol), NBS (0.36 mmol), THF.
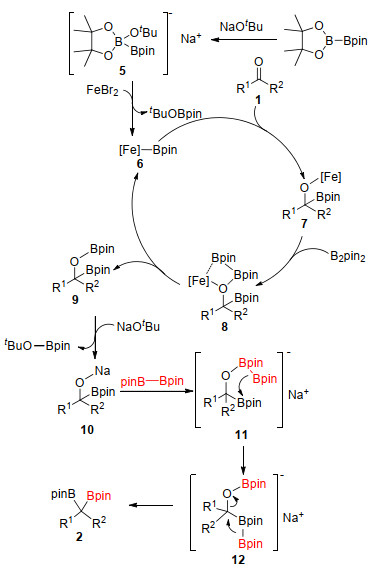
Based on the previous reports[16] and our works, [13, 14] the reaction pathway was proposed (Scheme 4). Initially, 'ate' complex 5, which was generated by interaction of NaOtBu with B2pin2, reacted with FeBr2 to generate iron boryl species 6. Then the ketones inserted into the Fe—B bond of 6 to afford complex 7. Subsequently, σ-bond metathesis with B2pin2 occurred to afford the α-OBpin boronates 9 and regenerated 6. The α-OBpin boronates 9 could interact with NaOtBu to generate 10, followed by the reaction with B2pin2 to afford adduct 11. Then the intramolecular transfer of the boryl group occurred to produce tetracoordinate boryl species 12, which underwent 1, 2-migration to afford the final internal gem-diboronates 2.
In summary, a Fe-catalyzed/ base-promoted deoxygenative diborylation of ketones was demonstrated. Various aliphatic ketones were gem-diborylated under current reaction conditions. More importantly this novel strategy shows great potential in synthetic chemistry through the gram scale experiments and the elaboration of the final internal gem-diboronate products.
All reactions were isolated from moisture and oxygen by a nitrogen atmosphere with a sealed tube. All glassware was oven dried at 110 ℃ for hours and cooled down under vacuum. Toluene was purified using Pure Solv 7-SDS solvent drying system. Unless otherwise noted, materials were obtained from commercial suppliers and used without further purification. Thin layer chromatography (TLC) employed glass 0.25 mm silica gel plates. Flash chromatography columns were packed with 100~200 mesh silica gel or through SepaBeamTM Machine SPB-3006012. Gas chromatographic analyses were performed on a GC-2010 Plus gas chromatography instrument with an FID detector. GC-MS spectra were recorded on a GCMS-QP2010 SE. The High Resolution MS analyses were performed on an Agilent 6530 Accurate-Mass Q-TOF LC/MS with ESI mode. NMR spectra were recorded on a 400 MHz for 1H NMR and 101 MHz for 13C NMR, using tetramethylsilane as an internal reference and CDCl3 as the solvent. Chemical shift values for protons were reported downfield from tetramethylsilane and were referenced to residual proton of TMS. Carbon nuclear magnetic resonance spectra (13C NMR) were recorded at 101 MHz. Chemical shifts for carbons were reported downfield from tetramethylsilane and were referenced to the carbon resonance of CDCl3 (δ 77.0). The boron-bound carbon was not detected due to quadrupolar relaxation.
In glove box, a 25 mL resealable reaction tube of solvent flask equipped with a stirrer bar was charged with NaOtBu (124.9 mg, 1.3 mmol), B2pin2 (711.2 mg, 2.8 mmol), FeBr2 (21.6 mg, 0.1 mmol), and 5 mL of toluene. The tube was sealed with a Teflon screw cap and taken out of the glove box. The mixture was stirred at room temperature for 5 min. Subsequently, ketone (1.0 mmol) was added under nitrogen atmosphere. Then, the tube was sealed and the mixture was heated at 100 ℃ with stirring for 5 h. Upon completion, ethyl acetate was added to the mixture to quench the reaction. The pure product was obtained by flash column chromatography on silica gel.
2, 2'-(4-Phenylbutane-2, 2-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2a):[12b] White solid, 265.4 mg, 69% yield. m.p. 104~105 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.26~7.18 (m, 4H), 7.15~7.11 (m, 1H), 2.60~2.56 (m, 2H), 1.85~1.80 (m, 2H), 1.23 (s, 24H), 1.16 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 143.8, 128.5, 128.1, 125.3, 82.9, 36.7, 34.2, 24.7, 24.6, 16.0.
2, 2'-(4-(Benzo[d][1, 3]dioxol-5-yl)butane-2, 2-diyl)bis-(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2b): White solid, 267.9 mg, 62% yield. m.p. 129~130 ℃; 1H NMR (400 MHz, CDCl3) δ: 6.72~6.63 (m, 3H), 5.89 (s, 2H), 2.51~2.47 (m, 2H), 1.80~1.76 (m, 2H), 1.23 (s, 24H), 1.13 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 147.3, 145.3, 137.8, 121.1, 109.1, 107.9, 100.6, 83.0, 37.0, 34.0, 24.8, 24.6, 16.0; HRMS (ESI) calcd for C23H36B2O6Na [M+ Na]+ 453.2596, found 453.2600.
2, 2'-(4-(4-Methoxyphenyl)butane-2, 2-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2c): White solid, 257.7 mg, 62% yield. m.p. 108~109 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.11 (d, J=8.4 Hz, 2H), 6.79 (d, J=8.4 Hz, 2H), 3.75 (s, 3H), 2.52 (t, J=8.8 Hz, 2 H), 1.80 (t, J=8.8 Hz, 2H), 1.22 (s, 24H), 1.15 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 157.4, 135.7, 129.2, 113.4, 82.8, 55.0, 36.8, 33.1, 24.6, 24.5, 15.9; HRMS (ESI) calcd for C23H38B2- O5Na [M+Na]+ 439.2803, found 439.2808.
2, 2'-(1-(4-Chlorophenyl)propane-2, 2-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2d): White solid, 288.6 mg, 71% yield. m.p. 79~80 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.20~7.15 (m, 4H), 2.84 (s, 2H), 1.23 (s, 12H), 1.20 (s, 12H), 0.98 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 140.2, 131.2(3), 131.1(7), 127.6, 83.1, 38.5, 24.8, 24.6, 15.8; HRMS (ESI) calcd for C21H33B2ClO4Na [M+Na]+ 429.2151, found 429.2158.
2, 2'-(1-(3, 4-Dimethoxyphenyl)propane-2, 2-diyl)bis-(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2e): Colorless oil, 174.9 mg, 40% yield. 1H NMR (400 MHz, CDCl3) δ: 6.82~6.72 (m, 3H), 3.85 (s, 3H), 3.83 (s, 3H), 2.83 (s, 2H), 1.24 (s, 12H), 1.20 (s, 12H), 1.03 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 148.0, 146.9, 134.3, 121.8, 113.6, 110.5, 83.0, 55.7, 55.6, 38.7, 24.8, 24.5, 15.9; HRMS (ESI) calcd for C23H38B2O6Na [M+Na]+ 455.2752, found 455.2757.
2, 2'-(1-(3-(Trifluoromethyl)phenyl)propane-2, 2-diyl)bis-(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2f): Colorless oil, 262.2 mg, 60% yield. 1H NMR (400 MHz, CDCl3) δ: 7.61~7.60 (m, 1H), 7.41~7.38 (m, 2H), 7.33~7.29 (m, 1H), 2.93 (s, 2H), 1.24 (s, 12H), 1.20 (s, 12H), 1.00 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 142.7, 133.5, 130.0 (q, J=31.6 Hz), 127.8, 126.5 (q, J=3.8 Hz), 124.5 (q, J=273.7 Hz), 122.4 (q, J=3.9 Hz), 83.3, 39.1, 24.8, 24.5, 15.8; HRMS (ESI) calcd for C22H33B2F3O4Na [M+Na]+ 463.2415, found 463.2417.
2, 2'-(1-(4-Methoxyphenyl)propane-2, 2-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2g):[13] White solid, 220.5 mg, 55% yield. m.p. 69~70 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.16 (d, J=8.0 Hz, 2H), 6.75 (d, J=8.0 Hz, 2H), 3.75 (s, 3H), 2.82 (s, 2 H), 1.23 (s, 12H), 1.20 (s, 12H), 0.98 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 157.5, 133.8, 130.7, 112.9, 83.0, 55.0, 38.2, 24.7, 24.6, 15.8.
2, 2'-(Cyclopentane-1, 1-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2h):[11a] White solid, 130.2 mg, yield 40%. m.p. 50~51 ℃(lit.[11a] m.p. 52~53 ℃); 1H NMR (400 MHz, CDCl3) δ: 1.81 (t, J=6.8 Hz, 4H), 1.53~1.49 (m, 4H), 1.22 (s, 24H); 13C NMR (101 MHz, CDCl3) δ: 82.8, 30.5, 27.2, 24.5.
2, 2'-(2-Methylcyclopentane-1, 1-diyl)bis(4, 4, 5, 5-tetra-methyl-1, 3, 2-dioxaborolane) (2i): Colorless oil, 135.6 mg, 40% yield. 1H NMR (400 MHz, CDCl3) δ: 2.44~2.36 (m, 1H), 1.95~1.81 (m, 2H), 1.74~1.60 (m, 2H), 1.53~1.42 (m, 1H), 1.23~1.20 (m, 25H), 0.98 (d, J=6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 82.7(2), 82.7(0), 38.2, 35.5, 28.8, 24.8, 24.6, 24.4, 19.6; HRMS (ESI) calcd for C18H34B2O4Na [M+Na]+ 359.2541, found 359.2545.
2, 2'-(Cyclohexane-1, 1-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2j): White solid, 111.3 mg, 66% yield. m.p. 59~60 ℃ (lit.[11a] 59~60 ℃); 1H NMR (400 MHz, CDCl3) δ: 1.66~1.63 (m, 4H), 1.45~1.37 (m, 6H), 1.22 (s, 24H); 13C NMR (101 MHz, CDCl3) δ: 82.7, 29.5, 26.7, 26.4, 24.
2, 2'-(4-Methylcyclohexane-1, 1-diyl)bis(4, 4, 5, 5-tetra-methyl-1, 3, 2-dioxaborolane) (2k): White solid, 191.6 mg, 55% yield. m.p. 45~46 ℃; 1H NMR (400 MHz, CDCl3) δ: 2.02~1.96 (m, 2H), 1.63~1.58 (m, 2H), 1.38~1.18 (m, 27H), 0.98~0.87 (m, 2H), 0.82 (d, J=6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 82.9, 82.7, 35.1, 32.5, 29.2, 24.7, 24.6, 23.0; HRMS (ESI) calcd for C19H36B2O4Na [M+Na]+ 373.2697, found 373.2702.
2, 2'-(3-Methylcyclohexane-1, 1-diyl)bis(4, 4, 5, 5-tetra-methyl-1, 3, 2-dioxaborolane) (2l): White solid, 217.8 mg, 62% yield. m.p. 57~58 ℃; 1H NMR (400 MHz, CDCl3) δ: 1.96~1.93 (m, 2H), 1.62~1.56 (m, 2H), 1.37~1.26 (m, 3H), 1.21 (s, 12H), 1.18 (s, 12H), 0.95~0.89 (m, 1H), 0.82 (d, J=6.4 Hz, 3H), 0.78~0.71 (m, 1H); 13C NMR (101 MHz, CDCl3) δ: 82.7, 82.5, 37.8, 35.3, 32.1, 28.8, 26.3, 24.5(4), 24.4(9), 24.4(5), 23.2; HRMS (ESI) calcd for C19H40B2NO4 [M+NH4]+ 368.3143, found 368.3146.
2, 2'-(Cycloheptane-1, 1-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2m): White solid, 185.4 mg, 53% yield. m.p. 36~37 ℃ (lit.[11a] 36~37 ℃); 1H NMR (400 MHz, CDCl3) δ: 1.82~1.79 (m, 4H), 1.60~1.57 (m, 4H), 1.51~1.49 (m, 4H), 1.23 (s, 24H); 13C NMR (101 MHz, CDCl3) δ: 82.7, 31.6, 28.9, 27.8, 24.6.
2, 2'-(4-Phenylcyclohexane-1, 1-diyl)bis(4, 4, 5, 5-tetra-methyl-1, 3, 2-dioxaborolane) (2n): White solid, 265.7 mg, 64% yield. m.p. 90~91 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.28~7.13 (m, 5H), 2.46~2.40 (m, 1H), 2.17~2.15 (m, 2H), 1.84~1.83 (m, 2H), 1.54~1.46 (m, 4H), 1.28 (s, 12H), 1.22 (s, 12H); 13C NMR (101 MHz, CDCl3) δ: 148.4, 128.2, 126.8, 125.7, 83.1, 82.9, 44.5, 34.1, 29.7, 24.8, 24.7; HRMS (ESI) calcd for C24H38B2O4Na [M+ Na]+ 435.2854, found 435.2859.
2, 2'-(1, 2, 3, 4-Tetrahydronaphthalene-2, 2-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2o):[13] White solid, 274.9 mg, 72% yield. m.p. 106~107 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.14~7.05 (m, 4H), 2.99 (s, 2H), 2.89 (t, J=6.6 Hz, 2H), 2.05 (t, J=6.6 Hz, 2H), 1.22 (s, 12H), 1.19 (s, 12H); 13C NMR (101 MHz, CDCl3) δ: 138.2, 136.7, 128.9, 128.8, 125.0, 124.8, 83.0, 32.6, 28.0, 26.0, 24.5, 24.4.
2, 2'-(7-Methoxy-1, 2, 3, 4-tetrahydronaphthalene-2, 2-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2p): White solid, 243.8 mg, 59% yield. m.p. 98~99 ℃; 1H NMR (400 MHz, CDCl3) δ: 6.90 (d, J=8.0 Hz, 1H), 6.64~6.60 (m, 2H), 3.74 (s, 3H), 2.90 (s, 2H), 2.75 (t, J=6.6 Hz, 2H), 1.97 (t, J=6.4 Hz, 2H), 1.17 (s, 12H), 1.14 (s, 12H); 13C NMR (101 MHz, CDCl3) δ: 156.8, 139.1, 129.6, 128.8, 113.3, 111.5, 82.9, 55.0, 32.9, 27.2, 26.2, 24.4(2), 24.4(0); HRMS (ESI) calcd for C23H40B2NO5 [M+NH4]+ 432.3093, found 432.3097.
2, 2'-(2, 3-Dihydro-1H-indene-2, 2-diyl)bis(4, 4, 5, 5-tetra-methyl-1, 3, 2-dioxaborolane) (2q):[13] Pale yellow solid, 59.3 mg, 32% yield. m.p. 109~110 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.18~7.16 (m, 2H), 7.08~7.06 (m, 2H), 3.23 (s, 4H), 1.18 (s, 24H); 13C NMR (101 MHz, CDCl3) δ: 144.2, 125.6, 124.0, 83.3, 37.2, 24.6.
2, 2'-(1, 4-Dioxaspiro[4.5]decane-8, 8-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2r):[2a] White solid, 247.0 mg, 63% yield. m.p. 139~140 ℃; 1H NMR (400 MHz, CDCl3) δ: 3.92 (s, 4H), 1.82 (t, J=6.0 Hz, 4H), 1.61 (t, J=6.2 Hz, 4H), 1.21 (s, 24H); 13C NMR (101 MHz, CDCl3) δ: 109.2, 83.0, 64.0, 34.9, 26.8, 24.6.
2, 2'-(9, 9-Dimethyl-1, 5-dioxaspiro[5.5]undecane-3, 3-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2s): White solid, 271.7 mg, 62% yield. m.p. 129~130 ℃; 1H NMR (400 MHz, CDCl3) δ: 3.47 (s, 4H), 1.74 (s, 6H), 1.22 (s, 26H), 0.94 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 97.7, 82.9, 69.6, 32.3, 30.1, 25.2, 24.6, 22.6; HRMS (ESI) calcd for C23H43B2O6 [M+H]+ 437.3246, found 437.3251.
2, 2'-(6-Methylhept-5-ene-2, 2-diyl)bis(4, 4, 5, 5-tetra-methyl-1, 3, 2-dioxaborolane) (2t): Colorless oil, 217.2 mg, 60% yield. 1H NMR (400 MHz, CDCl3) δ: 5.16 (t, J=7.4Hz, 1H), 1.99~1.93 (m, 2H), 1.67 (s, 3H), 1.61 (s, 3H), 1.58~1.53 (m, 2H), 1.23 (s, 24H), 1.09 (s, 3H); 13C NMR (101 MHz, CDCl3) δ: 130.5, 125.4, 82.7, 34.0, 26.1, 25.6, 24.6, 24.5, 17.4, 15.8; HRMS (ESI) calcd for C20H39B2O4 [M+H]+ 365.3034, found 365.3041.
2, 2'-(Pentane-3, 3-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2u):[13] Colorless oil, 68.3 mg, 42% yield. 1H NMR (400 MHz, CDCl3) δ: 1.66 (q, J=7.6 Hz, 4H), 1.22 (s, 24H), 0.83 (t, J=7.2 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ: 82.8, 24.7, 21.1, 11.2.
2, 2'-(Pentane-2, 2-diyl)bis(4, 4, 5, 5-tetramethyl-1, 3, 2-dioxaborolane) (2v):[13] Colorless oil, 85.0 mg, 52% yield. 1H NMR (400 MHz, CDCl3) δ: 1.53~1.49 (m, 2H), 1.35~1.17 (m, 26H), 1.05 (s, 3H), 0.88 (t, J=7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 82.8, 36.3, 24.6, 20.6, 15.9, 14.9.
2, 2'-(Propane-2, 2-diyl-1, 1, 1, 3, 3, 3-d6)bis(4, 4, 5, 5-tetra-methyl-1, 3, 2-dioxaborolane) (2w): Colorless oil, 71.1 mg, 47% yield. 1H NMR (400 MHz, CDCl3) δ: 1.21 (s, 24H); 13C NMR (101 MHz, CDCl3) δ: 82.8, 24.6; HRMS (ESI) calcd for C15H24D6B2O4Na [M+Na]+ 325.2605, found 325.2606.
In glove box, a 250 mL resealabel reaction tube of solvent flask equipped with a stirrer bar was charged with NaOtBu (1.25 g, 13.0 mmol), B2pin2 (7.11 g, 28.0 mmol), FeBr2 (215.7 mg, 1 mmol), and 50 mL of toluene. The tube was sealed with a Teflon screw cap and taken out of the glove box. The mixture was stirred at room temperature for 5 min. Subsequently, 3, 3-dimethyl-1, 5-dioxaspiro[5.5]- undecan-9-one (1.98 g, 10.0 mmol) was added under nitrogen atmosphere. Then, the tube was sealed and the mixture was heated at 100 ℃ with stirring for 5 h. Upon completion, ethyl acetate was added to the mixture to quench the reaction. The pure product 2s was obtained by flash column chromatography on silica gel.
Compound 3a was prepared according literature.[11] In glove box, a 25 mL resealable reaction tube of solvent flask equipped with a stirrer bar was charged with NaOtBu (86.5 mg, 0.9 mmol), 2wa (115.4 mg, 0.39 mmol), and 3 mL of tetrahydrofuran (THF). The tube was sealed with a Teflon screw cap and taken out of the glove box. After that, 1-bromooctane (0.3 mmol, 57.9 mg) was added under N2 atmosphere. Then the mixture was stirred at room temperature for 12 h. Upon completion, ethyl acetate was added to the mixture to quench the reaction. The pure product was obtained by flash column chromatography on silica gel. 4, 4, 5, 5-Tetra-methyl-2-(2-methyldecan-2-yl)- 1, 3, 2-dioxaborolane (3a): colorless oil, 70.3 mg, 83% yield. 1H NMR (400 MHz, CDCl3) δ: 1.26~1.22 (m, 26H), 0.91 (s, 6H), 0.88 (t, J=6.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 82.8, 41.3, 31.9, 30.5, 29.6, 29.3, 26.5, 24.9, 24.7, 22.7, 14.1.
Compound 3b was prepared according literature.[1e] In glove box, a 25 mL resealable reaction tube of solvent flask equipped with a stirrer bar was charged with benzoic acid (30.5 mg, 0.25 mmol), 2wa (111.0 mg, 0.375 mmol), and 2 mL of THF. The tube was sealed with a Teflon screw cap and taken out of the glove box. Then the mixture was cooled to -30 ℃ and the MeLi (0.625 mmol, 1.6 mol/L in Et2O) was added and stirred for 5 min. Then the cooling bath was removed and the reaction mixture was heated at 100 ℃ for 8 h. After that, the mixture was cooled to room temperature and Methyl bromoacetate (0.5 mmol, 76.5 mg) was added and stirred at 100 ℃ for 6 h. Upon completion, ethyl acetate was added to the mixture to quench the reaction. The pure product was obtained by flash column chromatography on silica gel. Methyl 3, 3-dimethyl-4-oxo-4-phenylbutanoate (3b): colorless oil, 43.3 mg, 78% yield. 1H NMR (400 MHz, CDCl3) δ: 7.62 (d, J=8.0 Hz, 2H), 7.47~7.37 (m, 3H), 3.63 (s, 3H), 2.79 (s, 2H), 1.42 (s, 6H); 13C NMR (101 MHz, CDCl3) δ: 208.6, 171.9, 139.1, 130.5, 128.0, 127.3, 51.5, 45.9, 45.0, 26.4; HRMS (ESI) calcd for C13H16O3Na [M+Na]+ 243.0997, found 243.0994.
Compound 4a was prepared according literature.[15] At N2 atmosphere, a 25 mL resealable reaction tube of solvent flask equipped with a stirrer bar was charged with 2-bromothiophene (58.7 mg, 0.36 mmol) and 2 mL of THF. The mixture was cooled to -78 ℃, then nBuLi (0.36 mmol, 1.6 mol/L in hexane) was added. Removing the cooling bath and the mixture was stirred at room temperature for 1 h. After that, the mixture was cooled at -78 ℃ again, and 3a (0.3 mmol, 84.7 mg) was added as a solution in THF (1 mL) and stirred at this temperature for 1 h. Then the N-bromosuccinamide (NBS) (0.36 mmol, 64.1 mg) as a solution of THF (2 mL) was added and stirred for another 1 h. Saturated solution of Na2S2O3 (2 mL) was added and the mixture was allowed to warm to room temperature. Upon completion, ethyl acetate was used to extract the aqueous layer. The pure product 4a was obtained by flash column chromatography on silica gel. 2-(2-Methyldecan-2-yl)thiophene (4a):[15] colorless oil, 46.8 mg, 66% yield. 1H NMR (400 MHz, CDCl3) δ: 7.12~7.11 (m, 1H), 6.91~6.89 (m, 1H), 6.78~6.77 (m, 1H), 1.61~1.57 (m, 2H), 1.34 (s, 6H), 1.29~1.16 (m, 12H), 0.86 (t, J=6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ: 156.4, 126.2, 122.3, 121.9, 45.8, 37.5, 31.9, 30.2(2), 30.1(7), 29.5, 29.3, 24.7, 22.7, 14.1.
Supporting Information 1H NMR and 13C NMR spectra of all prodoucts. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
(a) Nallagonda, R.; Padala, K.; Masarwa, A. Org. Biomol. Chem. 2018, 16, 1050.
(b) Wu, C.; Wang, J. Tetrahedron Lett. 2018, 59, 2128.
(c) Zhang, Z.-Q.; Yang, C.-T.; Liang, L.-J.; Xiao, B.; Lu, X.; Liu, J.-H.; Sun, Y.-Y.; Marder, T. B.; Fu, Y. Org. Lett. 2014, 16, 6342.
(d) Zhang, Z.-Q.; Zhang, B.; Lu, X.; Liu, J.-H.; Lu, X.-Y.; Xiao, B.; Fu, Y. Org. Lett. 2016, 18, 952.
(e) Sun, W.; Wang, L.; Xia, C.; Liu, C. Angew. Chem., Int. Ed. 2018, 57, 5501.
(f) Brown, H. C.; Rhodes, S. P. J. Am. Chem. Soc. 1969, 91, 4306.
(g) Coombs, J. R.; Zhang, L.; Morken, J. P. Org. Lett. 2015, 17, 1708.
(h) Endo, K.; Hirokami, M.; Shibata, T. J. Org. Chem. 2010, 75, 3469.
(i) Endo, K.; Ohkubo, T.; Hirokami, M.; Shibata, T. J. Am. Chem. Soc. 2010, 132, 11033.
(j) Endo, K.; Ohkubo, T.; Ishioka, T.; Shibata, T. J. Org. Chem. 2012, 77, 4826.
(k) Endo, K.; Ohkubo, T.; Shibata, T. Org. Lett. 2011, 13, 3368.
(l) Hong, K.; Liu, X.; Morken, J. P. J. Am. Chem. Soc. 2014, 136, 10581.
(m) Jo, W.; Kim, J.; Choi, S.; Cho, S. H. Angew. Chem., Int. Ed. 2016, 55, 9690.
(n) Kim, J.; Park, S.; Park, J.; Cho, S. H. Angew. Chem., Int. Ed. 2016, 55, 1498.
(o) Li, H.; Zhang, Z.; Shangguan, X.; Huang, S.; Chen, J.; Zhang, Y.; Wang, J. Angew. Chem., Int. Ed. 2014, 53, 11921.
(p) Matteson, D. S.; Moody, R. J. J. Am. Chem. Soc. 1977, 99, 3196.
(q) Matteson, D. S.; Moody, R. J. Organometallics 1982, 1, 20.
(r) Matteson, D. S.; Moody, R. J.; Jesthi, P. K. J. Am. Chem. Soc. 1975, 97, 5608.
(s) Park, J.; Lee, Y.; Kim, J.; Cho, S. H. Org. Lett. 2016, 18, 1210.
(t) Potter, B.; Edelstein, E. K.; Morken, J. P. Org. Lett. 2016, 18, 3286.
(u) Potter, B.; Szymaniak, A. A.; Edelstein, E. K.; Morken, J. P. J. Am. Chem. Soc. 2014, 136, 17918.
(v) Sun, C.; Potter, B.; Morken, J. P. J. Am. Chem. Soc. 2014, 136, 6534.
(w) Xu, S.; Shangguan, X.; Li, H.; Zhang, Y.; Wang, J. J. Org. Chem. 2015, 80, 7779.
(x) Zhan, M.; Li, R.-Z.; Mou, Z.-D.; Cao, C.-G.; Liu, J.; Chen, Y.-W.; Niu, D. ACS Catal. 2016, 6, 3381.
(y) Iacono, C. E.; Stephens, T. C.; Rajan, T. S.; Pattison, G. J. Am. Chem. Soc. 2018, 140, 2036.
(z) Kim, J.; Shin, M.; Cho, S. H. ACS Catal. 2019, 8503.
(aa) Kim, J.; Lee, E.; Cho, S. H. Asian J. Org. Chem. 2019, 8, 1664.
(ab) Lee, H.; Lee, Y.; Cho, S. H. Org. Lett. 2019, 21, 5912.
(ac) Kim, J.; Hwang, C.; Kim, Y.; Cho, S. H. Org. Process Res. Dev. 2019, 23, 1663.
(ad) Kim, J.; Cho, S. H. ACS Catal. 2019, 9, 230.
(ae) Park, J.; Choi, S.; Lee, Y.; Cho, S. H. Org. Lett. 2017, 19, 4054.
(af) Kim, J.; Ko, K.; Cho, S. H. Angew. Chem., Int. Ed. 2017, 56, 11584.
(ag) Lee, Y.; Park, J.; Cho, S. H. Angew. Chem., Int. Ed. 2018, 57, 12930.
(a) Zheng, P.; Zhai, Y.; Zhao, X.; Xu, T. Chem. Commun. 2018, 54, 13375.
(b) Zheng, P.; Zhai, Y.; Zhao, X.; Xu, T. Org. Lett. 2019, 21, 393.
(a) Zhao, H.; Tong, M.; Wang, H.; Xu, S. Org. Biomol. Chem. 2017, 15, 3418.
(b) Hwang, C.; Jo, W.; Cho, S. H. Chem. Commun. 2017, 53, 7573.
(c) Liu, X.; Deaton, T. M.; Haeffner, F.; Morken, J. P. Angew. Chem., Int. Ed. 2017, 56, 11485.
(a) Harris, M. R.; Wisniewska, H. M.; Jiao, W.; Wang, X.; Bradow, J. N. Org. Lett. 2018, 20, 2867.
(b) Sandford, C.; Aggarwal, V. K. Chem. Commun. 2017, 53, 5481.
(c) Liu, Y.; Zhang, W. Chin. J. Org. Chem. 2016, 36, 2249 (in Chinese).
(刘媛媛, 张万斌, 有机化学, 2016, 36, 2249.)
(d) Chen, D.; Xu, M.-H. Chin. J. Org. Chem. 2017, 37, 1589 (in Chinese).
(陈雕, 徐明华, 有机化学, 2017, 37, 1589.)
(e) Xu, X.; Cheng, R.; Qiu, Z.; Pan, C. Chin. J. Org. Chem. 2018, 38, 3078 (in Chinese).
(许新彬, 程若飞, 邱早早, 潘成岭, 有机化学, 2018, 38, 3078.)
Li, Z.; Wang, Z.; Zhu, L.; Tan, X.; Li, C. J. Am. Chem. Soc. 2014, 136, 16439. doi: 10.1021/ja509548z
Masaki, S.; Michael, S.; Ikuhiro, N.; Katsuhiro, S.; Takuya, K.; Tamejiro, H. Chem. Lett. 2006, 35, 1222. doi: 10.1246/cl.2006.1222
(a) Edelstein, E. K.; Grote, A. C.; Palkowitz, M. D.; Morken, J. P. Synlett 2018, 29, 1749.
(b) Li, X.; Hall, D. G. Angew. Chem., Int. Ed. 2018, 57, 10304.
(a) Palmer, W. N.; Zarate, C.; Chirik, P. J. J. Am. Chem. Soc. 2017, 139, 2589.
(b) Zhang, L.; Huang, Z. J. Am. Chem. Soc. 2015, 137, 15600.
(a) Brown, H. C.; Scouten, C. G.; Liotta, R. J. Am. Chem. Soc. 1979, 101, 96.
(b) Espinal-Viguri, M.; Woof, C. R.; Webster, R. L. Chem.-Eur. J. 2016, 22, 11605.
(c) He, T.; Li, B.; Liu, L.-C.; Wang, J.; Ma, W.-P.; Li, G.-Y.; Zhang, Q.-W.; He, W. Chem.-Eur. J. 2019, 25, 966.
(d) Yukimori, D.; Nagashima, Y.; Wang, C.; Muranaka, A.; Uchiyama, M. J. Am. Chem. Soc. 2019, 141, 9819.
(e) Yuma, N.; Naofumi, T. Lett. Org. Chem. 2017, 14, 243.
Lee, S.; Li, D.; Yun, J. Chem.-Asian J. 2014, 9, 2440. doi: 10.1002/asia.201402458
(a) Li, H.; Shangguan, X.; Zhang, Z.; Huang, S.; Zhang, Y.; Wang, J. Org. Lett. 2014, 16, 448.
(b) Cuenca, A. B.; Cid, J.; García-López, D.; Carbó, J. J.; Fernández, E. Org. Biomol. Chem. 2015, 13, 9659.
(a) Abu Ali, H.; Goldberg, I.; Kaufmann, D.; Burmeister, C.; Srebnik, M. Organometallics 2002, 21, 1870.
(b) Wommack, A. J.; Kingsbury, J. S. Tetrahedron Lett. 2014, 55, 3163.
Wang, L.; Zhang, T.; Sun, W.; He, Z.; Xia, C.; Lan, Y.; Liu, C. J. Am. Chem. Soc. 2017, 139, 5257. doi: 10.1021/jacs.7b02518
He, Z.; Zhu, Q.; Hu, X.; Wang, L.; Xia, C.; Liu, C. Org. Chem. Front. 2019, 6, 900. doi: 10.1039/C9QO00007K
Bonet, A.; Odachowski, M.; Leonori, D.; Essafi, S.; Aggarwal, V. K. Nat. Chem. 2014, 6, 584. doi: 10.1038/nchem.1971
(a) Bedford, R. B.; Brenner, P. B.; Carter, E.; Gallagher, T.; Murphy, D. M.; Pye, D. R. Organometallics 2014, 33, 5940.
(b) Laitar, D. S.; Tsui, E. Y.; Sadighi, J. P. J. Am. Chem. Soc. 2006, 128, 11036.
(c) Nakagawa, N.; Hatakeyama, T.; Nakamura, M. Chem.-Eur. J. 2015, 21, 4257.
(d) Zhao, H.; Dang, L.; Marder, T. B.; Lin, Z. J. Am. Chem. Soc. 2008, 130, 5586.
(e) Zhou, Y.; Wang, H.; Liu, Y.; Zhao, Y.; Zhang, C.; Qu, J. Org. Chem. Front. 2017, 4, 1580.

Scheme 2 Gram scale synthesis of internal gem-diboronates
Conditions and regents: 1s (10.0 mmol), B2pin2 (2.8 equiv. 28.0 mmol), NaOtBu (1.3 equiv. 13.0 mmol), FeBr2 (10 mol%, 1.0 mmol), toluene (50.0 mL), 100 ℃ for 5 h in N2 atmosphere. Yields are based on isolated products

Scheme 3 Further application of internal gem-diboronates
Reaction conditions: (a) 2wa (0.39 mmol), 1-bromoocatane (0.3 mmol), NaOtBu (0.9 mmol), THF, r.t., 12 h; (b) 2wa (0.375 mmol), MeLi (0.625 mmol, 1.6 mol/L in diethyl ether), benzoic acid (0.25 mmol), methyl bromoacetate (0.5 mmol), THF; (c) 2-bromothiophene (0.36 mmol), nBuLi (0.36 mmol, 1.6 mol/L in hexanes), 3a (0.3 mmol), NBS (0.36 mmol), THF.
Table 1. Reaction parameters of the deoxygenative gem- diborylation of ketonesa
|
|
||
| Entry | Variation from "standard conditions" | Yieldb/% |
| 1 | None | 73 |
| 2 | FeF2 instead of FeBr2 | 14 |
| 3 | FeCl2 instead of FeBr2 | 39 |
| 4 | Fe(OAc)2 instead of FeBr2 | 59 |
| 5 | No FeBr2 | n.d. |
| 6 | NaOtBu (0.4 equiv.) | Trace |
| 7 | NaOtBu (2.0 equiv.) | 59 |
| 8 | NaOtBu (3.0 equiv.) | 32 |
| a Conditions: 1a (0.5 mmol), B2pin2 (2.8 equiv. 1.4 mmol), NaOtBu (1.3 equiv., 0.65 mmol), catalyst (10 mol%, 0.05 mmol), toluene (3.0 mL), 100 ℃, 5 h; b Determined by gas chromatography (GC) analysis using naphthalene as the internal standard. | ||
 下载: 导出CSV
下载: 导出CSV
Table 2. Substrates scope of the iron catalyzed deoxygenative diborylation of ketonesa
|
|
|
|
| a Conditions: 1 (1.0 mmol), B2pin2 (2.8 equiv. 2.8 mmol), NaOtBu (1.3 equiv. 1.3 mmol), FeBr2 (10 mol%, 0.1 mmol), toluene (5.0 mL), 100 ℃ for 5 h in N2 atmosphere, yields are based on isolated products; b 0.5 mmol scale, toluene (3.0 mL); c B2pin2 (2.0 mmol), NaOtBu (1.5 mmol). |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们