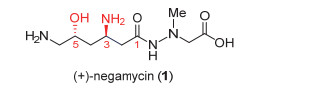
图 1.
耐格霉素的结构
Figure 1.
Structure of (+)‒negamycin

抗生素通过抑制致病菌的生长, 使得许多感染性疾病得到有效治疗, 延长了人类的寿命, 但是同时也促进了细菌的进化, 导致耐药菌株的产生[1].目前, 细菌抗药性成为威胁人类健康的严重问题, 且预计到2050年, 每年由于细菌抗药性问题引起的人死亡数将达到上千万并将耗费100万亿美元[2].而在众多抗药性细菌中, 由于革兰氏阴性菌的细胞壁上包裹一层具有保护作用的外膜, 从而阻滞一些抗生素、染料等进入细胞, 且其引起的抗药性最为严重.过去半个世纪, 临床上没有引入有效的新类型抗生素. 2017年世界卫生组织宣布, 寻找新的抗生素抑制革兰氏阴性菌感染是当前最为迫切的任务之一[3], 而天然产物结构丰富多样, 一直以来是人们发现通过新作用机制抵御耐药细菌的新药物分子的重要灵感源泉[2].
1970年, Umezawa小组[4]从绛红褐链霉菌(Strepto- myces purpeofuscus)的培养基中分离得到一种具有抗革兰氏阴性菌包括铜绿假单胞菌(P. aeruginosa)以及克雷白氏肺炎杆菌(K. pneumoniae)的抗生素‒耐格霉素(negamycin, 或称负霉素)(图 1).进一步的研究发现表明, 耐格霉素通过影响蛋白RNA的翻译启动、链增长以及终止引起密码子错译进而抑制细菌蛋白的合成, 达到抑制细菌生长的作用[5].此外, 耐格霉素具有对提前终止密码子(PTCs)通读的生理活性, 在杜氏肌营养不良症(DMD)小鼠模型显示出抑制抗肌营养不良蛋白无义基因突变的作用[6].无义基因突变可导致基因解读提前终止, 从而影响蛋白的合成.耐格霉素对无义突变基因的通读作用可以促使蛋白完整地表达, 是杜氏肌营养不良症的潜在对症药物分子.
正因为耐格霉素是一个结构简单且具有良好的广谱抗革兰氏阴性菌活性的天然产物, 其结构一经报道就引起了合成化学家和药物化学家极大的研究兴趣[7]. 1972年, Shibahara小组以D-半乳糖醛酸为原料, 首次完成了耐格霉素的不对称合成, 并确定了其绝对构型[7a], 随后有二十多条合成路线相继报道.在众多的合成路线中, 以Oliver小组[7d]的八步合成法路线最短, 他们以手性的伯醇衍生物为原料, 利用手性辅基介导的不对称Mannich反应构建手性C3中心.但是由于其Mannich反应选择性低(dr≈5/1)以及路线初期使用易爆物叠氮化钠, 限制了该路线的可操作性和实验室放大.与此同时, 有些小组进行了该化合物的结构类似物的构效关系研究, 以筛选出具有更优良抗菌活性的先导化合物[8].为了进一步提高合成效率、促进该类型化合物活性分子库的快速建立, 便于药物分子的高通量筛选, 本文在已有文献的基础上[7d], 设计改进合成步骤, 以便发展高效的合成策略, 实现高对映体纯的耐格霉素的合成.
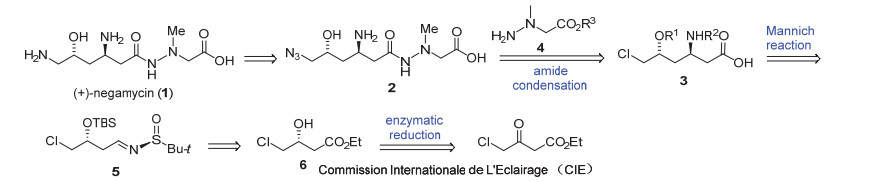
虽然耐格霉素结构并不复杂, 但是众多的合成路线由于操作繁琐, 不易放大, 而且难以获得高光学纯度的目标化合物.其中通过还原叠氮引入C-6位的氨基是文献合成路线中常用的构建氨基官能团的方法之一, 很多合成路线在全合成的早期就引入叠氮官能团.为了积累后续中间体, 沿用相似的策略, 放大合成时就会在操作上带来一定的安全隐患.为了减少易致爆叠氮原料(如NaN3)的使用, 本策略采用后期引入叠氮官能团的方法完成C-6位氨基的引入(Scheme 1).而C-1位的酰胺肼片段则由羧酸3与文献已知的肼衍生物4[9]通过酰胺缩合方式构建.关键的合成中间体3可以通过不对称的Mannich反应得到, 通过底物亚胺上的手性亚砜(Ellman试剂)[10]取代基控制反应的立体选择性, 从而建立C-3位的手性中心.亚胺中间体5可由生物酶催化还原大量获得的手性化合物6 [11, 8f]通过简单的还原和氨基缩合来制备.
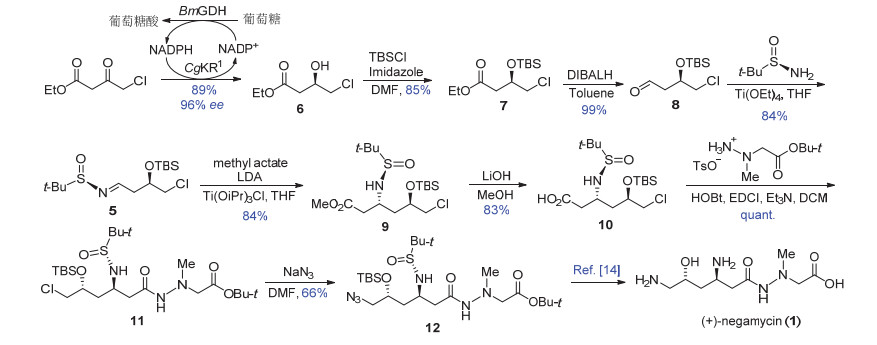
尽管利用过渡金属催化不对称还原是很成熟的方法[12], 生物催化由于其条件温和及高度立体专一性更符合绿色化学的要求, 并在一些药物合成中成功地应用[13].例如, 酶促不对称还原3-羰基-4-氯丁酸乙酯已成功应用于他汀类药物侧链的工业生产[14].我们受此启发, 将来源于平滑假丝酵母(Candida glabrata)的具备相反立体选择性的大肠杆菌重组酮还原酶CgKR1作为催化剂[11], 耦合来源于巨大芽孢杆菌(Bacillus megaterium)葡萄糖脱氢酶BmGDH进行NADPH辅酶再生, 在中性条件下避免了低温反应, 以及硼烷还原法后处理过程中底物酯基水解的问题.利用该方法, 可大量制得手性羟基化合物6 (96% ee).以6原料, 在碱性条件下利用二甲基叔丁基硅基氯(TBSCl)保护羟基, 随后在低温条件下, 二异丙基铝化氢(DIBALH)选择性地将酯基还原为醛基(Scheme 2).所得丁醛衍生物在四乙氧基钛作用下与(R)-(-)-叔丁基亚磺酰胺(Ellman试剂)发生缩合反应[15], 以84%的收率制备得到Mannich反应前体亚胺衍生物5.
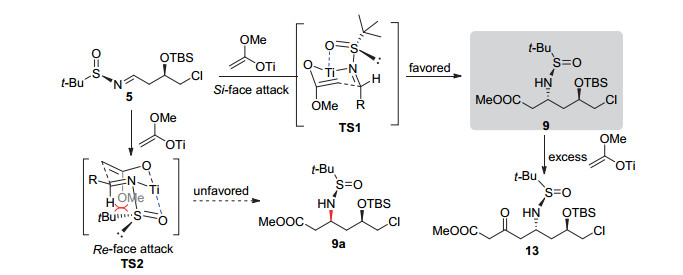
在获得亚胺衍生物5后, 首先利用在二异丙胺基锂(LDA)作用下, 对烯醇锂盐与亚胺的Mannich反应进行尝试, 并未检测到所需的加成产物生成.分析可能是烯醇锂盐与亚胺衍生物的酯基发生了Claisen缩合.根据文献报道, 烯醇锂盐能与三异丙氧基氯化钛发生转金属化反应生成烯醇钛盐, 该钛盐不仅能促进Mannich反应的进行, 并能提高该反应的立体选择性[13].随后Ellman等[16]发展了烯醇钛盐参与的Mannich反应条件, 将烯醇锂盐与三异丙氧基氯化钛在低温下搅拌40 min, 原位制备烯醇钛盐, 随后滴入亚胺衍生物5, 保持低温反应3 h, 所得产物分离鉴定, 得到了单一化合物13 (Scheme 3).我们推测烯醇钛盐首先与亚胺发生Mannich反应生成化合物9, 该化合物进一步与体系中过量的烯醇钛盐发生Claisen缩合转化为13.进一步控制加成反应1 h, 以84%的分离产率得到单一的非对映异构体9, 且未发现Claisen缩合产物13的生成.在该反应中, 烯醇钛盐与亚胺形成椅式六元环过渡态, 由于亚砜上叔丁基的立体位阻效应, 使得过渡态TS1中相邻亚胺的侧链(R)也处于直立键, 因此亚胺的Si面接受亲核试剂进攻, 产生单一的手性胺衍生物.显然, 六元环过渡态中, 两个相邻大位阻取代基均处于平伏键反而是不利的, 因此过渡态TS2 不是优势过渡态.而文献中类似反应过程只取得5/1的非对映选择性和41%的分离收率[7d], 可见远端的官能团(C-6取代基)可能对反应的控制产生一定的影响.
在碱性条件下, Mannich产物的甲酯发生水解反应转化为相应的羧酸, 该羧酸在缩合条件下与文献已知的肼衍生物4发生酰胺缩合反应, 以两步83%总收率得到化合物11, 完成了所有碳骨架的建立.相对于目标产物, 需要在C-6位引入氨基, 本路线仍采用经典的叠氮还原法构建氨基.在加热条件下, 化合物11与叠氮化钠发生SN2取代反应, 以66%的收率制备得到文献已知关键中间体12[7d].化合物12可通过简单的氢化、酸脱除保护两步转化即可得到天然产物耐格霉素.
从商品化的的3-羰基-4-氯丁酸乙酯出发, 首先利用双酶耦法方便地构建出C-5位的手性羟基, 进而利用Ellman辅基控制的不对称Mannich反应构建C-3位的手性中心, 并在合成后期引入具有潜在制备风险的叠氮官能团, 以8步29%的总收率完成了耐格霉素的形式合成.该合成策略为快速建立耐格霉素类似物分子库提供了一种新的合成路线, 有助于开展类似物的合成, 推动构效关系研究, 加快寻找高活性抗菌药物的步伐.
核磁数据用Bruker AM-400 (400 MHz)核磁共振仪测定的; 红外光谱用Digital FT-IRspectrometer或Bruker-Tensor 27测定; 质谱用Shimadzu LCMS-2010EV (ESI) mass spectrometer或Agilent G6100 LC/MSD(ESI)质谱仪测定; 高分辨质谱采用Bruker Daltonics, Inc. APEXIII 7.0 TESLA FTMS (ESI)质谱仪测定; 比旋光值通过JASCO P-1030 polarimeter测定.
所有试剂除了特殊说明都是直接使用; 四氢呋喃与甲苯都是经金属钠和二苯甲酮回流至蓝色后蒸出; 二氯甲烷是由氢化钙干燥新蒸.
含有5 g重组pET-28(a)+-CgKR1大肠杆菌湿细胞的细胞裂解液(47 mL, 总活力3000 U, 细胞悬浮于500 mmol•L-1磷酸钾缓冲液, pH 6.0, 冰浴超声破碎)与无水葡萄糖(27 g), BmGDH(葡萄糖脱氢酶)冻干酶粉(1.5 g, 总活力4500 U), 底物3-羰基-4-氯丁酸乙酯酮(15 g, 91.5 mmol)及乙醇(3.0 mL)在25 ℃搅拌36 h.期间通过pH滴定仪以碳酸钠水溶液恒定pH 6.0.反应结束后体系加入氯化钠(27 g), 以乙酸乙酯萃取, 以离心机分相.合并有机相, 无水硫酸镁干燥, 水泵真空除溶剂, 柱层析纯化[V(乙酸乙酯)/V(石油醚)=1/6]得淡黄色油状液体6[11] 13.5 g, 产率89%, 96% ee. [α]D25-21.4 (c 1.0 CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.23~4.30 (m, 1H), 4.19 (q, J=7.1 Hz, 2H), 3.57~3.66 (m, 2H), 3.21 (d, J=5.2 Hz, 1H), 2.57~2.70 (m, 2H), 1.29 (t, J=7.2 Hz, 3H).
在100 mL茄形瓶中加入咪唑(6.80 g, 100 mmol), 氮气保护下溶于20 mL超干N, N-二甲基甲酰胺(DMF)中, 用冰水浴冷却至0 ℃后, 滴加仲醇化合物6 (3.3 g, 20 mmol), 并加入TBSCl (3.9 g, 26 mmol), 加料完成后常温下搅拌反应24 h.反应结束后加入饱和碳酸氢钠水溶液(5 mL)淬灭反应, 加入20 mL水充分搅拌, 分离有机相, 用乙醚(30 mL×3)萃取水相, 合并有机相, 用饱和氯化钠水溶液洗涤, 无水硫酸钠干燥, 过滤, 浓缩, 柱层析纯化[V(乙酸乙酯)/V(石油醚)=1/100]得到黄色油状物[17] 4.82 g, 产率85%. [α]D25+18.0 (c 1.03, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 4.34~4.28 (m, 1H), 4.20~4.08 (m, 2H), 3.55~3.47 (m, 2H), 2.68 (dd, J=15.2, 4.8 Hz, 1H), 2.52 (dd, J=15.2, 7.2 Hz, 1H), 1.27 (t, J=7.2 Hz, 3H), 0.87 (s, 9H), 0.09 (d, J=15.6 Hz, 6H).
在氮气气氛下, 将化合物7 (11.6 mmol, 3.28 g)置于250 mL茄形瓶中, 用100 mL新蒸甲苯溶解, 并将反应温度降至-78 ℃, 待温度稳定后滴加二异丁基氢化铝(1.5 mol•L-1甲苯溶液, 8.5 mL, 12.8 mmol), 反应1 h后, 加入无水甲醇(3 mL)淬灭反应.恢复至室温, 向体系中加入饱和酒石酸钠钾溶液(50 mL), 充分搅拌2 h至体系澄清.分离有机相, 水相用乙醚(30 mL×3)萃取, 合并有机相, 用饱和氯化钠水溶液洗涤, 无水硫酸钠干燥, 过滤, 浓缩, 柱层析纯化[V(乙酸乙酯)/V(石油醚)=1/40], 得到黄色油状化合物8[17] 2.73 g, 产率99%. [α]D25+17.1 (c 1.01, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 9.80 (t, J=1.6 Hz, 1H), 4.41~4.37 (m, 1H), 3.54 (dd, J=11.2, 4.8 Hz, 1H), 3.47 (dd, J=10.8, 6.4 Hz, 1H), 2.81~2.65 (m, 2H), 0.87 (s, 9H), 0.10 (d, J=14.4Hz, 6H).
在100 mL圆底烧瓶中加入(R)-(+)叔丁基亚磺酰胺(1.7 g, 14.2 mmol), 氮气保护下溶于40 mL新蒸四氢呋喃中, 置于冰水浴中冷却至0 ℃后, 依次滴加化合物8 (2.8 g, 11.8 mmol)的四氢呋喃(THF) (12 mL)的溶液, 四乙氧基钛(5 mL, 23.6 mmol), 加料完成后恢复至室温, 加热回流反应1 h.停止加热并冷却至室温, 加入饱和氯化钠水溶液(5 mL)淬灭反应, 硅藻土过滤, 用乙醚洗涤滤饼, 将滤液浓缩以除去大部分的THF.向所得的残余物中加入乙醚稀释, 分离有机相, 水相用乙醚(20 mL×3)萃取, 合并有机相, 用无水硫酸钠干燥, 过滤, 浓缩后用柱层析纯化[V(乙酸乙酯)/V(石油醚)=1/20]得到黄色油状化合物5 3.4 g, 产率84%. [α]D25-117.0 (c 1.19, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 8.12 (t, J=4.8 Hz, 1H), 4.30~4.24 (m, 1H), 3.55 (dd, J=10.8, 4.8 Hz, 1H), 3.50 (dd, J=11.2, 6.4 Hz, 1H), 2.90~2.77 (m, 2H), 1.20 (s, 9H), 0.89 (s, 9H), 0.10 (d, J=8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 166.7, 70.1, 56.9, 48.0, 41.2, 25.8, 22.5, 18.1, -4.5, -4.6; IR (KBr) ν: 2956, 2929, 2898, 2858, 1737, 1622, 1089, 838 cm-1; ESI-LR m/z: 340.2 [M+H]+; HRMS (ESI) calcd for C14H31O2N- ClSSi [M+H]+ 340.1528, found 340.1527.
在氮气保护下, 于100 mL茄形瓶中加入二异丙胺(1.5 mL, 10.9 mmol), 10 mL新蒸四氢呋喃, 并冷却至-78 ℃, 缓慢滴入正丁基锂(2.4 mol•L-1正己烷溶液, 4.8 mL, 10.9 mmol), 滴加完后缓慢升至0 ℃搅拌1 h.将反应瓶再次冷却至-78 ℃, 滴入乙酸甲酯(778 mg, 10.5 mmol)的THF (10 mL)溶液, 滴加完成后保持该温度搅拌反应1 h.缓慢滴入三异丙氧基氯化钛(1 mol•L-1正己烷溶液, 21.7 mL, 21.7 mmol)后反应40 min, 滴入化合物5 (1.2 g, 3.5 mmol)的THF (7 mL)的溶液.薄层色谱(TLC)跟踪至原料反应完全, 低温下加入无水甲醇(3 mL)淬灭反应, 恢复至室温, 加10 mL水充分搅拌后硅藻土过滤, 乙醚洗涤滤饼, 将滤液浓缩以除去大部分的THF.向所得的残余物中加入乙醚稀释, 分离有机相, 水相用乙醚(20 mL×3)萃取, 合并有机相后用无水硫酸钠干燥, 过滤, 浓缩后柱层析纯化[V(乙酸乙酯)/V(石油醚)=1/4]得到黄色油状化合物9 1.2 g, 产率84%. [α]D25-48.3 (c 0.33, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 4.34 (d, J=10.0 Hz, 1H), 4.02~4.00 (m, 1H), 3.68 (s, 3H), 3.46 (dd, J=10.8, 3.6 Hz, 1H), 3.39 (dd, J=10.8, 6.4 Hz, 1H), 2.96 (dd, J=16.4, 5.6 Hz, 1H), 2.62 (dd, J=16.8, 4.4 Hz, 1H), 1.99~1.92 (m, 1H), 1.61~1.54 (m, 1H), 1.23 (s, 9H), 0.89 (s, 9H), 0.09 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 172.6, 69.4, 56.3, 51.7, 51.0, 48.9, 41.7, 41.0, 25.9, 22.9, 18.1, -4.0, -4.2; IR (KBr) ν: 3206, 2955, 2929, 2857, 1740, 1092, 1044, 837 cm-1; ESI-LR m/z: 414.1 [M+H]+; HRMS (ESI) calcd for C17H37O4NClSSi [M+H]+ 414.1896, found 414.1893.
实验操作同化合物9的方法, 反应时间延长至3.5 h, 分离得到黄色油状物13. 1H NMR (400 MHz, CDCl3) δ: 4.10 (d, J=10.5 Hz, 1H), 4.02~3.99 (m, 1H), 3.73 (s, 3H), 3.48~3.44 (m, 2H), 3.42~3.37 (m, 1H), 3.18 (dd, J=18.2, 5.5 Hz, 1H), 2.94 (dd, J=18.2, 3.5 Hz, 1H), 2.22 (t, J=7.6 Hz, 1H), 2.08~2.00 (m, 2H), 1.22 (s, 9H), 0.88 (s, 9H), 0.10 (s, 6H).
将化合物9 (2.5 g, 6.0 mmol)置于100 mL茄形瓶中, 加入20 mL无水甲醇溶解, 放置于冰水浴中冷却至0 ℃, 滴加氢氧化锂水溶液(1 mol•L-1水溶液, 18 mL, 18.0 mmol), 滴加完成后恢复至室温反应, TLC跟踪至原料反应完全, 反应结束后滴加1 mol•L-1盐酸将反应液将pH调至酸性, 用乙醚(40 mL×4)萃取, 合并有机相用饱和氯化钠水溶液洗涤, 无水硫酸钠干燥, 过滤, 浓缩后柱层析纯[V(MeOH)/V(DCM)=1/50]得到白色固体10 2.0 g, 产率83%. [α]D25+7.5 (c 1.91, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 4.80 (d, J=9.2 Hz, 1H), 3.96 (s, 1H), 3.69~3.63 (m, 1H), 3.48~3.37 (m, 2H), 3.06~3.01 (m, 1H), 2.52 (d, J=16.4 Hz, 1H), 2.16~2.09 (m, 1H), 1.64~1.57 (m, 1H), 1.28 (s, 9H), 0.89 (s, 9H), 0.09 (s, 6H); 13C NMR (100 MHz, CDCl3) δ: 173.8, 69.8, 57.1, 51.7, 48.9, 41.8, 40.5, 25.9, 23.1, 18.1, -3.9, -4.2; IR (KBr) ν: 3212, 2956, 2929, 2857, 1718, 1255, 1197, 1094, 837 cm-1; ESI-LR m/z: 398.2 [M-H]-; HRMS (ESI) calcd for C16H33O4NClSSi [M-H]- 398.1594, found 398.1591.
将化合物10 (563 mg, 1.4 mmol), N-氨基-N-甲基甘氨酸叔丁酯对甲磺酸盐(515 mg, 1.55 mmol)和HOBt (378 mg, 2.8 mmol)置于50 mL茄形瓶中, 氮气保护下加入15 mL新蒸二氯甲烷溶解, 放置于冰水浴中冷却至0 ℃后, 滴加三乙胺(0.4 mL, 2.8 mmol)并加入EDCI (537 mg, 2.8 mmol), 然后恢复至室温搅拌反应. TLC跟踪至原料反应完全, 加入10%柠檬酸水溶液(30 mL)充分搅拌, 分离有机相, 水相用乙酸乙酯(30 mL×3)萃取, 合并有机相, 依次用饱和碳酸氢钠水溶液和饱和氯化钠水溶液洗涤, 无水硫酸钠干燥, 浓缩后柱层析纯化[V(丙酮)/V(石油醚)=1/5]得到黄色油状化合物11 762 mg, 产率定量. [α]D25-4.7 (c 0.85, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ: 5.28~4.96 (m, 1H), 4.02~3.96 (m, 1H), 3.67~3.32 (m, 5H), 3.03~2.84 (m, 1H), 2.71 (s, 1.8H, major isomer), 2.70 (s, 1.2H, minor isomer), 2.23~1.90 (m, 2H), 1.67~1.52 (m, 1H), 1.45 (s, 9H), 1.24 (s, 5.7H, major isomer), 1.21 (s, 3.8H, minor isomer), 0.89 (s, 3.6H, minor isomer), 0.87 (s, 5.4H, major isomer), 0.09 (s, 1.2H, minor isomer) 0.084 (s, 1.2H, minor isomer), 0.076 (s, 1.8H, major isomer), 0.07 (s, 1.8H, major isomer); The ratio of rotamers is 1.5:1; IR (KBr) ν: 3236, 2956, 2929, 2857, 1744, 1667, 1253, 1225, 1072, 837 cm-1; ESI-LR m/z: 542.2 [M+H]+; HRMS (ESI) calcd for C23H48O5- N3ClNaSSi [M+Na]+ 564.2665, found 564.2668.
将化合物11 (279 mg, 0.5 mmol)置于反应管中, 氮气保护下加入3 mL超干DMF溶解, 加入碘化钾(83 mg, 0.5 mmol)和叠氮化钠(98 mg, 1.5 mmol), 体系加热至90 ℃搅拌反应, 1H NMR跟踪至原料反应完全, 停止加热, 冷却至室温, 加入5 mL水充分搅拌, 分离有机相, 用乙醚(10 mL×5)萃取水相, 合并有机相, 用饱和氯化钠水溶液洗涤, 无水硫酸钠干燥, 浓缩后柱层析纯化[V(MeOH)/V(DCM)=1/50]得到黄色油状化合物12[7d] 179 mg, 产率66%. [α]D25-10.9 (c 1.51, CHCl3); 1H NMR (400 MHz, CDCl3) δ: 5.08~4.97 (m, 1H), 4.05~3.91 (m, 1H), 3.65~3.28 (m, 4H), 3.17~2.86 (m, 2H), 2.72 (s, 1.7H, major isomer), 2.71 (s, 1.3H, minor isomer), 2.24~1.94 (m, 1H), 1.85~1.56 (m, 1H), 1.47 (s, 3.8H, minor isomer), 1.46 (s, 5.3H, major isomer), 1.24 (s, 5.3H, major isomer), 1.22 (s, 3.8H, minor isomer), 0.90 (s, 3.8H, minor isomer), 0.89 (s, 5.3H, major isomer), 0.10 (s, 3.4H, major isomer), 0.08 (s, 2.5H, minor isomer); The ratio of rotamers is 1.3:1; ESI-LR m/z: 549.2 [M+ H]+.
辅助材料(Supporting Information) 所有合成化合物的核磁共振原始谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
(a) Antibacterial agents in clinical development: an analysis of the antibacterial clinical development pipeline, including tuberculosis, World Health Organization, Geneva, 2017 (WHO/EMP/IAU/ 2017.12). License: CCBY-NC-SA 3.0 IGO.
(b) Payne, D. J.; Gwynn, M. N.; Holmes, D. J.; Pompliano, D. L. Nat. Rev. Drug Discovery 2007, 6, 29.
(a) Kupferschmidt, K. Science 2016, 352, 758.
(b) Liu, Y.; Li, R.; Xiao, X.; Wang, Z. Crit. Rev. Microbiol. 2019, 45, 301.
World Health Organization WHO Annual Report on Global Priority List of Antibiotic-Resistant Bacteria, 2017.
(a) Hamada, M.; Takeuchi, T.; Kondo, S.; Ikeda, Y.; Naganawa. H. J. Antibiot. 1970, 23, 170.
(b) Kondo, S.; Shibahara, S.; Takahashi, S.; Maeda, K.; Umezawa, H. J Am Chem Soc. 1971, 93, 6305.
(a) Uehara, Y.; Hori, M.; Umezawa, H. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1974, 374, 82.
(b) Uehara, Y.; Hori, M.; Umezawa, H. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1976, 447, 406.
(c) Uehara, Y.; Hori, M.; Umezawa, H. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1976, 442, 251.
(d) Mizuno, S.; Nitta, K.; Umezawa, H. J. Antibiot. 1970, 23, 589.
(e) Mizuno, S.; Nitta, K.; Umezawa, H. J. Antibiot. 1970, 23, 581.
(a) Arakawa, M.; Shiozuka, M.; Nakayama Y.; Hara, T.; Hamada, M.; Kondo, A.; Ikeda, D.; Takahashi, Y.; Sawa, R.; Nonomura, Y.; Sheykholeslami, K.; Kondo, K.; Kaga, K.; Suzuki-Miyagoe, Y.; Takeda, S.; Matsuda, R. J. Biochem. 2003, 134, 751.
(b) Allamand, V.; Bidou, L.; Arakawa, M.; Floquet, C.; Shiozuka, M.; Paturneau-Jouas, M.; Gartioux, C.; Butler-Browne, G. S.; Mouly, V.; Rousset, J.-P.; Matsuda, R.; Ikeda, D.; Guicheney, P. J. Gene Med. 2008, 10, 217.
(c) Taguchi, A.; Nishiguchi, S.; Shiozuka, M.; Nomoto, T.; Ina, M.; Nojima, S.; Matsuda, R.; Nonomura, Y.; Kiso, Y.; Yamazaki, Y.; Yakushiji, F.; Hayashi, Y. ACS Med. Chem. Lett. 2012, 3, 118.
(d) Taguchi, A.; Hamada, K.; Hayashi, Y. J. Antibiot. 2018, 71, 205.
Select asymmetric synthesis literature:
(a) Shibahara, S.; Kondo, S.; Maeda, K.; Umezawa, H.; Ohno, M. J. Am. Chem. Soc. 1972, 94, 4353.
(b) Masters, J. J.; Hegedus, L. S. J. Org. Chem. 1993, 58, 4547.
(c) Davies, S. G.; Ichihara, O. Tetrahedron: Asymmetry 1996, 7, 1919.
(d) Olivier, N. B.; Altman, R. B.; Noeske, J.; Basarab, G. S.; Code, E.; Ferguson, A. D.; Gao, N.; Huang, J.; Juette, M. F.; Livchak, S.; Miller, M. D.; Prince, D. B.; Cate, J. H. D.; Buurman, E. T.; Blanchard, S. C. Proc. Natl. Acad. Sci. 2014, 111, 16274.
(e) For a recent summary about the total synthesis of negamycin, see: Zhu, L.; Hong, R. Tetrahedron Lett. 2018, 59, 2112.
(a) Kondo, S.; Iinuma, K.; Yoshida, K.; Yokose, K.; Ikeda, Y.; Shimazaki, M.; Umezawa, H. J. Antibiot. 1976, 29, 208.
(b) Uehara, Y.; Hori, M.; Kondo, S.; Hamada, M.; Umezawa, H. J. Antibiot. 1976, 29, 937.
(c) Raju, B.; Mortell, K.; Anandan, S.; O'Dowd, H.; Gao, H.; Gomez, M.; Hackbarth, C.; Wu, C.; Wang, W.; Yuan, Z.; White, R.; Trias, J.; Patel, D. V. Bioorg. Med. Chem. Lett. 2003, 13, 2413.
(d) Raju, B.; Anandan, S.; Gu, S.; Herradura, P.; O'Dowd, H.; Kim, B.; Gomez, M.; Hackbarth, C.; Wu, C.; Wang, W.; Yuan, Z.; White, R.; Trias, J.; Patel, D. V. Bioorg. Med. Chem. Lett. 2004, 14, 3103.
(e) McKinney, D. C.; Basarab, G. S.; Cocozaki, A. I.; Foulk, M. A.; Miller, M. D.; Ruvinsky, A. M.; Scott, C. W.; Thakur, K.; Zhao, L.; Buurman, E. T.; Narayan, S. ACS Med. Chem. Lett. 2015, 6, 930.
(f) Schmidt, U.; Stäbler, F.; Lieberknecht, A. Synthesis 1992, 482.
Shibahara, S.; Kondo, S.; Maeda, K.; Umezawa, H.; Ohno, M. J. Am. Chem. Soc. 1972, 94, 4353. doi: 10.1021/ja00767a059
Liu, G.; Cogan, D. A.; Owens, T. D.; Tang, T. P.; Ellman, J. A. J. Org. Chem. 1999, 64, 1278. doi: 10.1021/jo982059i
Ma, H. M.; Yang, L. L.; Ni, Y.; Zhang, J.; Li, C. X.; Zheng, G. W.; Yang, H. Y.; Xu, J. H. Adv. Synth. Catal. 2012, 354, 1765.
(a) Reetz, M. T.; Li, X. G. J. Am. Chem. Soc. 2006, 128, 1044.
(b) Ohkuma, T.; Tsutsumi, K.; Utsumi, N.; Arai, N.; Noyori, R.; Murata, K. Org. Lett. 2007, 9: 255.
(a) Xu, J.; Lin, X. I. Chin. J. Org. Chem. 2007, 27, 1473 (in Chinese).
(许建明, 林贤福, 有机化学, 2007, 27, 1473.)
(b) Liao, X.; Jiang, Y.; Lai, S.; Liu, Y.; Wang, S.; Xiong, X. Chin. J. Org. Chem. 2019, 39, 668 (in Chinese).
(廖旭, 蒋岩, 赖石林, 刘源岗, 王士斌, 熊兴泉, 有机化学, 2019, 39, 668.)
Müller, M. Angew. Chem., Int. Ed. 2005, 44, 362. doi: 10.1002/anie.200460852
Liu, G.; Cogan, D. A.; Owens, T. D.; Tang, T. P.; Ellman, J. A. J. Org. Chem. 1999, 64, 1278. doi: 10.1021/jo982059i
(a) Tang, T. P.; Ellman, J. A. J. Org. Chem. 2002, 67, 7819.
(b) Siegel C.; Thornton, E. R. J. Am. Chem. Soc. 1989, 111, 5722.
(c) Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600.
Makino, K.; Jiang, H.; Suzuki, T.; Hamada, H. Tetrahedron: Asymmetry 2006, 17, 1644. doi: 10.1016/j.tetasy.2006.06.004

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:


