引用本文:
张潮, 刘志军, 李艳冰, 何业谱, 林晓洪, 陈河如. 靶向抗肿瘤药N-(N'-苄氧羰基甘氨酰脯氨酰)丙卡巴肼(Z-GP-Pcb)的环境友好合成及其透过血脑屏障活性评价[J]. 有机化学,
2020, 40(2): 536-540.
doi:
10.6023/cjoc201908015 Citation:
Zhang Chao, Liu Zhijun, Li Yanbing, He Yepu, Lin Xiaohong, Chen Heru. Environment-Friendly Synthesis of Targeted Anticancer Drug N-(N'-Carbobenzoxyglycylprolyl)procarbazine (Z-GP-Pcb) and the Evaluation of Its Activity to Penetrate Blood-Brain Barrier[J]. Chinese Journal of Organic Chemistry,
2020, 40(2): 536-540.
doi:
10.6023/cjoc201908015
Environment-Friendly Synthesis of Targeted Anticancer Drug N-(N'-Carbobenzoxyglycylprolyl)procarbazine (Z-GP-Pcb) and the Evaluation of Its Activity to Penetrate Blood-Brain Barrier
Institute of Traditional Chinese Medicine and Natural Products, College of Pharmacy, Jinan University, Guangzhou 510632
b.
Guangzhou PharmCherub Medical Sci. &Tech. Incorporated Corporation, Guangzhou 510663
c.
Guangdong Province Key Laboratory of Pharmacodynamic Constituents of Traditional Chinese Medicine and New Drugs Research, Jinan University, Guangzhou 510632
Received Date:
10 August 2019 Revised Date:
21 September 2019 Available Online:
25 February 2020
Fund Project:
Project supported by the Natural Science Foundation of Guangdong Province (No. 2018B030311020)
Abstract:
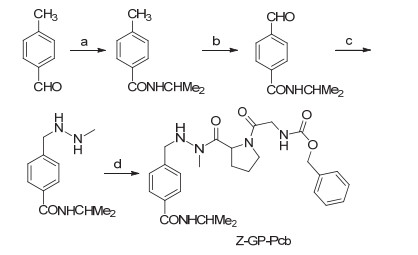
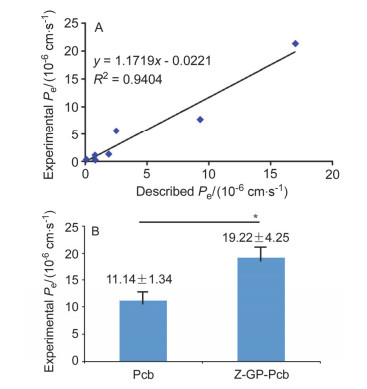
Anti-cancer drug N-(N'-carbobenzoxyglycylprolyl)procarbazine (Z-GP-Pcb) has been designed based on targeting strategy and a 3-step method has been developed for the synthesis of procarbazine (Pcb). Firstly, 4-methylbenaldehyde, as starting material, was transformed into N-isopropyl-4-methylbenzamide using dibromoisocyanuric acid (DBI). This compound was then directly oxidized to N-isopropyl-4-formylbenzamide by 2-iodoxybenzoic acid (IBX). A reductive amination reaction was then followed leading to Pcb. At last, Pcb was condensed with N-carbobenzoxyglycylproline resulting in Z-GP-Pcb. The overall yield was 49.9%. Furthermore, an in vitro blood-brain barrier (BBB) permeation assay (PAMPA-BBB) was set up to evaluate the permeation activity of Z-GP-Pcb. It was found that the permeation constant (Pe) was (19.22±4.25)×10-6 cm·s-1, which was more than that of the parent drug Pcb[(11.14±1.34))×10-6 cm·s-1]. This evidence suggests that Z-GP-Pcb possesses high activity to penetrate BBB.
(A) Lineal correlation between experimental and reported permeability of commercial drugs using the PAMPA-BBB assay; (B) experimental Pe values of Pcb and Z-GP-Pcb. *p < 0.05 vs Pcb group (n=3).
Weiss, H. D.; Walker, M. D.; Wiernik, P. H. New Engl. J. Med.1974, 291, 128.
[24]
Di, L.; Kerns, E. H.; Fan, K.; McConnell, O. J.; Carter, G. T. Eur. J. Med. Chem.2003, 38, 223. doi: 10.1016/S0223-5234(03)00012-6
[25]
Camps, P.; Formosa, X.; Galdeano, C.; Munoz-Torrero, D.; Ramírez, L.; Gomez, E.; Isambert, N. S.; Lavilla, R.; Badia, A.; Clos, M. V. R.; Bartolini, M.; Mancini, F.; Andrisano, V.; Arce, M. P.; Rodríguez-Franco, M. I.; Huertas, O. S.; Dafni, T.; Luque, F. J. J. Med. Chem.2009, 52, 5365. doi: 10.1021/jm900859q
(A) Lineal correlation between experimental and reported permeability of commercial drugs using the PAMPA-BBB assay; (B) experimental Pe values of Pcb and Z-GP-Pcb. *p < 0.05 vs Pcb group (n=3).

 下载:
下载:


 下载:
下载:



