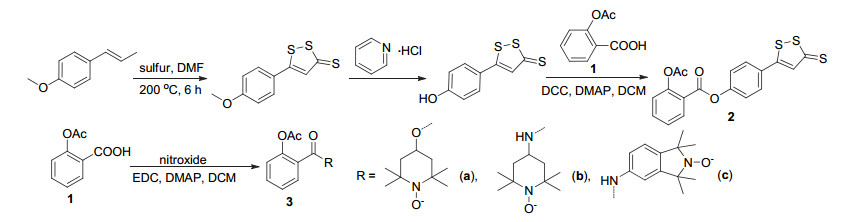
图式 1.
阿司匹林衍生物2和3的合成路线
Scheme 1.
Synthetic route of aspirin derivatives 2 and 3

阿司匹林(Aspirin, ASP, 1), 别名乙酰水杨酸, 化学名2-乙酰氧基苯甲酸, 是一种常用的解热抗炎药物, 其前体水杨酸最初是从白杨柳中提取而来. 1899年, 费利克斯•霍夫曼成功地将水杨酸合成为乙酰水杨酸, 使其成为第一个人工合成的药物[1].阿司匹林具有抗血栓及抗炎等多种药理活性, 近年来还发现阿司匹林具有抗肿瘤等其他活性, 目前抗肿瘤活性已成为其研究热点.阿司匹林的抗血栓机制为阿司匹林通过不可逆地乙酰化血小板环氧化酶, 主要是乙酰化环氧化酶- 1 (COX-1)丝氨酸530位点, 抑制血栓素A2 (TXA2)的形成, 从而达到抗血小板聚集和抗血栓的作用[2].抗炎机制为阿司匹林能够抑制细胞环氧化酶从而抑制前列腺素的合成, 发挥解热、镇痛及抗炎作用[3].其抗肿瘤机制尚不明确, 但随着研究深入, 越来越多的实验结果证明了在众多肿瘤组织中都存在环氧合酶-2 (COX-2)的过度表达[4, 5].当环氧化酶-2表达过度时致使机体过量合成前列腺素E2 (PGE2), 而PGE2含量的增加使机体的免疫系统功能遭到削弱.这种情况下, 免疫系统对肿瘤细胞的监控能力就不可避免地减弱了, 致使肿瘤组织不能被免疫系统及时清除, 所以阿司匹林抑制COX-2的活性能够产生抗癌作用[6].虽然阿司匹林具有多种优良活性, 但长期使用阿司匹林会造成对胃、肝、肾以及神经系统造成伤害.此外, 对特异体质的人群有可能引发过敏反应及贫血等症状.为了克服阿司匹林的副作用, 并提高阿司匹林的疗效, 大量科学家不断地对阿司匹林进行结构修饰, 并获得了一定的成果.
阿司匹林的结构修饰位点相对较少, 其中对C(1)- COOH进行修饰是主要的衍生方式.对C(1)-COOH修饰可利用羧基与醇、酚或胺等一步反应制得酯或酰胺(Scheme 1), 如阿司匹林C(1)-COOH直接与5-(4-羟基苯基)-3H-1, 2-二硫杂环戊烯-3-硫酮酯化, 获得衍生物2[7].衍生物2可通过影响纤维蛋白原受体的活化, 增加细胞内环磷酸腺苷(cAMP)的含量, 从而减少动脉血栓的形成, 抑制血小板聚集而显示出抗血栓活性, 并且还具有保护胃粘膜的作用[8].与醇或胺反应制得的衍生物3a~3c (Scheme 1)对非小细胞肺癌细胞株A549 (NSCLC A549)细胞显示出抗炎和抗肿瘤活性, 且成酯的衍生物3a比成酰胺的衍生物3b和3c活性更高, 其中3a对于NSCLC A549细胞的IC50值为130 μmol•L-1, 并在较高含量(180 μmol•L-1)时对非小细胞肺癌细胞株H1299 (NCI-H1299)细胞显示出强烈的抑制作用[9].
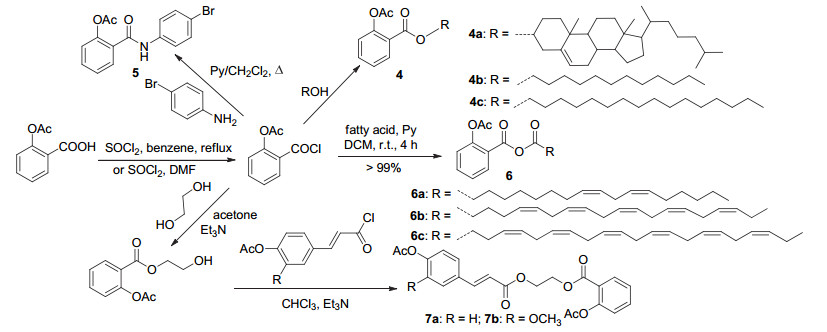
阿司匹林除可直接与醇、酚或胺等一步反应制得衍生物之外, 更常将其C(1)-COOH酰化, 再与醇、酚或胺等反应获得衍生物(Scheme 2).阿司匹林酰氯与十二醇、十六醇及胆固醇酰化获得衍生物4a~4c, 4a~4c降低血栓素B2 (TXB2)浓度的程度随化合物酰基链长度的增加而减小[10].与4-溴苯胺酰化获得衍生物5, 其具有较好的抗真菌活性, 作用于核盘菌及灰葡萄孢菌的ED50值分别为8.6和1.8 mg/L[11].通过与不饱和脂肪酸反应得到的衍生物6a~6c (Scheme 2)可通过抑制COX-1-TXAS途径, 减少血栓素的合成, 从而具有抑制血小板聚集的抗血栓活性, 其活性大小为6a>6b>6c[12].与乙二醇酯化, 再与对乙酰氧基苯丙烯酸酰氯或3-甲氧基-4-乙酰氧基苯丙烯酸酰氯反应获得的化合物7a和7b (Scheme 2), 均显示出小鼠耳部巴豆油炎症的抑制作用, 其耳肿胀抑制率分别为43.5%和37.1%, 高于耳肿胀抑制率为22.4%的阿司匹林[13].
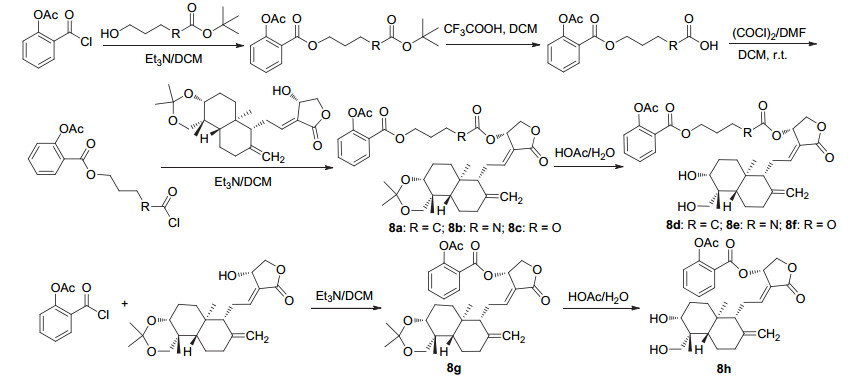
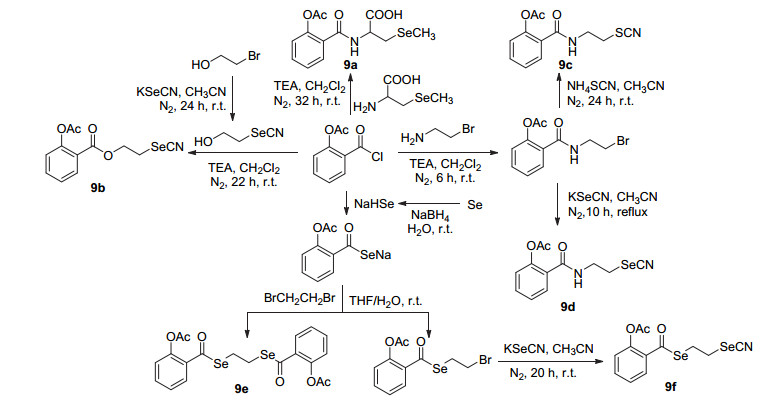
阿司匹林酰氯经成酯和脱叔丁基, 随后酰化, 再与双羟基被保护的穿心莲内酯酯化, 最后水解开环生成衍生物8a~8f, 或直接与双羟基被保护的穿心莲内酯酯化后水解开环生成衍生物8g和8h (Scheme 3).其中8a、8e、8f和8g可通过抑制乙酰肝素酶的活性来抑制肿瘤的血管新生和转移, 体外测试中四者对PC-3、MCF-7、MDA- MB-231及A549癌细胞均显示抑制活性, IC50值在8~30 μmol•L-1, 强于阿司匹林[14].同样地, 阿司匹林酰氯与含硒化合物酰化制得衍生物9a~9f (Scheme 4), 化合物9b、9c和9e具有显著的抗癌活性, 对HT29、DA-MB231、PANC及UACC903细胞均有抑制活性, 对HT29细胞的IC50值分别为(3.4±1.1)、(10.2±13.3)和(18.6±3.8) μmol•L-1.其中化合物9c具有优良的抗癌活性, 研究表明该衍生物可通过抑制细胞增殖的G1期和G2/M期来抑制癌细胞的生长, 并且选择性较高, 是潜在的化疗药物[15].
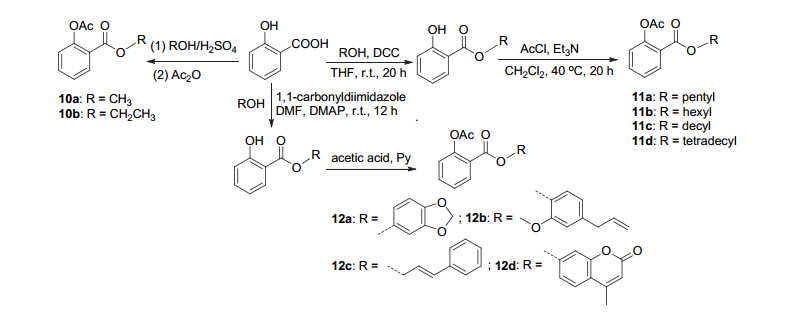
设计阿司匹林衍生物时, 除了从阿司匹林本体结构为出发点之外, 还常以水杨酸、水杨醛等其他物质出发合成设计阿司匹林的衍生物.如水杨酸C(1)- COOH与相应的醇酯化生成衍生物10a和10b (Scheme 5), 两者能够抑制NMDA引起的神经毒性[16].将C(1)-COOH与戊醇、己醇、癸醇及十四醇酯化后, 再将酚羟基酰化得到衍生物11a~11d (Scheme 5), 化合物11a~11c在体外实验中显示出与阿司匹林相同的抗炎活性[17].
氧化应激与炎症的产生是与年龄相关的机体功能下降的主要影响因素, 阿司匹林可通过减少类胰岛素信号的释放, 从而防止氧化应激, 延缓与年龄相关的机体功能衰退, 达到抗衰老的作用[18, 19].将水杨酸C(1)- COOH醇酯化, 随后用乙酸修饰酚羟基获得衍生物12a~12d (Scheme 5), 其中12a对大鼠肝匀浆中丙二醛的生成显示出良好的抑制活性, 抑制率高于同等剂量下食品防腐剂羟基茴香醚, 达84.95%, 抗氧化效果优异[20].
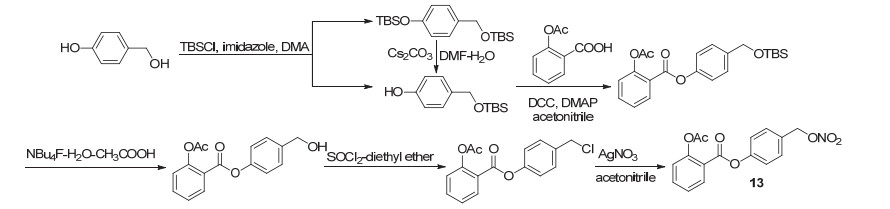
NO、H2S和CO是一组具有生理活性的气体, 被称为气体信号因子, 在适当的浓度下, 它们在调节多种细胞的生理功能中起着重要作用.例如, 它们可通过心血管系统引起血管扩张, 促进血管生成, 并在心脏保护中发挥关键作用[21].其中内皮源性舒张因子NO是一种具有生理活性的气体, 具有维持血管稳态、调节血管张力及血压及舒张血管等功能[22, 23], 此外还可抗血小板聚集, 具有胃肠道保护作用.把含有NO供体的化合物与阿司匹林组合形成的NO-ASP, 可在体内同时释放NO和阿司匹林, 在抗血栓、抗肿瘤及抗炎方面显示出药理活性[24], 是提高阿司匹林生物活性的有效选择.以该设计原理为出发点, 将经叔丁基二甲基保护的对羟基苯甲醇与阿司匹林酯化, 随后脱去保护基团, 再氯代后硝化, 获得的衍生物13 (Scheme 6), 作为一种治疗人结肠癌抗癌药物正在临床前实验中[25].
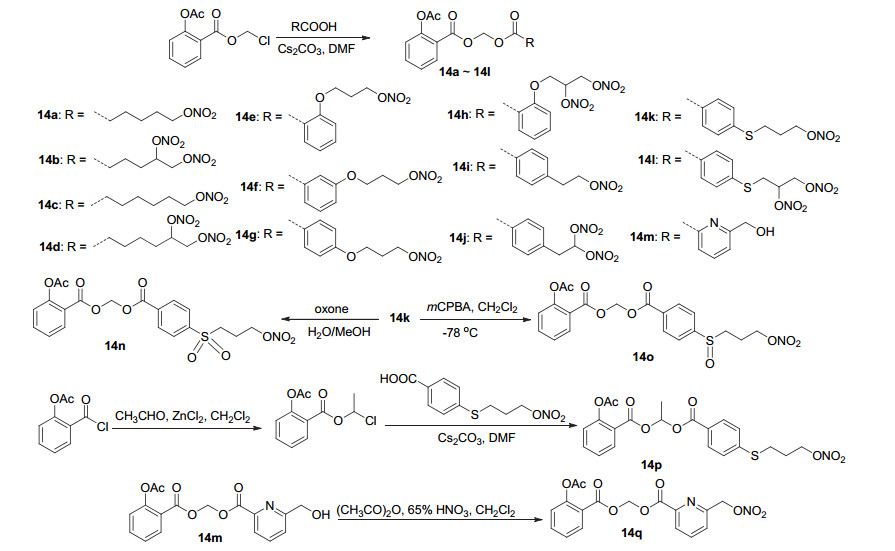
在2-(乙酰氧基)苯甲酸氯甲基酯的基础上, 将之与适当的酸酰化获得衍生物14a~14m, 并在14k的基础上氧化获得砜与亚砜衍生物14n与14o, 将阿司匹林酰氯与乙醛加成, 随后与酸偶联获得衍生物14p, 在14m的基础上与硝酸反应获得衍生物14q (Scheme 7).其中14i、14n与14q具有较好的抗血小板聚集活性, 对由胶原蛋白诱导的富血小板血浆(PRP)的血小板聚集抑制活性IC50值分别为41、38和20 μmol•L-1, 优于阿司匹林[26].
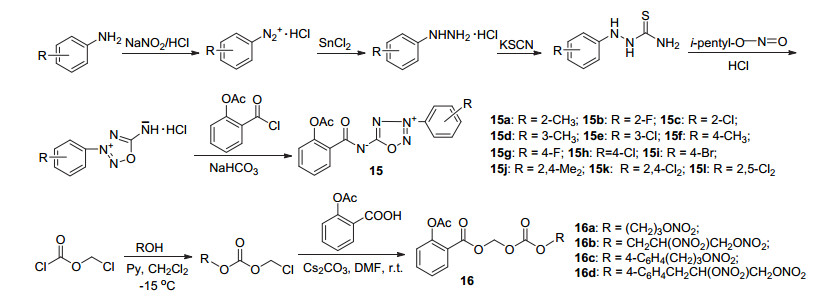
周洲等[27]在C(1)-COCl引入3-芳基-1, 2, 3, 4-噁三唑-5-亚胺作NO供体, 形成NO-ASP杂合体15a~15l (Scheme 8), 测试表明除15b、15c和15e外其他9种化合物均显示出一定的抑制血小板聚集的作用, 15f抑制ADP诱导的血小板聚集率达30.7%, 而15b、15f、15k和15l在体内实验中对由胶原蛋白引发的小鼠肺血栓也具有较好的抑制作用, 在0.33 mmol•L-1•kg-1)的剂量下, 小鼠最高存活率达70%.体外抗血小板聚集实验结果表明, 当NO供体苯环上的R基为单一取代基时, 取代基的位置与抗血小板聚集活性间存在一定相关性, 对位取代时活性最高, 其次为间位, 而邻位则较弱(15f>15d>15a, 15g>15b, 15h>15e>15c); 当苯环上的单一取代基位置固定时, 甲基取代物的活性强于氟代物, 氟代物强于氯代物(15a>15b>15c, 15d>15e, 15f>15g>15h), 苯环上为二氯取代时活性强于单一卤代物(15k, 15l>15b, 15c, 15e, 15g, 15i), 2, 4-二氯取代物活性略强于2, 5-二氯取代物.同样地, 以硝酸酯对阿司匹林C(1)-COOH进行修饰, 获得的NO-ASP衍生物16a~16d (Scheme 8), 均存在一定抗血小板聚集活性, 但相对较弱[28].
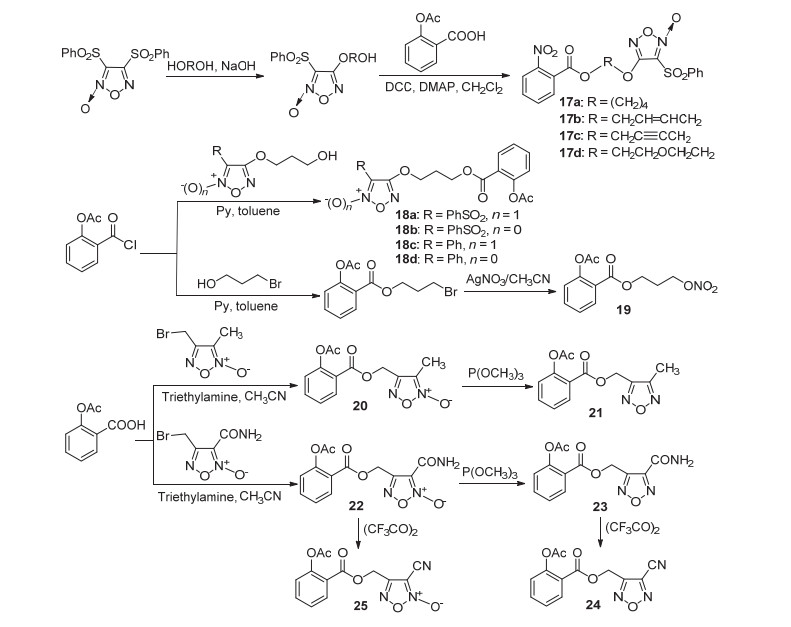
阿司匹林C(1)-COOH与3-苯磺酰基呋咱氮氧化物酯化修饰获得NO-ASP化合物17a~17d (Scheme 9), 这四种化合物均表现出一定抗炎活性, 在剂量9.5 mg/kg时对由二甲苯致小鼠肿胀的抑制率为17.8%~40.4%, 其中17a和17b的抗炎活性强于阿司匹林[29].阿司匹林或阿司匹林酰氯与NO供体呋咱氮氧化物酯化获得衍生物18a~18d和19~25 (Scheme 9), 体内实验显示这些衍生物均有一定的抗炎活性, 其中衍生物18c、22、25在剂量120 mg/kg下对由鹿角菜胶诱导的小鼠肿胀抑制率均高于50%, 抗炎活性与阿司匹林相当.同时所有衍生物的胃粘膜损伤均低于阿司匹林.此外衍生物18a、22和25还表现出同阿司匹林类似的抗血栓活性, 三者于富含血小板血浆(PRP)中对由ADP诱导的血小板聚集抑制活性pIC50值为(5.40±0.20)、(7.22±0.10)和(4.20±0.02) μmol•L-1[30].
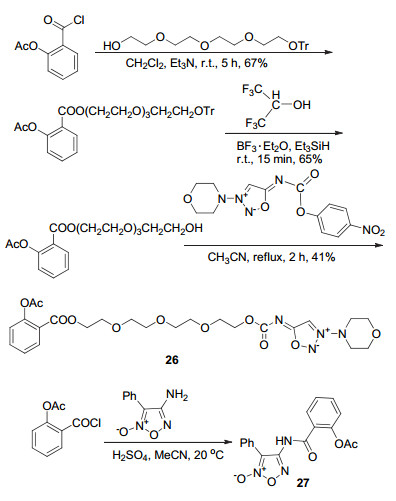
将阿司匹林酰氯通过取代, 再与NO供体酯化, 得到衍生物26 (Scheme 10), 该化合物具有舒张血管及抗心肌缺血活性, 推测可能是NO释放使得冠状动脉舒张, 有待进一步的活性研究[31]; 与4-氨基-3-苯基呋咱取代获得的衍生物27, 具有微弱的抗血小板聚集活性[32].
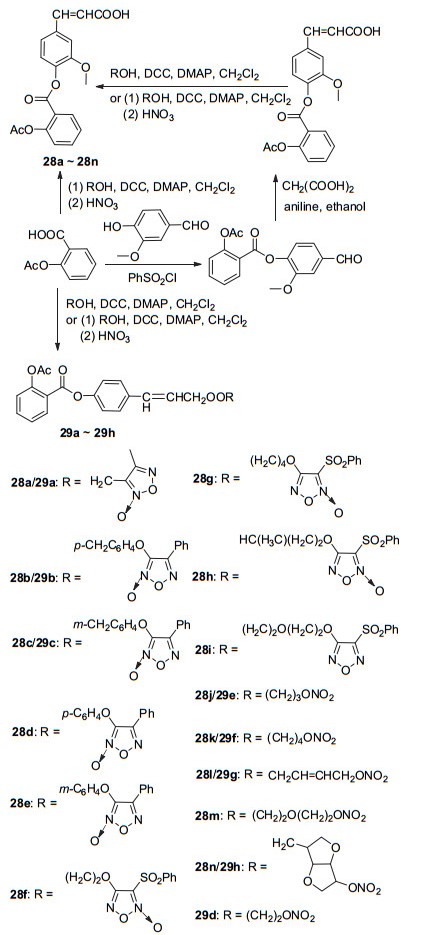
NO供体除可直接与阿司匹林C(1)-COOH相连以外, 还可在C(1)-COOH与NO供体间插入具有相似活性的化合物, 这样可通过协同作用使衍生物活性更高, 但其反应路线相对复杂.呋咱氮氧化物为NO供体, 可在体内通过巯基还原释放气体信使分子[33].以可抗血小板聚集的阿魏酸为中间连接基团, 分别以4-苯基呋咱氮氧化物、3-苯磺酰基呋咱氮氧化物或硝酸酯为NO供体, 形成杂合体28a~28n (Scheme 11).体内实验结果显示化合物28a~28c、28i、28l和28n能显著抑制血小板聚集, 其中抑制由ADP诱导的血小板聚集率最低为28.31%, 最高达43.81%[34].同样地, 将对羟基桂皮酸作为中间连接基团, 以4-苯基呋咱氮氧化物或硝酸酯为NO供体, 形成杂合体29a~29h (Scheme 11).其中化合物29f具有显著的抗血小板聚集作用, 其抑制小鼠体内血小板聚集率43.9%高于阿司匹林的28.05%, 此外该衍生物还能够抑制鼠脑血栓的形成, 降低血浆中TXB2的水平[35].
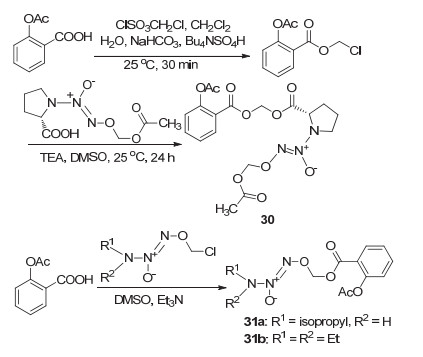
在NO-NSAIDs的基础上, Velázquez等[36]提出了NONO-NSAIDs的概念, NONO-NSAIDs与传统NO-NSAIDs的区别在于, NONO-NSAIDs可释放两分子的NO.以N-重氮-1-铵-1, 2-二酸酯基团为设计点, 阿司匹林C(1)-COOH位经磺酰氯酰化再与NO供体反应, 生成了NONO-ASP衍生物30 (Scheme 12), 该衍生物具有更优良的抗炎活性, ED50值为314 μmol/kg, 为阿司匹林的2.2倍.相似地, 阿司匹林C(1)-COOH经酯化生成了衍生物31a和31b, 体外实验表明, 衍生物31a在剂量121 mg/kg时同129 mg/kg的阿司匹林有相同的抗炎活性, 衍生物31b抗炎活性相对较弱[37].
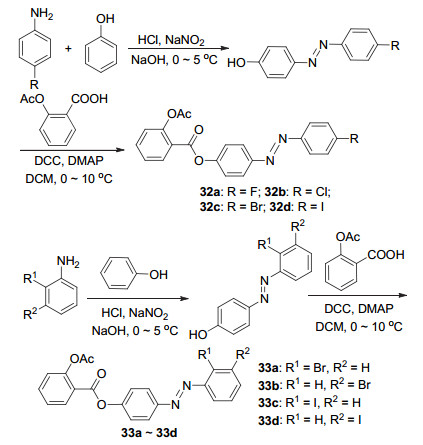
偶氮是另外一种具有抗糖尿病、抗肿瘤及抗菌等药理作用的活性化合物[38], 其抗癌和抑制蛋白质合成活性等得到了广泛研究[39, 40].将苯酚与4-卤代苯胺经过偶合反应制备得到4-[(4-卤代苯基)重氮基]苯酚, 再与阿司匹林C(1)-COOH酯化生成阿司匹林偶氮衍生物32a~32d (Scheme 13).这些衍生物在HK-1癌细胞中均显示出一定的抗癌活性, 其中化合物32a及32b的抗癌活性优于阿司匹林, 两者的IC50分别为(38.7±1.5)和(44.9±9.4) μmol•L-1[41].同样地, 阿司匹林与偶氮化合物酯化获得了衍生物33a~33d (Scheme 13), 该系列化合物对大肠杆菌和金黄色葡萄球菌显示出较差的抗菌活性, 最低抑菌浓度(MIC)>220 mg/L, 综合抗菌活性较弱.将化合物33a~33d与未酯化的偶氮化合物比较, 推测可能为阿司匹林与偶氮化合物酯化后失去了羟基, 使得与相应酶结合时缺乏氢键, 致使抗菌活性较低, 实验表明羟基、羧基和卤素基团的存在对提高化合物的抗菌活性起着重要作用[42].
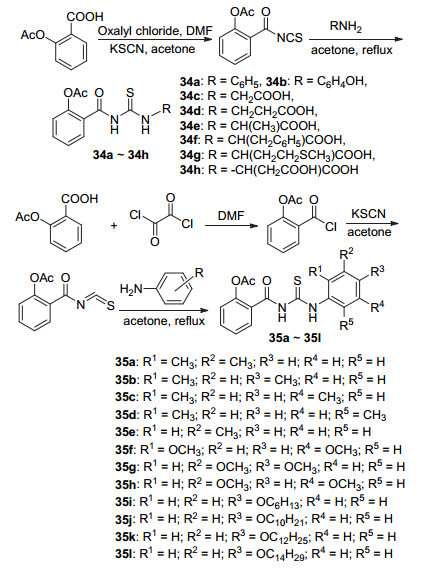
硫脲是一种反应性前体物质, 具有抗菌、抗癌、抗结核和抗HIV等多种生物学活性, 其存在的C=S和C=O在许多生物过程中发挥着重要作用[43].将阿司匹林C(1)-COOH经过酰化再于碱性条件下与氨基酸反应, 生成阿司匹林硫脲衍生物34a~34h (Scheme 14), 总收率为46%~73%.此8种化合物均对大肠杆菌显示出抗菌活性[44].类似地, Nordin等[43]合成了系列含硫脲的阿司匹林衍生物35a~35l, 与阿司匹林相比, 该系列化合物具有中等至优良的抗菌活性.其中取代基的位置与类型均对抗菌活性存在影响, 间位取代基相较于邻位和对位取代基具有更好的抗菌活性, 并且烷基链较短与烷基链较长的衍生物相比, 烷基链短的衍生物比烷基链长的衍生物活性高[43].
C(1)-COOH修饰是阿司匹林骨架衍生的主要方向, 其活性涉及抗血栓、抗癌、抗炎、抗衰老及抗菌等.将阿司匹林直接酯化和先制得乙酰水杨酸酰氯再与醇或胺反应, 是制备C(1)-COOH位衍生物的两种方法.酰氯相较于羧酸的反应活性更高, 因此第二种方法是主要的合成路径.而相较于酰胺化衍生物, 酯化物的脂水分配系数更佳, 更易透过血脑屏障而发挥药效. C(1)-COOH修饰的衍生物多为阿司匹林与活性物质结合为主, 如NO供体、偶氮和硫脲.这当中又以抗血栓化合物居多, 占比43.39%, 而抗血栓衍生物中93.75%为NO-ASP或NONO-ASP, 且大多活性优良, NO供体均为呋咱氮氧化物类或硝酸酯类.即用NO供体对阿司匹林进行C(1)-COOH结构修饰是制备抗血栓衍生物的主要方法.
用羟基磷灰石对阿司匹林进行结构修饰, 得到C(1)-COOH和C(2)-OAc以氢键闭合成环的衍生物36 (Eq. 1), 该衍生物与阿司匹林在相同剂量下具有类似的抗血栓活性, 由于羟基磷灰石具有上调PGE2的功效, 使得该衍生物降低了阿司匹林对胃粘膜的损伤[45].
|
|
(1) |
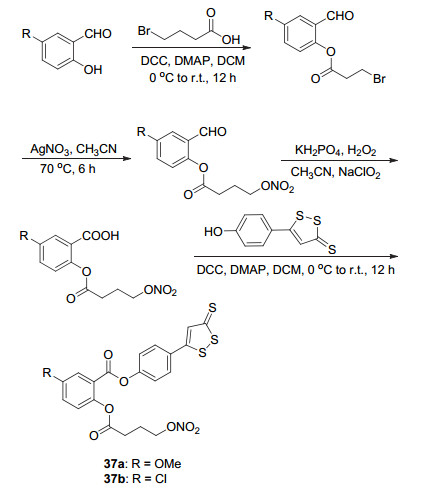
H2S作为一种重要的内源性调节剂, 对心血管系统具有与NO同样的有益作用, 并具有抗炎和抗过氧化物作用.如上述衍生物2在HT-29、SKBR3、MCF7等多种细胞中表现出强的抗癌活性, 其IC50值分别为(3.7±0.9)、(3.3±0.9)、(4.2±1.1) μmol•L-1[7].将NO和H2S两者结合的阿司匹林衍生物被称为NOSH-ASP, 此类衍生物能够在体内同时释放NO和H2S这两种活性气体.基于这种设计思路, 以硝酸酯为NO供体, 以5-(4-羟基苯基)-3H-1, 2-二硫杂环戊烯-3-硫酮为H2S供体, 将水杨醛C(1)位和C(2)位同时修饰得到两种NOSH-阿司匹林衍生物37a和37b (Scheme 15), 这两种衍生物表现出较好的抗癌活性, 体外实验测试得到它们在HT-29癌细胞中的IC50值分别为(200±50)和(380±150) μmol• L-1, 在HCT15癌细胞中的IC50值分别为(400±150)和(170±30) μmol•L-1, 活性远高于阿司匹林[46].
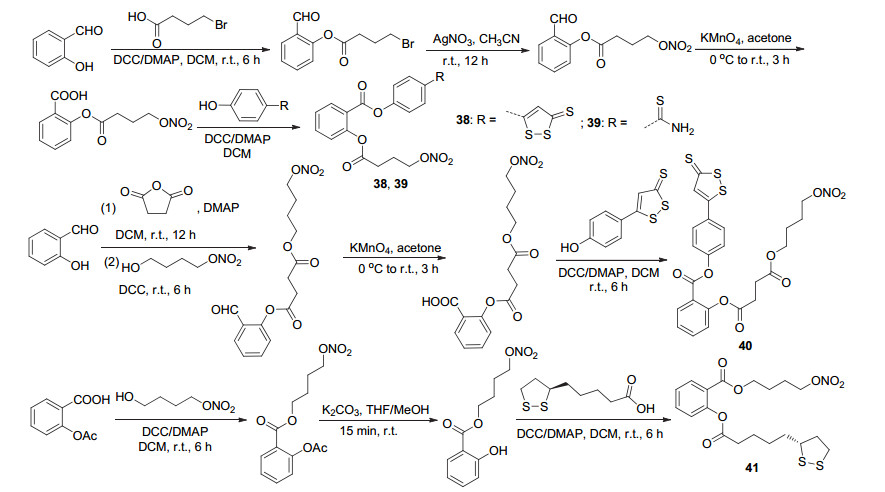
类似地, 在水杨醛和阿司匹林的基础上合成了四种NOSH-阿司匹林衍生物38~41 (Scheme 16), 四种衍生物在体内可同时释放出NO与H2S, 达到抗癌效果, 它们对结肠、乳腺、胰腺、前列腺、肺以及白血病中的11种癌细胞系均表现出强大的抗癌活性, 其中化合物38对HT-29细胞的抗癌活性至少是阿司匹林的十万倍, 且化合物39~41在同一细胞系中的增效作用分别为>6万倍、>600倍和>1.6万倍.此外, 化合物38还显示出一定的抗炎活性, 其抗炎能力与阿司匹林相近[47].
阿司匹林C(1)-COOH与C(2)-OAc为阿司匹林的重要活性位点, 目前同时修饰制备得到的衍生物仅7种.衍生物以抗癌活性为主, 效力强于阿司匹林.同C(1)- COOH修饰的衍生物类似, 此类衍生物也为阿司匹林与活性物质结合为主, 为NOSH-ASP, 相较于NO-ASP而言, NOSH-ASP由于引入两种活性物, 且需对C(1)- COOH与C(2)-OAc位同时修饰, 因此NOSH-ASP合成步骤相对复杂.但其活性更佳, 如衍生物38对HT-29细胞的IC50为(48±3) nmol•L-1, 而在纳摩尔级别阿司匹林与NO-ASP衍生物均无抗癌活性.
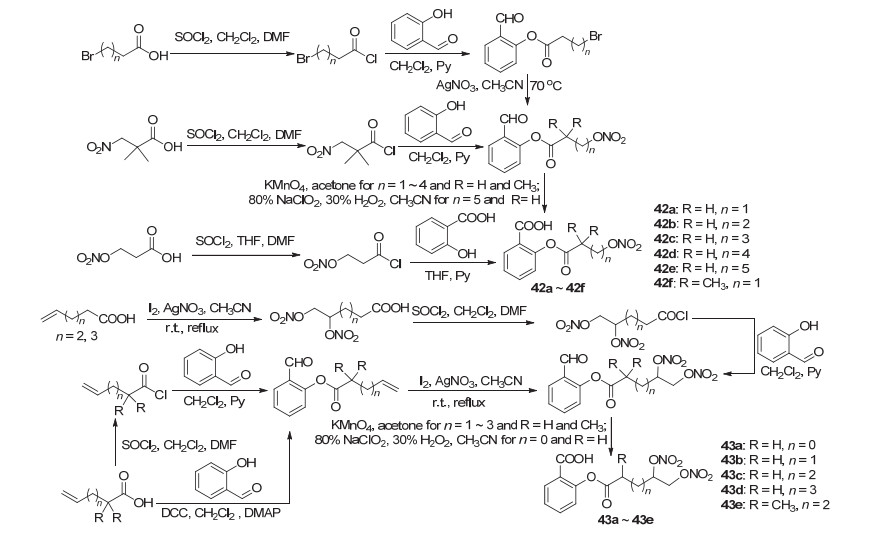
以水杨醛为底物, 将酚羟基分别与单硝基氧基和双硝基氧基取代化合物连接, 再氧化醛基获得NO-ASP衍生物42a~42f与43a~43e (Scheme 17), 体外实验结果显示, 衍生物42a~42c和43a显示出一定的抗血小板聚集活性, 它们IC50值分别为162、30、91和126 μmol• L-1, 其中42b活性高于阿司匹林.同时, 由角叉菜胶引起的小鼠爪水肿实验中, 各个衍生物均显示出不同程度的抑制水肿能力, 其中衍生物42c、42d、42f和43b~43d的抑制水肿能力同阿司匹林相近, 即显示出抗炎活性.此外, 在胃粘膜损伤实验中, 除衍生物42a~42c外, 其余衍生物的胃粘膜细胞的病变率均小于10%, 远低于胃粘膜细胞病变率 > 50%的阿司匹林, 副作用明显减少[48].
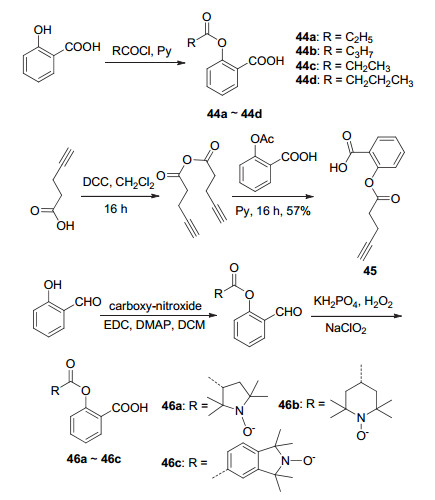
以水杨酸为底物, 将酚羟基酰化制得了阿司匹林衍生物44a~44d (Scheme 18), 44a与44d的活性均低于阿司匹林, 44a在含量为100 μmol•L-1时的血小板聚集抑制率为70%, 44b则在500 μmol•L-1时才显示出相近的血小板聚集抑制率[10, 16].戊炔酸在二环己基碳二亚胺(DDC)作用下制得戊炔酸酐, 再与阿司匹林的C(2)-OAc发生酯交换反应获得了衍生物45 (Scheme 18), 该化合物在HCT-15结肠癌细胞中显示出抗癌活性[49]. Thomas等[9]以药效团杂交的原理开发了双效抗炎/抗肿瘤类的阿司匹林化合物46a~46c (Scheme 18), 衍生物46a对NSCLC A549细胞显示出抗炎和抗肿瘤活性.
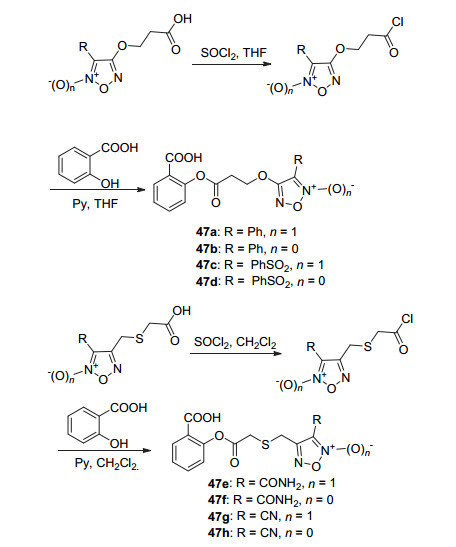
水杨酸与呋咱氮氧化物酰化生成了NO-ASP 47a~47h (Scheme 19), 它们在120 mg/kg的剂量下作用于由角叉菜胶引起的小鼠足肿胀时, 均显示出同阿司匹林类似的抗炎活性, 抑制率均在37%~55%之间, 其中衍生物47c和47g的抑制率最高达50%~55%.此外, 衍生物47c与47g还显示出较好的抗血小板聚集活性, IC50值分别为7.8与7.3 μmol•L-1[50].
阿司匹林C(2)-OAc修饰的衍生物相对较少, 仅27种, 常以其他底物如水杨酸和水杨醛等合成制备出具有阿司匹林母体结构的衍生物.衍生物活性涉及抗炎、抗血栓及抗癌, 其中NO-ASP占17种, 各种衍生物的活性大小与阿司匹林相近, 即阿司匹林C(2)-OAc修饰活性提高不明显.
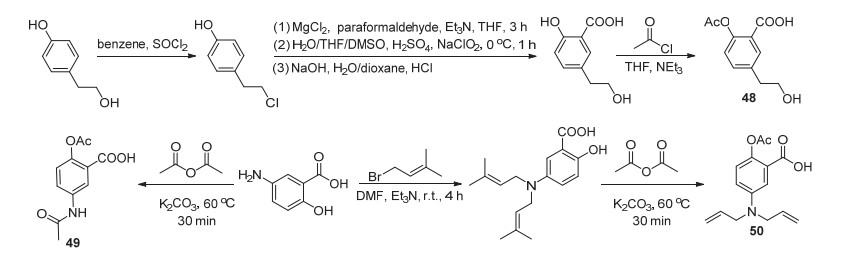
阿司匹林苯环上的结构修饰并不常见, 并且合成相对较难, 故常常从其他简单化合物出发制备其衍生物.对羟基苯乙醇经C(1)位氯代、C(3)位甲酰化、氧化和水解, 最后再使C(4)-OH酰化获得了化合物48 (Scheme 20).体外实验显示该化合物具有抗炎活性, 以剂量100 mg/kg给药, 4 h后其改善角叉菜胶诱导的肿胀率为(28.40±0.18)%[51]. Huang等[52]以5-氨基水杨酸与乙酸酐酰化或先与2-甲基-4-溴-2-丁烯制得中间产物, 再与乙酸酐酰化分别得到了衍生物49和50, 这两种衍生物经测试具有抗衰老活性的可能性较低, 但这预示了阿司匹林结构修饰的新方向.
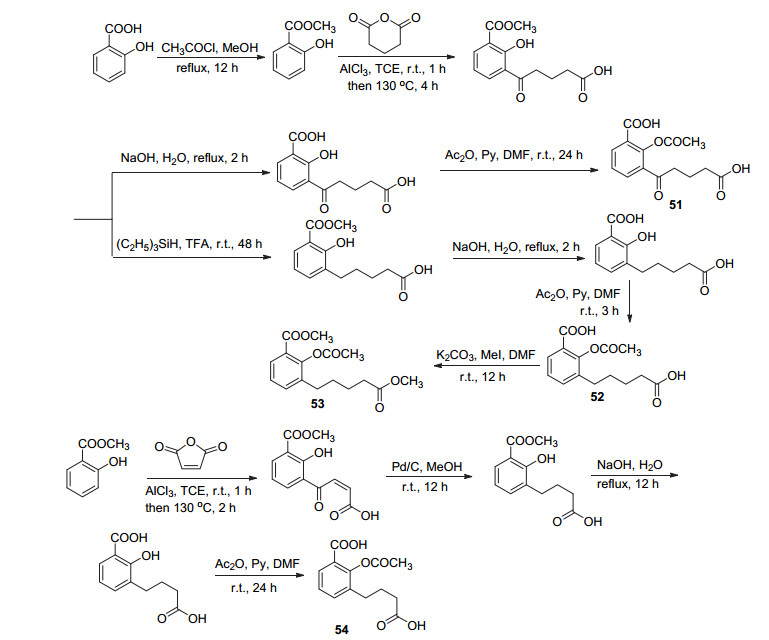
阿司匹林是一种有效的预防血栓药物, 它能够与COX-1上的Arg120或Tyr385相互作用, 使乙酰基传递到Ser530, 从而抑制前列腺素的合成. Alagha等[53]为了使阿司匹林能够进一步沿着COX活性位点通道前进, 同时在Arg120上保留一个盐桥, 利用分子对接模型在阿司匹林C(3)位引入了一条戊酸酯链, 制备了该类型化合物51~54 (Scheme 21).其中化合物52戊酸盐基团能够与活性位点Arg120相互作用, 同时水杨酸羧酸盐也能与Tyr385相互作用, 乙酰基与Ser530接近, 但无抗血栓活性; 进一步制备的化合物51、53、54中, 分子对接模型显示化合物53能够通过氢键与Arg120和Tyr385同时作用, 同时准确定位Ser530并将其乙酰化, 抑制血小板聚集, IC50值为(720±85) μmol•L-1.
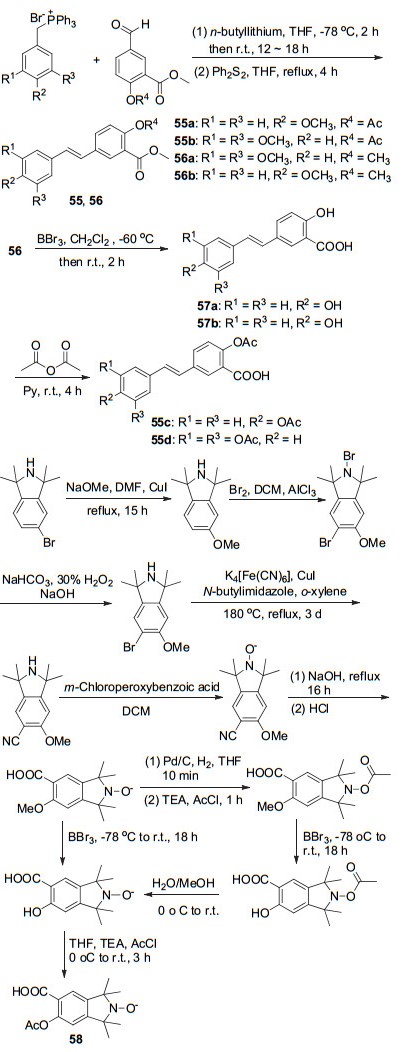
通过Wittig反应可制备阿司匹林的衍生物55a, 55b与56a, 56b, 其中56a和56b可与BBr3反应脱甲基得中间物57a和57b, 最后经乙酸酐酰化得到衍生物55c和55d (Scheme 22), 该系列化合物与白藜芦醇相比, 具有甲酯结构的化合物对致癌活性CYP1A1酶有中等抑制活性, 具有潜在的抗肿瘤活性[54].以药效团杂交的原理开发的阿司匹林衍生物58, 显示出较弱的抗炎活性[9].
阿司匹林苯环修饰不是阿司匹林的主要修饰方式.由于直接对苯环进行结构修饰较为困难, 因此通常选择结构更为简单的底物先进行苯环修饰, 再将结构调整为阿司匹林这一母体结构.制备的衍生物大多不具备活性或者活性低, 即从阿司匹林的苯环进行结构修饰不是优良的衍生方案.
阿司匹林的骨架衍生存在C(1)-COOH修饰、C(1)- COOH与C(2)-OAc同时修饰、C(2)-OAc修饰以及苯环修饰这四种, 其中以C(1)-COOH修饰为主.阿司匹林C(1)-COOH修饰的衍生物大多成酯或成酰胺, 以酯类化合物为佳.其中将阿司匹林与NO和H2S等活性供体结合是较好的选择方案, 制备获得的衍生物大多活性优良并以抗血栓为主. C(1)-COOH与C(2)-OAc同时修饰得到的衍生物活性高, 85.71%为NOSH-ASP, 且呈现出较好的抗癌活性, 但该类衍生物目前较少. C(2)-OAc修饰以及苯环修饰不是阿司匹林进行结构修饰的优选方案.
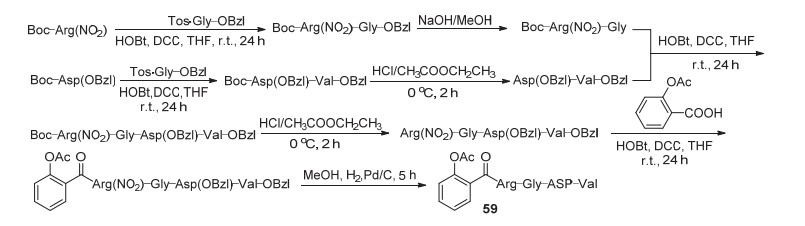
除了将阿司匹林进行结构衍生之外, 将阿司匹林制成前药也是改善其副作用与物化性质, 增强药物靶向性, 提高药理活性的较好方案.前药可分为载体前药和生物前体前药.药物载体是指能改变药物进入人体的方式和在体内的分布, 控制药物的释放速度, 并将药物输送到靶向器官的体系, 其中纳米级药物载体近年来得到迅速发展.纳米级药物载体是一种属于纳米级微观范畴的亚微粒药物载体输送系统, 可精确控制药物的生物分布、细胞靶向、体内药物稳定性、循环动力学和药物释放机制[55].阿司匹林C(1)-COOH与蛋白载体共价结合, 再还原获得衍生物59 (Scheme 23), 该化合物能够在血液中以纳米颗粒选择性地转运, 到达血栓形成部位之后与被激活的血小板表面GPIIb/Ha受体作用, 释放阿司匹林, 即通过靶向给药而发挥抗血栓活性[56].
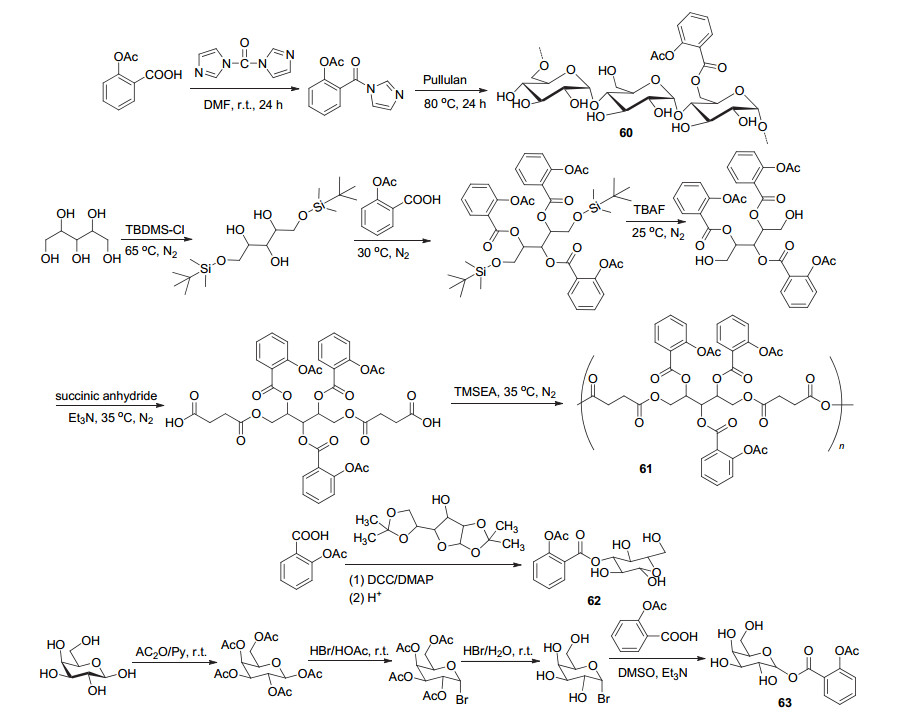
同样地, 将阿司匹林C(1)-COOH与支链淀粉载体结合获得了衍生物60 (Scheme 24), 该衍生物在阿司匹林原有的活性基础上, 还改善了阿司匹林的热稳定性与有机可溶性[57].将阿司匹林与木糖醇载体线性聚合获得衍生物61, 该化合物具有良好的细胞相容性, 并以可控的方式释放阿司匹林, 于四周之内可释放出20%的阿司匹林, 具有较好的抗炎活性[58].葡萄糖与阿司匹林偶联得到了葡萄糖-阿司匹林62, 其对SKBR3、PANC-1和PC3细胞系的抗癌活性为阿司匹林的8~9倍, 对WI38细胞系的抗癌活性与阿司匹林相近.阿司匹林由于水溶性差, 长期以来均为口服且副作用较多, 而由于葡萄糖的引入, 使得衍生物62的水溶性为阿司匹林的7倍, 为阿司匹林的注射液的发展提供了研究方向[59].阿司匹林与载体α-D-半乳糖基溴酰化制得衍生物63.体外测试显示, 该衍生物在剂量1.25~20 mmol/L时对人肺腺癌A549细胞的抑制率为6.3%~90.2%, 高于同剂量下的阿司匹林抑制率[60].
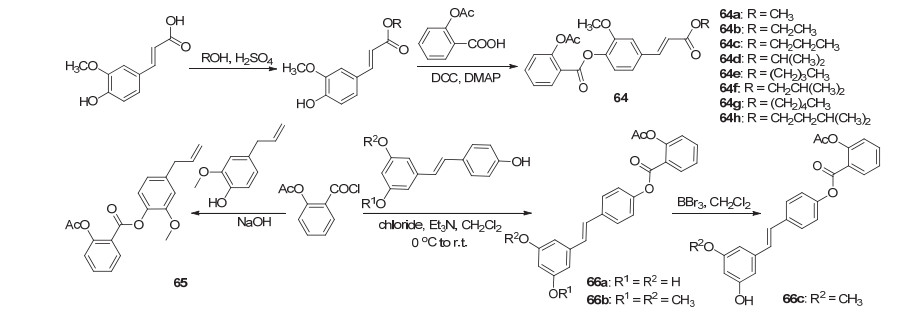
生物前体前药不同于载体前药, 活性物质无需与载体暂时性结合, 而是通过自身分子结构的改变来发挥作用.阿司匹林直接与阿魏酸单酯酯化可得衍生物64a~64h (Scheme 25), 其中64a~64c和64e~64h具有一定的抗血小板聚集活性, 它们对二磷酸腺苷体外诱导血小板聚集抑制的IC50值分别为87.4、44.7、107.4、43.1、27.6、79.9、35.8 mmol/L, 同时研究了其抗血小板聚集活性大小与取代基碳链长度以及是否含有支链的关系, 结果表明, 碳链越长其抗血小板聚集活性越好, 相同碳链的衍生物在带有支链时的抑制血小板聚集活性更佳[61].同丁香酚酯制得的阿司匹林丁香酚酯65能降低血脂水平和血管壁厚度, 防止血管内壁脂质沉积及管腔狹窄, 从而预防或冶疗动脉粥样硬化疾病[62].与此同时, 阿司匹林丁香酚酯与阿司匹林于相同剂量下有相近的抗炎活性, 两者对于由二甲苯引起的小鼠耳肿胀的抑制率均在45%左右[63]; 与白藜芦醇酯化, 再脱甲基得到了衍生物66a~66c, 其中衍生物66a表现出一定的抗癌活性, 对HCT-116、HT-29细胞的IC50值分别为(39.39±0.55)和(34.37±0.79) μmol•L-1[64].
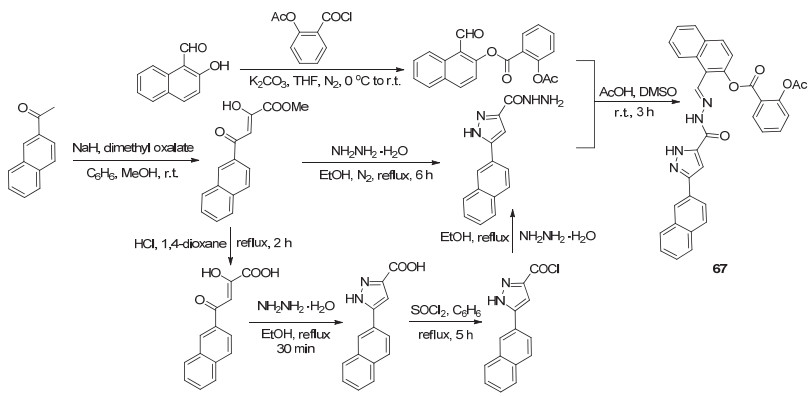
阿司匹林酰氯和2-羟基萘醛反应获得的中间产物再与可抑制肿瘤细胞生长的一种鞘氨醇激酶抑制剂酰化, 获得了阿司匹林衍生物67, 或者将阿司匹林酰氯与另一种鞘氨醇激酶抑制剂直接酰化得到了衍生物68 (Scheme 26), 体外实验显示这两种衍生物在多种肿瘤细胞均具有较好的抑制活性, 如对人脑组织中的U87MG癌细胞的IC50值分别为(1.5±0.5)和(8.1±1.6) μmol•L-1, 对肺组织中的H460、H226、H441及A549癌细胞的IC50值分别为(1.2±0.5)与(9.1±0.8)、(1.7±0.3)与(6.7±1.6)、(0.6±0.2)与(5.7±1.9)和(0.9±0.1)与(5.9±2.1) μmol•L-1[65].
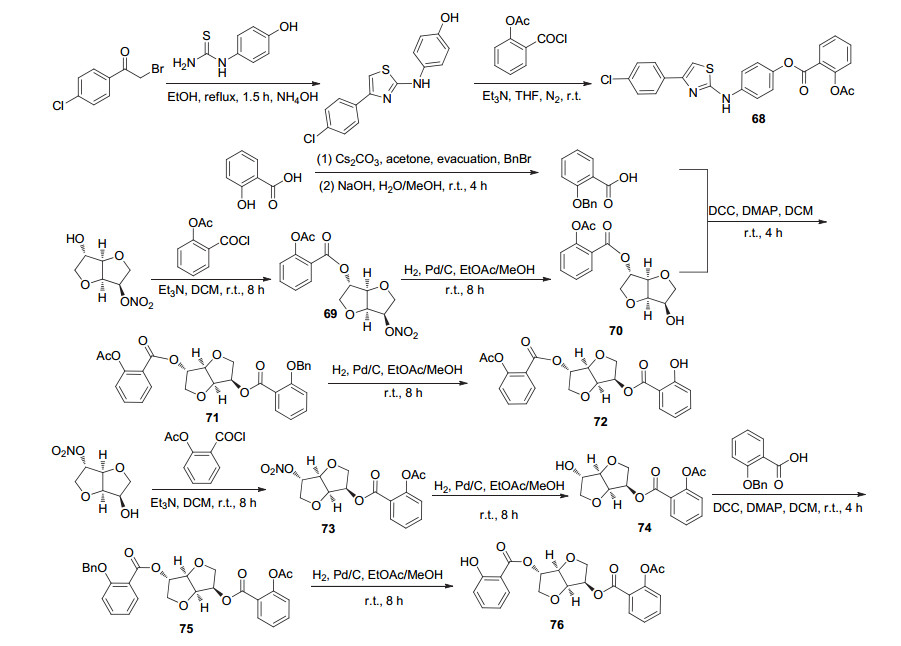
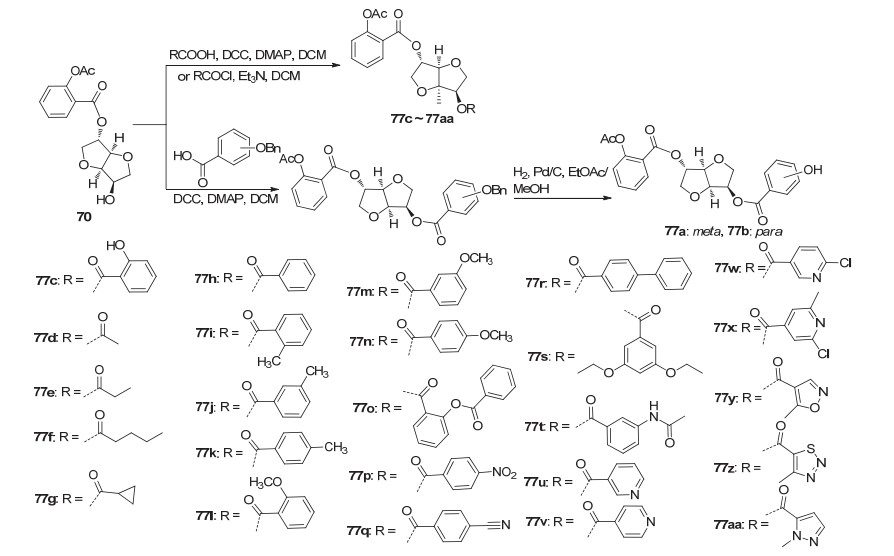
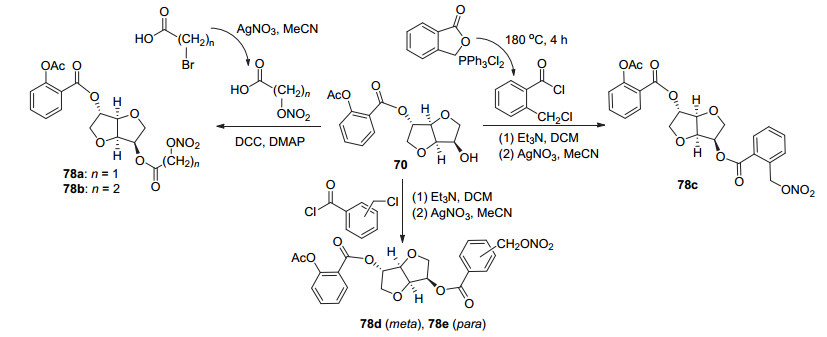
阿司匹林酰氯C(1)-COCl位与单硝酸异山梨醇于碱性条件下取代, 随后还原脱NO2, 再与2-苄氧基苯甲酸酯化, 最后还原脱苄基, 制得化合物69~76 (Scheme 27).其中化合72是一种有效的阿司匹林前药, 该化合物可通过释放阿司匹林而具有抗血栓活性[66]. Jones等[24]在化合物70的基础上, 使C(1)位衍生物的羟基与适当的酸在DCC和4-二甲氨基吡啶(DMAP)的催化下酯化获得化合物77a~77aa (Scheme 28), 其中3/4-羟基苯甲酸与3/4-羟基苯甲酸反应时, 羟基与羧基存在竞争反应, 故用苄基对3/4-羟基苯甲酸的羟基进行保护, 再参与酯化反应, 随后还原脱苄基获得化合物77a、77b; 与酸类化合物结合获得78a与78b (Scheme 29), 与2/3/4-氯甲基苯甲酰氯先取代再硝化获得化合物78c~78e.动力学研究发现, 衍生物苯甲酸基团上具有邻位取代基和空间位阻小的间位取代基时, 在人体血浆中有较好的阿司匹林释放特性, 其中衍生物78c和78d可分别释放出30%和50%的阿司匹林, 是有效的阿司匹林前药.同时在活性研究中发现, 衍生物77c、78c和78d在PRP中抑制由胶原蛋白诱导的血小板聚集的IC50值分别为20.6、90.3和17.1 μmol•L-1, 即具有较好的抗血小板聚集活性.
阿司匹林前药衍生中载体前药及生物前体前药均有涉及, 其中以生物前体前药为主.载体前药中载体多为蛋白质或糖类, 两者在提高阿司匹林水溶性、脂水分配系数及稳定性方面作用突出, 同时靶向性增加使得部分载体前药活性得到了提升.改变阿司匹林结构获得的生物前体前药, 仅为具有抗癌及抗血栓活性这两类化合物, 其中以抗血栓为主, 占比83%.
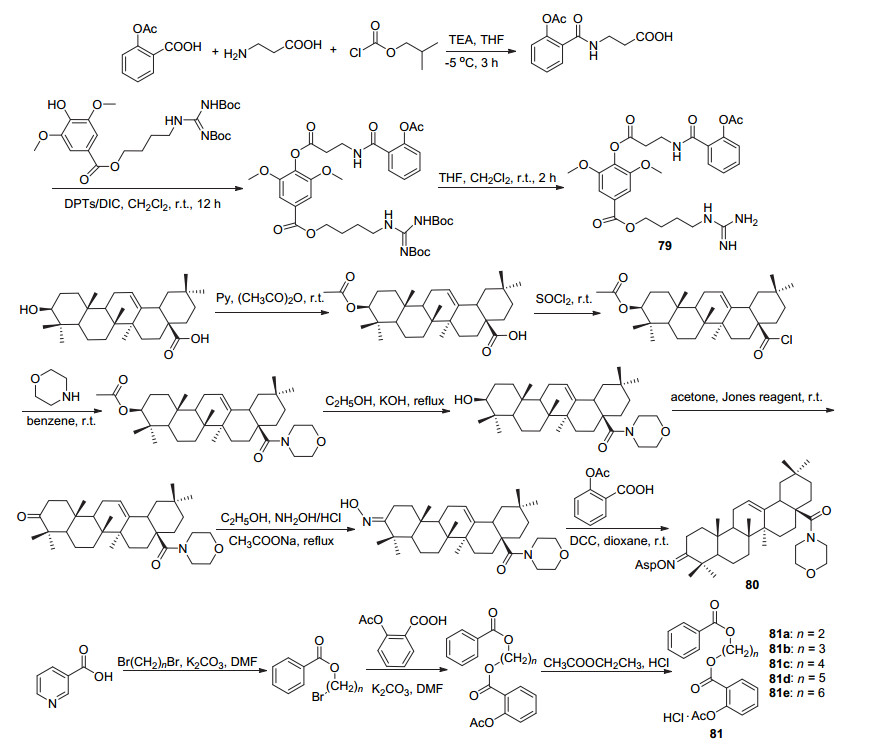
孪药是指两个或两个以上具有相似或不同治疗效果的化合物通过共价键结合在一起, 在生物体内代谢生成两种(或更多)不同的药物而产生协同作用, 用以增强活性或者产生新的药理作用以及提高药物的选择性.阿司匹林因其具有的多种药理活性, 常常与其他药物依据拼合原理进行孪药设计, 同样地, 结构修饰上主要是将C(1)-COOH成酯或成酰胺.如阿司匹林C(1)-COOH与丙氨酸在碱性条件下酰化, 再与具有心脏保护作用的益母草碱酯化获得衍生物79 (Scheme 30).在H9c2心肌细胞中, 衍生物79与益母草碱相比其药效提高了10倍左右, 并且该化合物还可通过抑制促炎介质保护心肌细胞[67]; 与3-羟基亚胺齐墩果酸在DDC作用下酯化获得化合物80, 该化合物具有抗炎镇痛活性, 其抗炎活性与该衍生物剂量呈倒“U”型关系, 在剂量30 mg/kg时显示出最大的抗炎活性[68]; 通过与烟酸酯化物取代获得化合物81a~81e, 五种衍生物的体外实验测试均呈现出一定的抗血小板聚集活性, 但其最高抑制率28.23%仍低于阿司匹林, 活性较低[69].
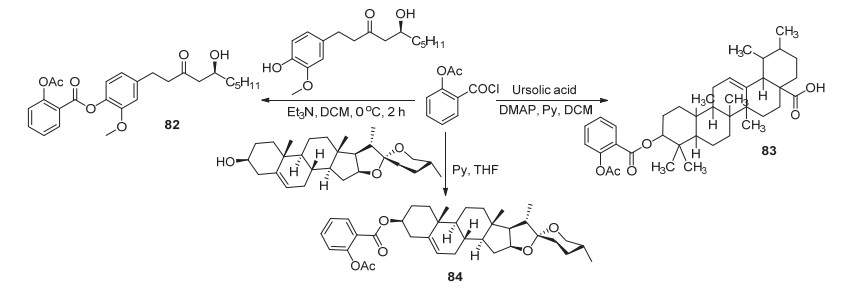
阿司匹林酰氯与6-姜酚酰化制得衍生物82 (Scheme 31), 其在HCT-116及HT-29癌细胞的IC50值分别为75.97和84.49 μmol•L-1, 抗癌活性优于阿司匹林, 且在浓度为20、40和60 μmol•L-1时均能显著抑制HCT-116和HT-29癌细胞的增殖, 60 μmol•L-1时对这两种癌细胞的抑制率高达100%和97%.除此之外, 衍生物82相较于阿司匹林而言, 具有显著的胃粘膜保护作用[70]; 与熊果酸取代制得的衍生物83对于MCF-7细胞的IC50值为70 μmol•L-1, 活性明显高于阿司匹林[71]; 与薯蓣皂苷元发生取代制得衍生物84[72], 该化合物能明显抑制血栓的形成, 其对血小板的抑制率为(29.42±3.1)%, 与阿司匹林相当[73].此外, 衍生物84还具有抗炎活性, 其在剂量为21.9 mg/kg时对小鼠耳朵的肿胀抑制率为18.03%, 并随剂量的增加抑制率不断上涨[72].
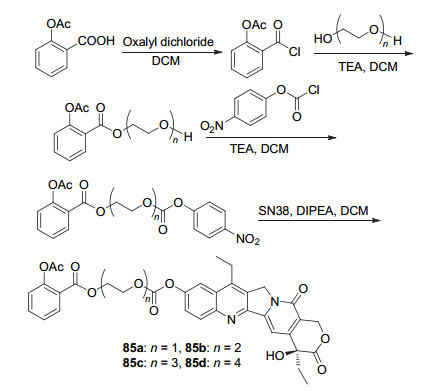
阿司匹林C(1)-COOH经酰化及酯化后, 再与7-乙基-10-羟基喜树碱(SN38)酯交换获得四种SN38-阿司匹林85a~85d (Scheme 32), 这四种衍生物均可同时释放SN38及阿司匹林, 体外实验都显示出明显的抗癌活性, 它们对于HepG2细胞的IC50值均小于0.21 μmol•L-1, 对于BEL-7404细胞的IC50值均小于6 μmol•L-1, 其中活性最强的衍生物85b对这两种癌细胞的IC50值分别为(0.1208±0.0081)和(2.5295±0.7884) μmol•L-1, 抗肿瘤活性较高[74].
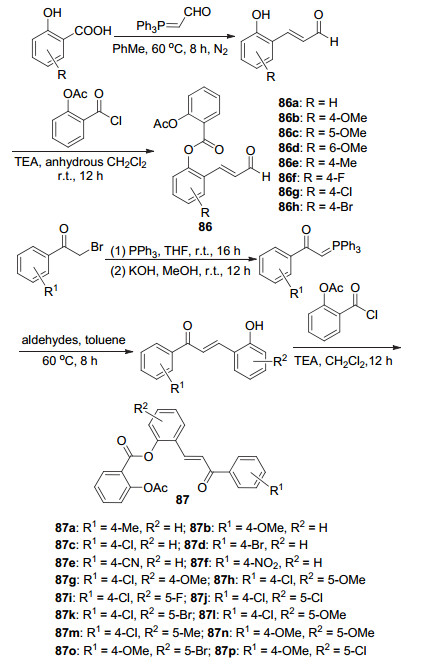
同样地, 阿司匹林酰氯与肉桂醛类化合物酰化制得了衍生物86a~86h (Scheme 33), 其中化合物86b和86e~86h对HTC-8人结直肠癌细胞的IC50值(9.3±0.7)、(8.5±0.4)、(2.6±0.6)、(4.3±0.9)和(5.6±0.7) μmol•L-1, 明显高于阿司匹林的IC50(>100 μmol•L-1), 活性最高的衍生物86f经体外测试显示出可阻碍结直肠癌细胞增殖的G1期, 同时对于DLD-1癌细胞IC50为(4.3±0.5) μmol•L-1, 相较于非癌细胞CCDB41的IC50 (30.7±2.9) μmol•L-1, 其抗增殖选择较好, 此外还可诱导癌细胞凋亡, 这些生物作用暗示衍生物86f可作为抗结直肠癌的化疗药物[75].而将通过Wittig反应制得的查尔酮类化合物, 与阿司匹林酰氯反应获得了衍生物87a~87p (Scheme 33), 其中87c、87f、87h和87i在10 μmol•L-1时对CRC癌细胞显示出强的抑制活性, 而对比衍生物结构与癌细胞抑制率发现衍生物R1取代基为Cl或NO2 (87c和87f)或R2取代基为5-OMe或5-F时的衍生物抑制率更高.其中衍生物87h和87i的抑制率最高, 活性测试得出这两种衍生物对HCT-8癌细胞的IC50值分别为(2.4±0.1)和(2.7±0.5) μmol•L-1, 对DLD-1癌细胞的IC50值分别为(2.7±0.2)和(3.5±0.4) μmol•L-1, 同样地, 87h也可阻碍结直肠癌细胞增殖的G1期, 且抗增殖选择较好, 可诱导癌细胞凋亡[76].
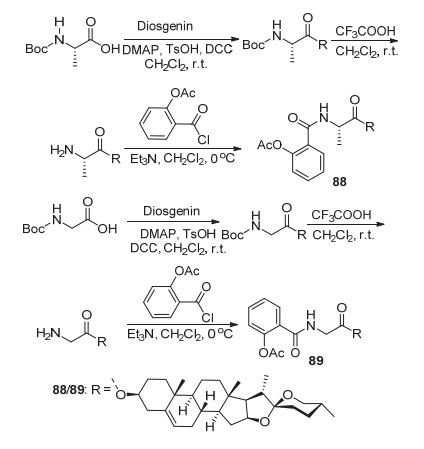
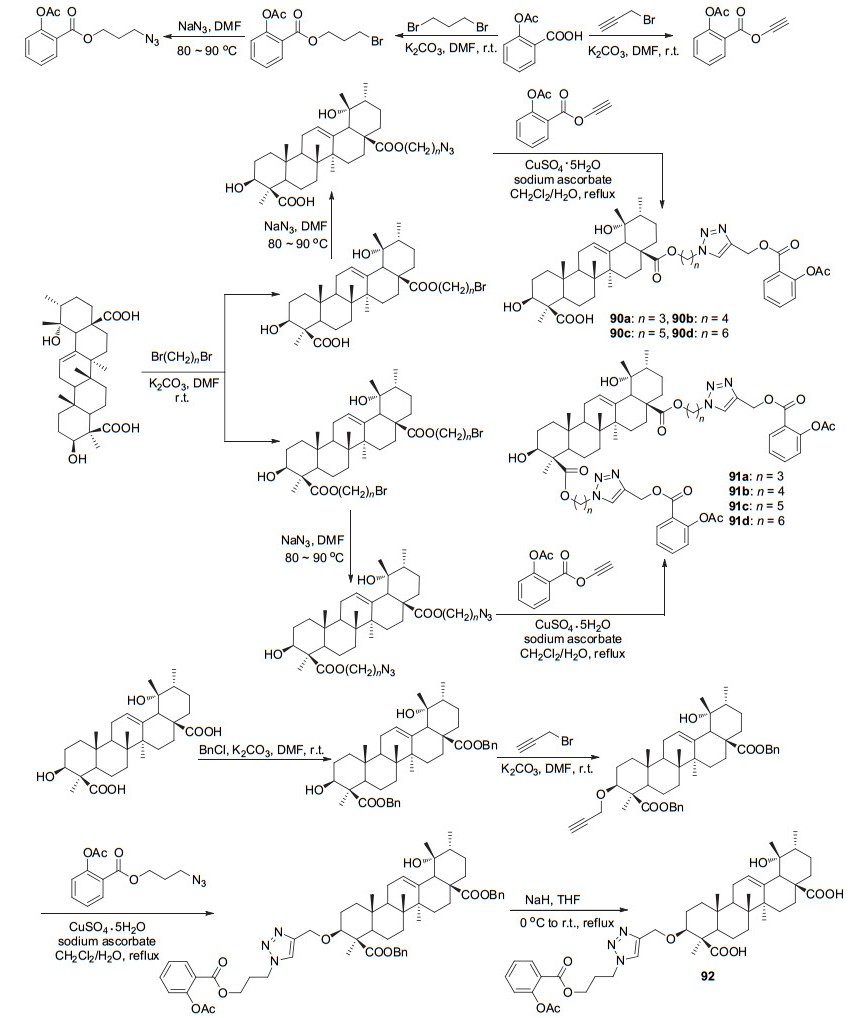
将阿司匹林酰氯C(1)-COCl与薯蓣皂苷衍生物酯化获得衍生物88和89 (Scheme 34), 两者在100 mg/kg时对水肿的抑制率分别为46.5%和39.1%, 具有抗炎活性[77].将1, 3/1, 4/1, 5/1, 6-二溴烷基作为阿司匹林和冬青素A的连接基团, 制备了衍生物90~92 (Scheme 35), 体外实验测试显示衍生物91a~91d在浓度为0.25 mmol/L时, 对由ADP诱导的血小板聚集抑制率为41.3%~77.2%, 其中衍生物91d的抑制率77.2%是阿司匹林的8倍[78].
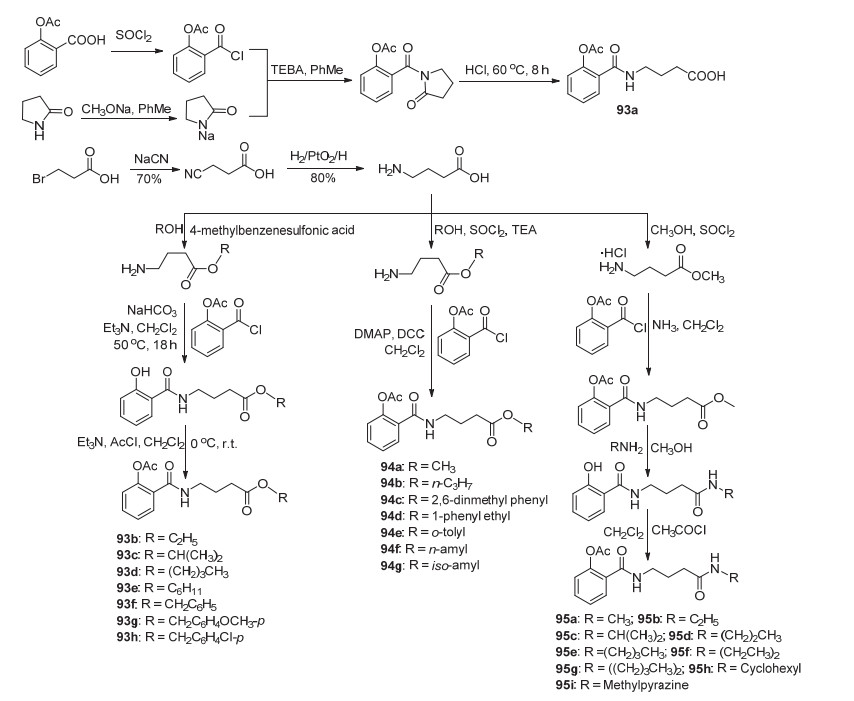
近年来发现阿司匹林具有改善精神疾病的作用, 如长期服用低剂量阿司匹林可能保护神经[79], 有预防阿尔兹海默病及改善抑郁的可能性[80]. He等[81]通过合成了系列阿司匹林抗癫痫衍生物93~95 (Scheme 36), 这些化合物均显示出一定的抗癫痫的活性.其中93a、93e~93h、94c~94h和95e~95h这13种化合物在剂量为240 mg/kg时显示出显著的抗癫痫活性, 并且有效比超过60%, 其中化合物93e~93f、93h、94c, 94d和95f~95h有效比高达83%, 93e、95h、95e和95g是活性最高的化合物; 化合物93h的ED50为0.4189 mmol/kg, LD50为1.321 mmol/kg, 化合物95g的ED50为0.3684 mmol/kg, LD50为1.1487 mmol/kg, 这两种化合物有待于开发成抗癫痫药物; 此外, 化合物95h与95g能够进入中枢神经系统释放GABA和阿司匹林从而达到抗癫痫作用.同时构效关系测试表明酯类化合物比酰胺类化合物更容易通过血脑屏障途径而释放阿司匹林.活性评价表明, 不同结构的目标化合物对活性影响较大:当端部与空间位阻较大的酯连接时, 化合物活性较高; 相反, 当空间位阻较小时, 活性较差并且毒性增加; 苯环上带有给电子基团取代基的化合物, 其抗癫痫作用降低, 毒性增加, 而吸电子基团呈现相反的效果; 脂水分配系数与ED50、LD50及治疗指数TI具有明显的相关性, 尤其是TI与脂水分配系数具有明显的平行关系.
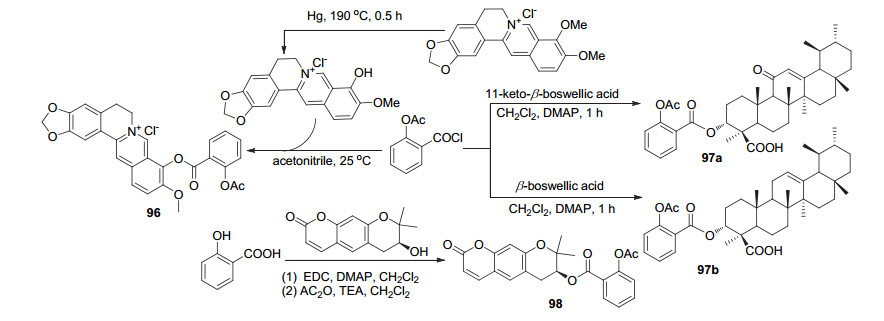
阿司匹林酰氯C(1)-COCl与脱甲基的小檗碱酯化获得衍生物96 (Scheme 37), 体外实验显示该衍生物可抑制由二甲苯引起的小鼠耳肿胀[82].阿司匹林酰氯C(1)- COCl与两种乳香脂酸反应获得衍生物97a和97b, 体内实验显示这两种衍生物在10 mg/kg剂量时, 2 h的水肿抑制率分别为58.51%和56.22%[83].水杨酸C(1)-COOH位与紫花前胡醇酯化, 再将酚羟基与乙酸酐取代获得阿司匹林衍生物98, 该衍生物20 mg/kg时可保护神经元免受短暂局灶性和全身性脑缺血损伤, 远高于同剂量下是阿司匹林治疗效果; 此外可降低缺血诱导的神经胶质增生, 维持相应损伤区域的抗氧化水平[84].
阿司匹林的孪药衍生可有效提高药理活性.阿司匹林孪药衍生物乃是阿司匹林与活性相同或相似的化合物依据拼合原理制备所得, 研发新的衍生物具有一定目的性, 可缩短新药研发的进程.孪药衍生物以同孪药为主, 少数为异孪药, 衍生物活性在抗炎、抗癌、抗血栓及保护神经系统等方面均有涉及, 其中绝大部分衍生物活性得到了极大的提高, 如衍生物91d的血小板聚集抑制率77.2%是阿司匹林的8倍.
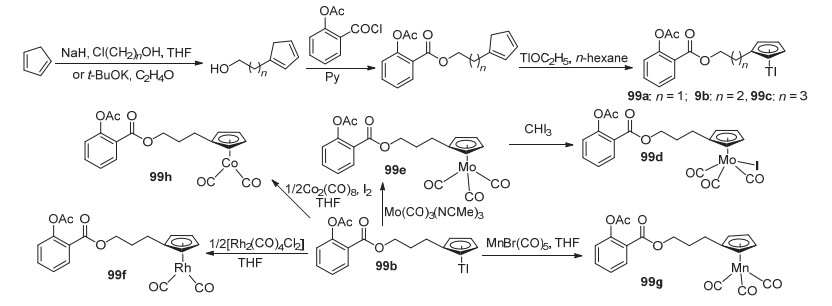
顺铂的发现意味着金属抗癌药物时代的到来[85].此后, 多种金属配合物, 如Pt、Pd、Au与Ru等相继被设计为抗癌药物并应用于临床[86].而金属化合物与阿司匹林的结合, 也成为了开发新型抗肿瘤药物的途径.将由环戊二烯制得的环戊二烯醇与阿司匹林酰氯C(1)-COCl取代得到中间产物, 随后与乙醇铊反应获得阿司匹林金属配合物99a~99c, 再在衍生物99b的基础上与Co、Mn等金属发生金属交换反应获得化合物99d~99h (Scheme 38).除衍生物99a以外, 衍生物99b~99h对于MCF-1、MDA-MB-231及HT-29癌细胞系均显示出抗癌活性, 且活性均高于阿司匹林, 如99f对三种癌细胞的IC50值分别为(10.2±3.4)、(26.6±0.5)和(9.2±1.7) μmol•L-1, 但其抗癌机制有待进一步研究.此外, 所有衍生物均能抑制COX-1和COX-2, 对COX-1的抑制率为30%, 同阿司匹林相近.但99f对COX-1的抑制率达(57.3±1.4)%, 而对COX-2的抑制率均高于阿司匹林[87].
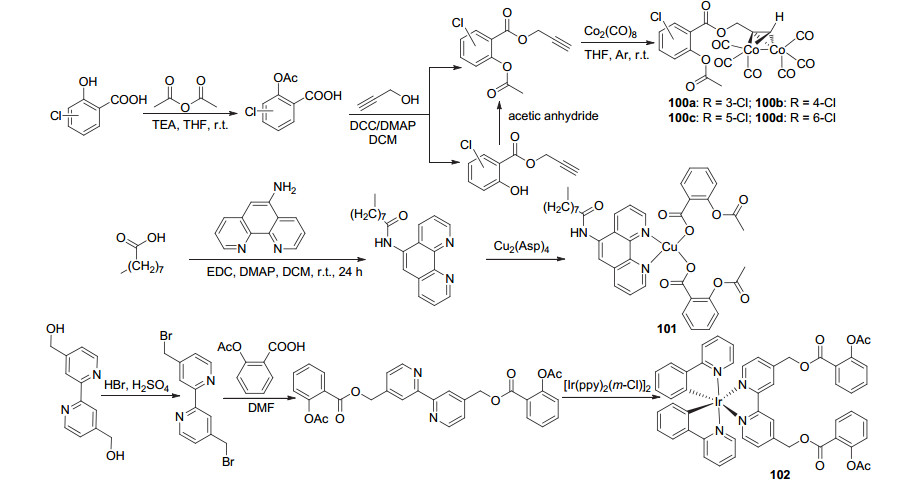
将3/4/5/6-氯水杨酸与乙酸酐结合获得3/4/5/6-氯乙酰水杨酸, 随后与丙炔醇进行酯化, 再与羰基钴结合, 由此产生阿司匹林金属抗癌衍生物3/4/5/6-Cl-Co-ASS, 100a~100d (Scheme 39), 这四种化合物在HT-29、MDA-MB-231和MCF-7癌细胞系中显示出较好的生长抑制能力, 对COX-2的抑制率达60%~80%, 如100b对这三种癌细胞的IC50值分别为(1.51±0.12)、(5.24±0.33)和(15.2±1.3) μmol•L-1[88, 89].将阿司匹林制得CuLA (101), 该化合物对SKOV-3、HeLa与HK-2三种癌细胞有强抗癌活性, 尤其是对于SKOV-3癌细胞, IC50值为(1.1±0.6) μmol•L-1, 同时, 该化合物增加了癌细胞与癌细胞线粒体中的铜含量, 使得其中SKOV-3癌细胞的ATP产量显著降低, 表明该化合物干扰了线粒体功能与细胞存活.此外, 化合物还显示出抗炎潜能, 可见具有抗肿瘤和抗炎活性的铜配合物可能代表一种新型的多功能金属配合物, 预示着抗癌药物的新趋势[90].阿司匹林与Ir配合物反应制得金属衍生物102, 其在PC3、CT26、HT29癌细胞中的IC50值分别为(4.5±0.5)、(4.4±0.7)和(2.8±0.3) μmol•L-1, 具有抗癌活性[91].
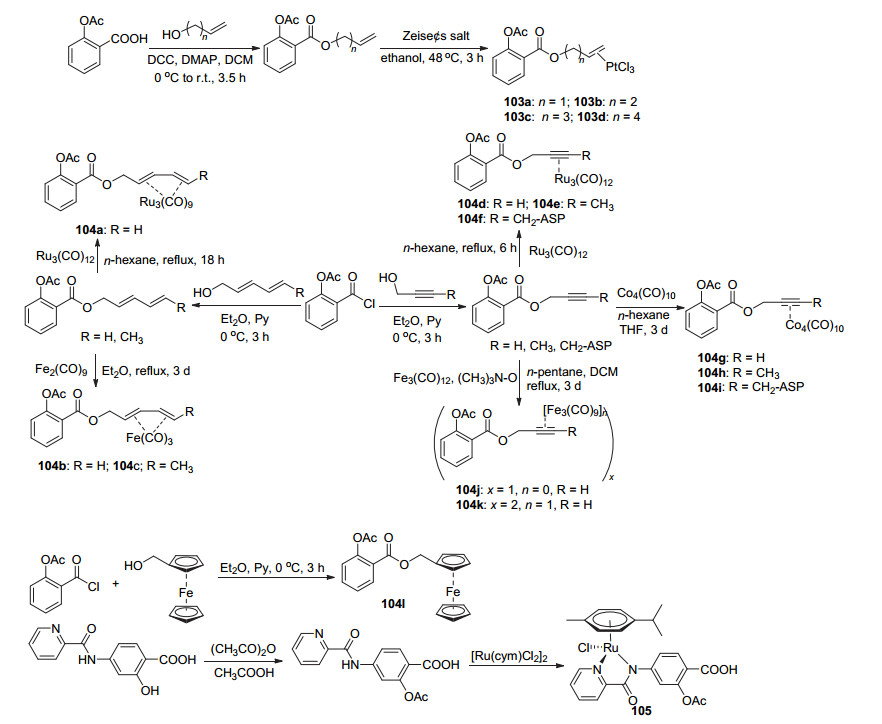
阿司匹林C(1)-COOH与脂肪醇酯化再与Pt金属配体反应制得衍生物103a~103d (Scheme 40), 这四种衍生物在10 μmol•L-1时均能够抑制COX-2与COX-1, 且对COX-2的抑制活性非常明显, 103c与103d对其抑制率近100%.此外, 衍生物103a~103d对HT-29及MCF-7癌细胞均显示出高于阿司匹林的抗癌活性, 其IC50值均在30~50 μmol•L-1[92].将阿司匹林酰氯经醇修饰, 再与金属配合物反应制得衍生物104a~104k, 或阿司匹林酰氯直接与二茂铁甲醇取代得到衍生物104l (Scheme 40), 其中104a和104d~104i对MCF-7、MDA-MB 231及HT-29癌细胞均呈现出优于阿司匹林的抗癌活性, 如104d对三种癌细胞的IC50值分别为(1.4±0.2)、2.4和(2.2±0.2) μmol•L-1[93]. Ashraf等[94]将2-羟基-4-吡啶酰氨基苯甲酸的2位羟基酰化再与金属化合物反应获得阿司匹林衍生物105, 该化合物对于NCI-H460、SiHa、HCT116和SW480癌细胞的IC50值均大于280 μmol•L-1.
金属配位衍生, 大多为阿司匹林先与简单醇类化合物进行成酯修饰, 再与无机金属配位制得.配位金属皆为Mn、Ti、Cu、Ir等过渡金属, 获得的衍生物均呈现出抗癌活性, 且活性较好, 但目前抗癌机制尚不明确.通过金属配位制备新型抗癌衍生物是阿司匹林衍生的可选方案.
阿司匹林作为人工合成药的鼻祖, 具有抗血栓、抗炎、抗肿瘤等多种生物活性, 作为非甾体抗炎药已使用上百年, 并具有良好的疗效. 2016年阿司匹林在中国的销售额为159838万元, 其中阿司匹林肠溶片及阿司匹林肠溶胶囊分别位列化学止痛药年销售第2位与第18位, 发展前景可观.阿司匹林目前有骨架衍生、前药衍生、孪药衍生及金属配位衍生四种方案, 根据修饰位点的不同, 骨架衍生进一步分为C(1)-COOH位、C(1)- COOH位与C(2)-OAc位同时修饰、C(2-)OAc位及苯环修饰, 其中C(1)-COOH位修饰的衍生物有306种, 占近20年阿司匹林衍生物总量的86%. C(1)-COOH位修饰的衍生物中抗血栓衍生物占比36%、抗炎衍生物占比11%、抗癌衍生物占比24%及其他活性衍生物15%.因此, C(1)-COOH位为结构修饰的主要位点, 并以抗血栓衍生物为主, 而在抗血栓衍生物当中又以NO-ASP为优, 如化合物14i对由胶原蛋白诱导的PRP血小板聚集抑制活性IC50值为14 μmol•L-1.前药衍生在改善阿司匹林理化性质及提高药物靶向性等方面具有突出作用, 如化合物62通过引入葡萄糖, 使得衍生物水溶性大大增加, 化合物59可选择性地于血栓部位释放出阿司匹林而发挥药效.孪药衍生使得衍生物制备目的性更强, 活性更高, 如由SN38和阿司匹林拼合所得的化合物85b, 其对HepG2细胞的IC50值为(0.1208±0.0081) μmol•L-1, 较阿司匹林相比活性大大提高.金属配位则是制备抗癌衍生物的较好选择, 涉及到的配位金属Tl、Mo、Mn、Rh、Co、Cu、Ir、Pt、Ru和Fe均为过渡金属.为改善阿司匹林的胃肠道副作用, 目前主要的解决方案有研发选择性的COX-2抑制剂, 将质子泵抑制剂与阿司匹林联合用药, 制备NO-ASP衍生物.其中采用NO-ASP方案较广, 现已报道102种NO-ASP衍生物, 占比29%.虽然阿司匹林的结构修饰取得了较好的进展, 但目前仍无法完全避免其副作用, 多数活性优良的衍生物暂未应用于临床.
Dreser, H. Pflugers Arch. 1899, 76, 306. doi: 10.1007/BF01662127
Van, F. J.; Buytenhek, M.; Nugteren, D. H.; Van, D. A. Eur. J. Biochem. 1980, 109, 1. doi: 10.1111/j.1432-1033.1980.tb04760.x
Vane, J. R. Nature (London), New Biol. 1971, 231, 232. doi: 10.1038/newbio231232a0
Tatsuguchi, A.; Matsui, K.; Shinji, Y.; Gudis, K.; Tsukui, K.; Kishida, K.; Fukuda, Y.; Sugisaki, Y.; Tokunaga, A.; Tajiri, T.; Sakamoto, C. Hum. Pathol. 2004, 35, 488. doi: 10.1016/j.humpath.2003.10.025
Denkert, C.; Winzer, K.; Hauptmann, S. Clin. Breast Cancer 2004, 4, 428. doi: 10.3816/CBC.2004.n.006
Zelenay, S.; Veen, A. G.; Böttcher, J. P.; Snelgrove, K. J.; Rogers, N.; Acton, S. E.; Chakravarty, P.; Girotti, M. R.; Marais, R.; Quezada, S. A.; Sahai, E.; Sousa, C. R. Cell 2015, 162, 1257. doi: 10.1016/j.cell.2015.08.015
Chattopadhyay, M.; Kodela, R.; Nath, N.; Dastagirzada, Y. M.; Velazquez-Martinez, C. A.; Boring, D.; Kashfi, K. Biochem. Pharmacol. 2012, 83, 715. doi: 10.1016/j.bcp.2011.12.018
Pircher, J.; Fochler, F.; Czermak, T.; Mannell, H.; Kraemer, B. F.; Wörnle, M.; Sparatore, A.; Soldato, P.D.; Pohl, U.; Krötz, F. Arterioscler., Thromb., Vasc. Biol. 2012, 32, 2884. doi: 10.1161/ATVBAHA.112.300627
Thomas, K.; Moody, T. W.; Jensen, R. T.; Tong, J.; Rayner, C. L.; Barnett, N. L.; Fairfull-Smith, K. E.; Ridnour, L. A.; Wink, D. A.; Bottle, S. E. Eur. J. Med. Chem. 2018, 147, 34. doi: 10.1016/j.ejmech.2018.01.077
Zavodnik, I. B.; Lapshina, E.; Sudnikovich, E.; Boncler, M.; Luzak, B.; Róalski, M.; Heliñska, M.; Watala, C. Pharmacol. Rep. 2009, 61, 476. doi: 10.1016/S1734-1140(09)70089-1
Zhang, Y.; Chen, S.; Wang, L.; Tang, X. Asian J. Chem. 2013, 25, 6550. doi: 10.14233/ajchem.2013.14355
Roy, J.; Adili, R.; Kulmacz, R.; Holinstat, M.; Das, A. J. Pharmacol. Exp. Ther. 2016, 359, 134. doi: 10.1124/jpet.116.234781
Li, Y. X.; Yu, L. B.; Zhang, Y. C.; Li, C. X. CN 1616404A 2005.
Liang, R. M.; Cheng, C. R.; Liu, Y.; Zheng, Z.; Xu, K.; Yang, R.; Ding, J.; Liang, X. Y. CN 108129468, 2018.
Plano, D.; Karelia, D.; Pandey, M.; Spallholz, J. E.; Amin, S. G.; Sharma, A. K. J. Med. Chem. 2016, 59, 1946. doi: 10.1021/acs.jmedchem.5b01503
Moon, H. S.; Nam, S. I.; Kim, S. D.; Kim, D. Y.; Gwag, B. J.; Lee, Y. A.; Yoon, S. H. J. Pharm. Pharmacol. 2002, 54, 935. doi: 10.1211/002235702760089054
Mancuso, R.; Ferlazzo, N.; Luca, G. D.; Amuso, R.; Piccionello, A. P.; Giofrè, S. V.; Navarra, M.; Gabriele, B. Med. Chem. Res. 2019, 28, 292. doi: 10.1007/s00044-018-02284-3
Ayyadevara, S.; Bharill, P.; Dandapat, A.; Hu, C.; Khaidakov, M.; Mitra, S.; Shmookler Reis, R. J.; Mehta, J. L. Antioxid. Redox Signaling 2013, 18, 481. doi: 10.1089/ars.2011.4151
Strong, R.; Miller, R. A.; Astle, C. M.; Floyd, R. A.; Flurkey, K.; Hensley, K. L.; Javors, M. A.; Leeuwenburgh, C.; Nelson, J. F.; Ongini, E.; Nadon, N. L.; Warner, H. R.; Harrison, D. E. Aging Cell 2008, 7, 641. doi: 10.1111/j.1474-9726.2008.00414.x
Cha, B. C.; Lee, S. B. Arch. Pharmacal Res. 2000, 23, 116. doi: 10.1007/BF02975499
Li, L.; Hsu, A.; Moore, P. K. Pharmacol. Ther. 2009, 123, 386. doi: 10.1016/j.pharmthera.2009.05.005
Godo, S.; Sawada, A.; Saito, H.; Ikeda, S.; Enkhjargal, B.; Suzuki, K.; Tanaka, S.; Shimokawa, H. Arterioscler., Thromb., Vasc. Biol. 2016, 36, 97. doi: 10.1161/ATVBAHA.115.306499
Liu, X.; El-Mahdy, M. A.; Boslett, J.; Varadharaj, S.; Hemann, C.; Abdelghany, T. M.; Ismail, R. S.; Little, S. C.; Zhou, D.; Thuy, L. T. T.; Kawada, N.; Zweier, J. L. Nat. Commun. 2017, 8, 1. doi: 10.1038/s41467-016-0009-6
Jones, M.; Inkielewicz, I.; Medina, C.; Santos-Martinez, M. J.; Radomski, A.; Radomski, M. W.; Lally, M. N.; Moriarty, L. M.; Gaynor, J.; Carolan, C. G.; Khan, D.; O'Byrne, P.; Harmon, S.; Holland, V.; Clancy, J. M.; Gilmer, J. F. J. Med. Chem. 2009, 52, 6588. doi: 10.1021/jm900561s
Xiao, M.; Yang, H.; Kleina, S. M.; Muenyib, C. M.; Stone, W. L.; Jiang, Y. L. Lett. Org. Chem. 2008, 5, 510. doi: 10.2174/157017808785982257
Lazzarato, L.; Donnola, M.; Rolando, B.; Chegaev, K.; Marini, E.; Cena, C.; Stilo, A. D.; Fruttero, R.; Biondi, S.; Ongini, E.; Gasco, A. J. Med. Chem. 2009, 52, 5058. doi: 10.1021/jm900587h
周洲, 赖宜生, 张奕华, 季晖, 李立文, 彭司勋, 有机化学, 2008, 28, 819. http://sioc-journal.cn/Jwk_yjhx/CN/Y2008/V28/I05/819Zhou, Z.; Lai, Y. S.; Zhang, Y. H.; Ji, H.; Li, L. W.; Peng, S. X. Chin. J. Org. Chem. 2008, 28, 819 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/Y2008/V28/I05/819
Lazzarato, L.; Chegaev, K.; Marini, E.; Rolando, B.; Borretto, E.; Guglielmo, S.; Joseph, S.; Stilo, A. D.; Fruttero, R.; Gasco, A. J. Med. Chem. 2011, 54, 5478. doi: 10.1021/jm2004514
项光亚, 周军, 陈述增, 吴俊, 罗智, 华中科技大学学报(医学版), 2007, 36, 23. doi: 10.3870/j.issn.1672-0741.2007.01.007Xiang, G. Y.; Zhou, J.; Chen, S. Z.; Wu, J.; Luo, Z. J. Huazhong Univ. Sci. Technol. (Health Sci.) 2007, 36, 23 (in Chinese). doi: 10.3870/j.issn.1672-0741.2007.01.007
Cena, C.; Lolli, M. L.; Lazzarato, L.; Guaita, E.; Morini, G.; Coruzzi, G.; McElroy, S. P.; Megson, I. L.; Fruttero, R.; Gasco, A. J. Med. Chem. 2003, 46, 747. doi: 10.1021/jm020969t
Szőke, K.; Czompa, A.; Lekli, I.; Szabados-Fürjesi, P.; Herczeg, M.; Csávás, M.; Borbás, A.; Herczegh, P.; Tósaki, A. Eur. J. Pharm. Sci. 2019, 131, 159. doi: 10.1016/j.ejps.2019.02.020
Larin, A. A.; Fershtat, L. L.; Ustyuzhanina, N. E.; Gening, M. L.; Nifantiev, N. E.; Makhova, N. N. Mendeleev Commun. 2018, 28, 595. doi: 10.1016/j.mencom.2018.11.010
Ferioli, R.; Folco, G. C.; Ferretti, C.; Gasco, A. M.; Medana, C.; Fruttero, R.; Civelli, M.; Gasco, A. Br. J. Pharmacol. 1995, 114, 816.
周洲, 蒋丽媛, 张奕华, 季晖, 孙易, 彭司勋, 药学学报, 2006, 41, 1050. doi: 10.3321/j.issn:0513-4870.2006.11.005Zhou, Z.; Jiang, L. Y.; Zhang, Y. H.; Ji, H.; Sun, Y.; Peng, S. X. Acta Pharm. Sin. 2006, 41, 1050 (in Chinese). doi: 10.3321/j.issn:0513-4870.2006.11.005
周洲, 蒋丽媛, 张奕华, 季晖, 孙易, 彭司勋, 有机化学, 2006, 26, 1403. doi: 10.3321/j.issn:0253-2786.2006.10.011Zhou, Z.; Jiang, L. Y.; Zhang, Y. H.; Ji, H.; Sun, Y.; Peng, S. X. Chin. J. Org. Chem. 2006, 26, 1403 (in Chinese). doi: 10.3321/j.issn:0253-2786.2006.10.011
Velázquez, C. A.; Chen, Q.; Citro, M. L.; Keefer, L. K.; Knaus, E. E. J. Med. Chem. 2008, 51, 1954. doi: 10.1021/jm701450q
Basudhar, D.; Bharadwaj, G.; Cheng, R. Y.; Jain, S.; Shi, S.; Heinecke, J. L.; Holland, R. L.; Ridnour, L. A.; Caceres, V. M.; Spadari-Bratfisch, R. C.; Paolocci, N.; Velázquez-Martínez, C. A.; Wink, D. A.; Miranda, K. M. J. Med. Chem. 2013, 56. 7804. doi: 10.1021/jm400196q
Samad, M. K.; Hawaiz, F. E. Bioorg. Chem. 2019, 85, 431. doi: 10.1016/j.bioorg.2019.01.014
Ispir, E.; Ikiz, M.; Inan, A.; Sunbul, A. B.; Tayhan, S. E.; Bilgin, S.; Kose, M.; Elmastas, M. J. Mol. Struct. 2019, 1182, 63. doi: 10.1016/j.molstruc.2019.01.029
Pradeepa, S. M.; Naik, H. S. B.; Kumar, B. V.; Priyadarsini, K. I.; Barik, A.; Prabhakara, M. C. Spectrochim. Acta, Part A 2015, 141, 34. doi: 10.1016/j.saa.2015.01.019
Ho, B. K.; Ngaini, Z.; Neilsen, P. M.; Hwang, S. S.; Linton, R. E.; Kong, E. L.; Lee, B. K. J. Chem. 2017, 2017, 1.
Ngaini, Z.; Mortadza, N. A. Nat. Prod. Res. 2019, 33, 3507. doi: 10.1080/14786419.2018.1486310
Nordin, N. A.; Chai, T. W.; Tan, B. L.; Choi, C. L.; Halim, A. N. A.; Hussain, H.; Ngaini, Z. J. Chem. 2017, DOI: 10.1155/2017/2378186.
Ngaini, Z.; Arif, M. A. M.; Hussain, H.; Mei, E. S.; Tang, D.; Halimatulzahrah, D.; Kamaluddin, A. Phosphorus, Sulfur Silicon Relat. Elem. 2012, 187, 1. doi: 10.1080/10426507.2011.562398
Zhen, X.; Zong, M.; Gao, S.; Cao, Y.; Jiang, L.; Chen, S.; Wang, K.; Sun, S.; Peng, H.; Bai, Y.; Li, S. PLoS One 2014, 9, 1.
Vannini, F.; MacKessack-Leitch, A. C.; Eschbach, E. K.; Chattopadhyay, E. K.; Kodela, R.; Kashfi, K. Bioorg. Med. Chem. Lett. 2015, 25, 4677. doi: 10.1016/j.bmcl.2015.08.023
Kodela, R.; Chattopadhyay, M.; Kashfi, K. ACS Med. Chem. Lett. 2012, 3, 257. doi: 10.1021/ml300002m
Lazzarato, L.; Donnola, M.; Rolando, B.; Marini, E.; Cena, C.; Coruzzi, G.; Guaita, E.; Morini, G.; Fruttero, R.; Gasco, A.; Biondi, S.; Ongini, E. J. Med. Chem. 2008, 51, 1894. doi: 10.1021/jm701104f
Bateman, L. A.; Zaro, B. W.; Miller, S. M.; Pratt, M. R. J. Am. Chem. Soc. 2013, 135, 14568. doi: 10.1021/ja408322b
Lazzarato, L.; Cena, C.; Rolando, B.; Marini, E.; Lolli, M. L.; Guglielmo, S.; Guaita, E.; Morini, G.; Coruzzi, G.; Fruttero, R.; Gasco, A. Bioorg. Med. Chem. 2011, 19, 5852. doi: 10.1016/j.bmc.2011.08.018
Aguiar, R. P.; Aldawsari, F. S.; Wiirzler, L. A. M.; Silva-Filho, S. E.; Silva-Comar, F. M. S.; Bersani-Amado, C. A.; Velázquez-Martínez, C. A.; Cuman, R. K. N. Curr. Pharm. Des. 2017, 23, 6841.
Huang, X. B.; Wu, G. S.; Ke, L. Y.; Zhou, X. G.; Wang, Y. H.; Luo, H. R. Molecules 2018, 23, 1359. doi: 10.3390/molecules23061359
Alagha, A.; Moman, E.; Adamo, M. F. A.; Nolan, K. B.; Chubb, A. J. Bioorg. Med. Chem. Lett. 2009, 19, 4213. doi: 10.1016/j.bmcl.2009.05.120
Aldawsari1, F. S.; Elshenawy, O. H.; Gendy, M. A. M. E; Aguayo-Ortiz, R.; Baksh, S.; El-Kadi, A. O. S.; Velazquez-Martınez, C. A. J. Enzyme Inhib. Med. Chem. 2014, 30, 884.
Caldorera-Moore, M.; Guimard, N.; Shi, L.; Roy, K. Expert Opin. Drug Delivery 2010, 7, 479. doi: 10.1517/17425240903579971
Peng, S. Q.; Zhao, M.; Wu, J. H.; Wang, Y. J.; Ma, H. P. CN 104211763, 2014
Hussain, M. A.; Abbas, K.; Lodhi, B. A.; Sher, M.; Ali, M.; Tahir, M. N.; Tremel, W.; Iqbal, S. Arabian J. Chem. 2017, 10, 1579.
Dasgupta, Q.; Movva, S.; Chatterjee, K.; Madras, K. Int. J. Pharm. 2017, 528, 732. doi: 10.1016/j.ijpharm.2017.06.065
Jacob, J. N.; Tazawa, M. J. Bioorg. Med. Chem. Lett. 2012, 22, 3168. doi: 10.1016/j.bmcl.2012.03.053
Huang, G.; Cheng, H.; Liu, Y.; Hu, J. Saudi Pharm. J. 2018, 26, 263. doi: 10.1016/j.jsps.2017.12.001
李宝泉, 李念光, 冯锋, 唐于平, 段金廒, 中国药科大学学报, 2009, 40, 486. http://www.zgykdxxb.cn/jcpu/ch/reader/view_abstract.aspx?file_no=20090602Li, B. Q.; Li, N. G.; Feng, F.; Tang, Y. P.; Duan, J. A. J. China Pharm. Univ. 2009, 40, 486 (in Chinese). http://www.zgykdxxb.cn/jcpu/ch/reader/view_abstract.aspx?file_no=20090602
Li, J. Y.; Liu, X. W.; Yang, Y. J.; Ma, N.; Sun, X. J.; Li, S. H.; Qin, Z.; Du, W. B.; Jiao, Z. H. CN 105796575, 2016.
Li, J.; Yu, Y.; Wang, Q.; Zhang, J.; Yang, Y.; Li, B.; Zhou, X.; Niu, J.; Wei, X.; Liu, X.; Liu, Z. Med. Chem. Res. 2012, 21, 995. doi: 10.1007/s00044-011-9609-1
Zhu, Y.; Fu, J.; Shurlknight, K. L.; Soroka, D. K.; Hu, Y.; Chen, X.; Sang, S. J. Med. Chem. 2015, 58, 6494. doi: 10.1021/acs.jmedchem.5b00536
Sharma, A. K.; Sk, U. S.; Gimbor, M. A.; Hengst, J. A.; Wang, X.; Yun, J.; Amin, S. Eur. J. Med. Chem. 2010, 45, 4149. doi: 10.1016/j.ejmech.2010.06.005
Moriarty, L. M.; Lally, M. N.; Carolan, C. G.; Jones, M.; Clancy, J. M.; Gilmer, J. F. J. Med. Chem. 2008, 51, 7991. doi: 10.1021/jm801094c
Gao, H.; Yang, X. H.; Gu, X. F.; Zhu, Y. Z. Bioorg. Med. Chem. Lett. 2016, 26, 4650. doi: 10.1016/j.bmcl.2016.08.058
Bednarczyk-Cwynar, B.; Wachowiak, N.; Szulc, M.; Kaminska, E.; Bogacz, A.; Bartkowiak-Wieczorek, J.; Zaprutko, L.; Mikolajczak, P. L. Front. Pharmacol. 2016, 7, 1.
何黎琴, 陈维珍, 王效山, 中国药物化学杂志, 2011, 21, 32. http://www.cnki.com.cn/Article/CJFDTotal-ZGYH201101007.htmHe, L. Q.; Chen, W. Z.; Wang, X. S. Chin. J. Med. Chem. 2011, 21, 32 (in Chinese). http://www.cnki.com.cn/Article/CJFDTotal-ZGYH201101007.htm
Zhu, Y.; Wang, F.; Zhao, Y.; Wang, P.; Sang, S. Sci. Rep. 2017, 7, 40119 doi: 10.1038/srep40119
Shao, J. W.; Tang, Q.; Liu, Y. J.; Yang, X. CN 105111271, 2015.
马茂华, 吴晓华, 何杨, 黄文, 四川大学学报(医学版), 2011, 42, 494. http://d.wanfangdata.com.cn/Periodical/hxykdxxb201104011Ma, M. H.; Wu, X. H.; He, Y.; Huang, W. J. Sichuan Univ. (Med. Sci. Ed.) 2011, 42, 494 (in Chinese). http://d.wanfangdata.com.cn/Periodical/hxykdxxb201104011
Zheng, H.; Wei, Z.; Xin, G.; Ji, C.; Wen, L.; Xia, Q.; Niu, H.; Huang, W. Bioorg. Med. Chem. Lett. 2016, 26, 3364. doi: 10.1016/j.bmcl.2016.05.032
Chen, Z.; Luo, Y.; Fang, A.; Fan, C.; Zeng, C. Turk. J. Chem. 2018, 42, 929.
Lu, S.; Obianom, O. N.; Ai, Y. Bioorg. Med. Chem. Lett. 2018, 28, 2869. doi: 10.1016/j.bmcl.2018.07.032
Lu, S.; Obianom, O. N.; Ai, Y. MedChemComm 2018, 9, 1722. doi: 10.1039/C8MD00284C
Huang, B.; Du, D.; Zhang, R.; Wu, X.; Xing, Z.; He, Y.; Huang, W. Bioorg. Med. Chem. Lett. 2012, 22, 7330. doi: 10.1016/j.bmcl.2012.10.086
Lin, L.; Wu, F.; Liang, J. Chin. Chem. Lett. 2013, 24, 723. doi: 10.1016/j.cclet.2013.05.021
Kern, S.; Skoog, I.; Östling, S.; Kern, J.; Börjesson-Hanson, A. BMJ Open. 2012, 2, 1288.
Berk, M.; Dean, O.; Drexhage, H.; McNeil, J. J.; Moylan, S.; Neil, A. O.; Davey, C. G.; Sanna, L.; Maes, M. BMC Med. 2013, 11, 74. doi: 10.1186/1741-7015-11-74
He, D.; Ma, J.; Shi, X.; Zhao, C.; Hou, M.; Guo, Q.; Ma, S.; Li, X.; Zhao, P.; Liu, W.; Yang, Z.; Mou, J.; Song, P.; Zhang, Y.; Li, J. Chem. Pharm. Bull. 2014, 62, 967. doi: 10.1248/cpb.c14-00329
Liu, Z.; Wang, X.; Zhang, H.; Zhang, S.; Li, Y.; Liu, L.; Peng, D. Med. Chem. Res. 2017, 26, 672. doi: 10.1007/s00044-017-1787-z
Chaturvedi, D.; Dwivedi, P. K.; Chaturvedi, A. K.; Mishra, N.; Siddiqui, H. H.; Mishra, Y. Med. Chem. Res. 2015, 24, 2799. doi: 10.1007/s00044-015-1331-y
Yan, B. C.; Park, J. H.; Shin, B. N.; Ahn, J. H.; Kim, I. H.; Lee, J. C.; Yoo, K. Y.; Hwang, I. K.; Choi, J. H.; Park, J. H.; Lee, Y. L.; Suh, H. W.; Jun, J. G.; Kwon, Y. G.; Kim, Y. M.; Kwon, S. H.; Her, S.; Kim, J. S.; Hyun, B. H.; Kim, C. K.; Cho, J. H.; Lee, C. H.; Won. M. H. PLoS One 2013, 8, 1.
Dasari, S.; Tchounwou, P. B. Eur. J. Pharmacol. 2014, 740, 364. doi: 10.1016/j.ejphar.2014.07.025
Trudu, F.; Amato, F.; Vanhara, P.; Pivetta, T.; Pena-Mendez, E. M.; Havel, J. J. Appl. Biomed. 2015, 13, 79. doi: 10.1016/j.jab.2015.03.003
Rubner, G.; Bensdorf, K.; Wellner, A.; Bergemann, S.; Ott, I.; Gust, R. Eur. J. Med. Chem. 2010, 45, 5157. doi: 10.1016/j.ejmech.2010.08.028
Obermoser, V.; Baecker, D.; Schuster, C.; Braun, V.; Kircherb, B.; Gust, R. Dalton Trans. 2018, 47, 4341. doi: 10.1039/C7DT04790H
Ott, I.; Kircher, B.; Bagowski, C. P.; Vlecken, D. H. W.; Ott, E. B.; Will, J.; Bensdorf, K.; Sheldrick, W. S.; Gust, R. Angew. Chem., Int. Ed. 2009, 48, 1160. doi: 10.1002/anie.200803347
Shi, X.; Fang, H.; Guo, Y.; Yuan, H.; Guo, Z.; Wang, X. J. Inorg. Biochem. 2019, 190, 38. doi: 10.1016/j.jinorgbio.2018.10.003
Wu, X. W.; Zheng, Y.; Wang, F. X.; Cao, J. J.; Zhang, H.; Zhang, D. Y.; Tan, C. P.; Ji, L. N.; Mao, Z. W. Chem.-Eur. J. 2019, 25, 7012. doi: 10.1002/chem.201900851
Weninger, A.; Baecker, D.; Obermoser, V.; Egger, D.; Wurst, K.; Gust, R. Int. J. Mol. Sci. 2018, 19, 1612. doi: 10.3390/ijms19061612
Rubner, G.; Bensdorf, K.; Wellner, A.; Kircher, B.; Bergemann, S.; Ott, I.; Gust, R. J. Med. Chem. 2010, 53, 6889. doi: 10.1021/jm101019j
Ashraf, A.; Hanif, M.; Kubanik, M.; Sohnel, T.; Jamieson, S. M. F.; Bhattacharyya, A.; Hartinger, C. G. J. Organomet. Chem. 2017, 837, 31.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:


 下载:
下载:






































