























































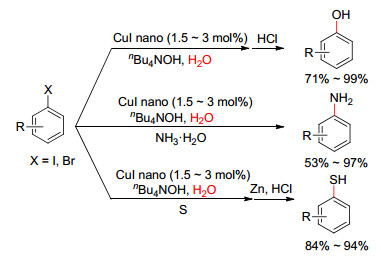
图式 1.
纳米铜催化合成苯酚、苯胺和苯硫酚
Scheme 1.
CuI-nanoparticles-catalyzed synthesis of phenols, anilines and thiophenols

芳香胺是一类重要的有机结构单元广泛存在于各种具有生理活性的天然产物、医药、农药及合成材料中[1].因此, 对于芳香胺的合成研究工作一直受到有机合成化学家的高度重视.其中, 过渡金属(特别是铜和钯)催化的偶联反应是高效、专一构建C—N键最重要的方法[2].
20世纪80年代初, 钯首次被用于催化溴苯的胺化反应[3].此后在Buchwald、Hartwig等众多科研小组的研究下, 该类反应可以在相对温和的条件下实现.然而该方法由于存在钯催化剂昂贵, 以及对有毒膦配体的强烈依赖等缺点, 限制了它的进一步应用.因此, 寻找廉价低毒的高效催化剂, 成为化学家发展金属催化C—N偶联反应的一个重要目标.
铜作为廉价低毒的金属, 已被用于经典的Ullmann反应中[4].然而, 该反应存在底物适应性窄, 催化剂铜盐用量大(等量甚至过量)、反应温度高(150~300 ℃)和后处理困难等问题.为了解决这些问题, 一些化学家发现向反应体系中加入有机双齿配体(一般为N, N型、N, O型和O, O型), 不仅避免了钯催化反应中的β-H消除[5]及双键移位[6]等副反应, 而且改善了铜催化剂在反应体系中的溶解性, 进而有效提高了铜催化偶联反应的反应效率.这使得铜/配体(copper/ligand)催化的Ullmann型(Ullmann-type)反应得到了快速的发展[7].
绿色化学是21世纪化工与化学发展的重要方向之一[8].选择环境友好的反应介质是绿色化学研究中的有效手段之一.水作为清洁, 安全和廉价的溶剂, 是绿色化学较为理想的介质.与有机溶剂相比, 水作为反应溶剂有其独特的优越性: (1)价廉易得, 安全性能高, 环境友好; (2)用水作反应溶剂有可能免除官能团的保护和去保护, 从而减少合成步骤, 提高合成效率; (3)在某些有机反应中, 水的存在可以有效促进反应发生及加快反应速率和提高反应的选择性[9].然而, 在水相反应中, 也存在一些挑战, 例如反应底物和催化剂的水溶性和稳定性、额外相转移催化剂的加入带来新的污染、产物分离仍需要有机溶剂等.近年来, 有不少的综述文章总结了铜催化Ullmann偶联反应的发展情况[7].从铜-配体催化体系角度, 总结了近些年在水介质或者纯水中, 铜催化C—N偶联反应的研究进展.
2007年, SanMartin课题组[10]以2-邻氨基苯取代苯并呋喃衍生物为原料, 在CuI/N, N'-二甲基乙二胺(L1)催化下, 实现了生物活性物质苯并呋喃吲哚杂环类物质的合成(Eq. 1).在该反应体系中, L1不仅作为配体, 还起到碱的作用.同时作者发现, 非水条件下, 需要两倍长的反应时间, 且产率只有73%.
|
|
(1) |
随后, 该课题组[11]又研究了纯水中, 链状及环状二胺类配体对CuI催化的邻溴芳基脲类化合物分子内关环反应的影响(Eq. 2).通过筛选, 确定了该反应最佳的催化体系为CuI/N, N, N', N'-四甲基乙二胺(L2), L2同样起到配体和碱的作用.在此催化体系下, 以51%~92%的产率制得多种苯并咪唑酮类衍生物.
|
|
(2) |
2008年, Taillefer和Majoral等[12]以氨基噻唑膦(L3)为配体, 在CuI催化下, 实现了溴苯或β-溴代苯乙烯与吡唑的偶联反应(Eq. 3).反应溶剂可以是水/乙腈或水/甲基异丁基酮混合体系.如果是纯水溶剂, 则需要添加20 mol%的相转移催化剂十六烷基三乙基溴化铵才能获得高产率的N-芳基化产物.
|
|
(3) |
2010年, Nageswar课题组[13]建立了CuI/反-1, 2-环己二胺(L4)催化体系, 并将其用于纯水相中芳基碘代物或溴代物与吲哚的偶联反应, 得到了一系列N-芳基吲哚类衍生物(Eq. 4).对于其它含氮化合物, 如吡唑、咪唑、苯胺、硫代苯甲酰胺和脂肪胺, 催化体系也表现出较高的催化活性.以碘苯和吲哚的偶联反应为模型, 反应结束后, 用乙酸乙酯萃取有机物, 水相溶液中再次加入反应底物, 可继续发生偶联反应, 如此循环四次, 催化活性没有明显减低.
|
|
(4) |
2011年, 彭进松等[14]以N-邻卤代芳基脒为原料, 在Cu2O催化下研究了水相中分子内C—N交叉偶联反应(Eq. 5).通过筛选配体, 发现以L1为配体时, 可以得到23%~99%产率的苯并咪唑衍生物.该研究还发现当苯胺环的对位有取代基时, 由于C=N双建的迁移, 会有不同比例的两种异构化产物出现.但为硝基取代时, 则产物结构单一.
|
|
(5) |
2012年, Beifuss课题组[15]报道了Cu2O/L1催化2-溴苄基溴与苯甲脒的串联反应, 得到了产率为57%~85%之间的喹唑啉衍生物(Eq. 6).在反应溶剂筛选时, 发现N, N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、MeCN和间二甲苯的效果都没有纯水好.
|
|
(6) |
2014年, 陈万芝等[16]报道了在CuI/L2催化体系下, 2-卤代芳基三氮烯(或2-卤代偶氮化合物)与叠氮化钠经C—N偶联以及氮烯分子内N=N键加成来合成苯并三唑类衍生物的反应(Eq. 7).该反应操作简单、原料易得、催化体系简单、区域选择性高, 为2-取代苯并三唑的合成提供了一条实用便利的路线.
|
|
(7) |
2016年, 吕荣文等[17]将13种配体分别缓慢滴加到溶有Cu(OAc)2的DMSO-H2O溶液中, 然后加入抗坏血酸钠, 从而制得催化体系溶液.随后, 将催化体系溶液滴加到溶有蒽醌类溴胺酸和芳香胺类化合物并预热到75 ℃的水溶液中, 并在此温度下继续反应5 h.结果显示, 配体L5对反应的催化效率和底物适应性最好(Eq. 8).作者进一步利用紫外可见(UV-Vis)和循环伏安法(CV)测试了铜和配体的结合能力, 结果表明催化体系的稳定性和催化活性和配体与铜的配位能力密切相关.
|
|
(8) |
2017年, Schmitt等[18]报道了一种在温和条件下(25~50 ℃), 水介质中CuBr2/反-N, N'-二甲基-1, 2-环己二胺(L6)催化的芳基碘代物和胺的偶联反应(Eq. 9).在反应中, D-葡萄糖起到还原剂的作用, 将Cu(Ⅱ)原位还原成Cu(Ⅰ), 随后再与L6形成配合物催化C—N偶联反应.该反应中, 各种含氮亲核试剂, 如酰胺、咔唑、吲哚及其他唑类等均能顺利地发生N-芳基化反应, 以较高产率得到对应的偶联产物.
|
|
(9) |
2017年, 任峰等[19]将CuI/L6体系用于苯并吡唑的选择性N-芳基化反应, 大多数芳香卤代物均可适应该体系, 体现了较强的底物适应性和较高的区域选择性(N-1:N-2芳基化产物最高可达99:1) (Eq. 10).该反应以质量分数为2%的吐温-20水溶液作为反应溶剂, 所形成的胶束可以有效改善反应底物的溶解性、增加局部反应物浓度, 从而提高反应速率, 因此该反应在60 ℃下反应2 h即可完成.
|
|
(10) |
二胺型配体是铜催化Ullmann型C—N偶联反应中应用最为广泛的一类配体类型, 无论是在水体系或非水体系中均有较多的文献报道.该类配体结构简单, 稳定性好, 适应于多种胺类的N-芳基化反应且催化效率较高.
2006年, 万一千课题组[20]报道了双环己酮草酰二腙(BCO, L7)作为配体用于水相铜催化Ullmann型C—N偶联反应(Eq. 11).该配体和CuO组成的催化体系展示了优异的催化活性, 在微波辅助加热下, 富电子或缺电子的芳基溴代物和碘代物和苯胺、苄胺、环状或链状脂肪胺反应, 5 min内就以较高的产率得到N-芳基化产物.他们还发现, 与微波辅助加热相比, 该反应在常规的加热回流条件下也能顺利发生反应, 只需要将反应时间延长至10 h即可.
随后, 该课题组[21]在利用GC-MS检测反应液时, 发现反应液中存在少量的环己酮和较大量的1, 2-二环己基亚肼.经过分析和查阅资料, 认为是BCO在反应体系中不稳定, 容易发生分解, 而实际与CuO一起起催化作用的可能是BCO分解产生的环己酮、草酰肼、联胺或1, 2-二环己基亚肼中的一种或多种组分.经过一系列实验, 作者建立了三元催化体系, 即CuO/草酰二肼(L8)/2, 5-己二酮, 可在水相中高效地实现Ullmann型C—N偶联反应.考虑到2, 5-己二酮和脂肪伯胺容易发生Paal-Knorr反应而影响目标产物的产率, 可以用环己酮代替2, 5-己二酮(Eq. 12).作者同样研究了微波加热和常规油浴加热对反应影响, 与之前文献报道的结果一致.与此同时, 将反应时间延长到96 h, 该反应还可以在室温条件下顺利发生.
|
|
(11) |
|
|
(12) |
2010年, 万一千课题组[22]又设计合成了一系列结构新颖的吡咯、呋喃、噻吩、吡啶、吲哚等杂环的2-取代酰肼配体, 并详细讨论了配体结构和催化活性的构-效关系.结果表明, 当以N-苯基-2-吡咯酰肼(L9)为配体, CuI为催化剂时, 在130℃微波加热的条件下芳基溴代物和碘代物可以与芳基胺或脂肪胺以优良的产率获得相应的N-芳基化产物(Eq. 13).同年, 该课题组[23]在前期工作基础上对已报道的草酰二酰肼进行修饰, 合成了二异丙基草酰二肼(L10), 并将其作为配体, 于水相中温和条件下(60 ℃, 24 h或120 ℃, 20~30 min)实现铜催化芳基溴代物或碘代物与氨水的交叉偶联反应合成芳香胺(Eq. 14).
2011年, 朱新海等[24]将“CuO/L8/2, 5-己二酮”三元催化体系扩展到水相芳基卤代物与氨水的C—N交叉偶联反应, 在90 ℃下反应不超过120 min即可得到很好的催化效果(Eq. 15).当反应时间延长至24~72 h时, 大部分反应还可在室温下顺利实现.同年, 该课题组[25]还发现在该三元催化体系下, 含不同取代基的芳基氯代物也能够顺利的与取代苯胺、取代苄胺、氨水及唑类的发生交叉偶联反应.此方法虽然底物适用范围广泛, 但该反应需使用大量的含氮亲核试剂(Eq. 16).
|
|
(13) |
|
|
(14) |
|
|
(15) |
|
|
(16) |
2015年, Kurandina等[26]在Wan报道的草酰肼和草酰腙配体的基础上, 建立了“Cu(OAc)2•H2O/N-苯基草酰基双肼基(L11)/2, 5-己二酮”三元催化体系(Eq. 17).在该催化体系下, 铜催化剂用量仅需2 mol%, 富电子的溴代芳烃可以以较高产率与脂肪胺发生偶联反应.但对于缺电子的溴代芳烃和二氯代芳烃则产率较低.
|
|
(17) |
为了实现配体的可循环使用, 万一千课题组基于先前报道的具有较好催化活性的N-苯基吡咯酰肼配体, 将其分别用聚苯乙烯[27]和改性壳聚糖[28]进行负载, 制成负载型配体PSP (polystyrene-supported pyrrole-2-car- bohydrazide, L12)和CSP(chitosan-supported 1H-pyrrole- 2-carbohydrazide, L13).其中,CuI/L12催化体系中, 芳基溴代物或碘代物与苯胺、苄胺、脂肪胺及咪唑等都得到较好的偶联产率(Eq. 18), 且L12在循环使用4次后, 依然保持较高的催化活性.该催化体系还可催化分子内的C—N交叉偶联反应, 用于具有生物活性的咪唑并喹喔啉类衍生物的合成.而对于CuO/L13催化体系, 也具有类似的催化效果(Eq. 19).由于CuO在反应中易于吸附到L13表面,因此在循环试验时, 无需另外加入CuO.循环使用六次后, 反应产率由90%下降到50%, 电感耦合等离子体发射光谱仪(ICP-OES)数据显示循环产率下降的原因来自于Cu负载量的逐渐减少.
|
|
(18) |
|
|
(19) |
酰肼类配体一般用于纯水相中的铜催化C—N偶联反应, 该类配体在反应体系中稳定性相对较差, 容易分解, 对脂肪胺(含氨水)的N-芳基化催化效果较好, 部分氯代物在该体系下也可顺利发生偶联反应.
2013年, Ulven课题组[29]筛选比较了邻菲罗啉及衍生物和其它二胺类配体, 活性结果表明4, 7-二吡咯啉-1, 10-邻菲罗啉(L14)为配体时催化活性最好, 并证实了配体结构中取代基的电子效应对催化活性的重要作用.在CuBr/L14催化体系中, 氨基取代的咪唑、吲哚、嘌呤、苯并咪唑均能与芳基卤代物的发生芳香N-芳基化反应, 并体现较好的化学选择性(Eq. 20).
|
|
(20) |
2016年, 周向葛课题组[30]采用邻菲罗啉(L15)为配体, 联合CuCl2于纯水中可催化2-碘苯胺、苄胺和硫粉的三组分反应, 通过一锅法得到2-芳基苯并噻唑类衍生物(Eq. 21).在该催化体系作用下, 含有各种官能团的2-碘苯胺及苄胺均能以优良的产率得到对应的环化产物.
|
|
(21) |
邻菲罗啉类配体按结构特点也属于芳香二胺类配体.但由于较大的芳香骨架结构, 其水溶性较差, 因此在水相中作为铜催化剂配体用于Ullmann型C—N偶联的报道相对较少.
糖是大自然含量最丰富的生物有机分子, 被广泛用于天然产物、药物和材料分子的合成, 但很少将糖作为有机配体用于偶联反应.受到糖分子含有多个氧原子的结构特征和优良的水溶性特点的启发, 2011年, Sekar课题组[31]首次采用CuI/氨基葡萄糖(L16)作为催化体系, 氨水作为氨源, 在丙酮/水中实现了芳基碘代物或溴代物的胺化反应(Eq. 22).值得注意的是, 该配体具有非常好的水溶性, 反应结束后, 它很容易通过水洗的方式从产物中去除.
|
|
(22) |
同年, 该课题组[32]利用相同的催化体系(CuI/L16), 将胺源变更为NaN3, KI为还原剂, 溶剂为DMF/水的混合溶液, 芳香卤代烃的胺化反应也能顺利进行(Eq. 23).作者固定对溴苯乙酮为反应底物, 分别加入苯乙烯、丙二酸二乙酯、吲哚、吗啡啉、苯酚和乙醇等含C-、N-、O-竞争性的亲核试剂时, 反应产物中均只检测到有对氨基苯乙酮, 这证明该催化体系具有高效的选择性.
|
|
(23) |
2012年, 万一千课题组[33]也报道了CuSO4/蔗糖(L17)催化的芳基碘或溴代物与氨水的胺化反应(Eq. 24).反应结束后, 用乙醚萃取反应物和产物, 并向体系中鼓入空气以去除乙醚和氨水.随后加入磷酸钾和反应底物, 催化体系便可循环使用, 循环三次之后催化剂仍然保持较高的催化活性.
|
|
(24) |
2015年, 章鹏飞等[34]也采用CuI/L16催化体系, 在DMSO-H2O中, 芳香溴代物/碘代物和咪唑、苯并咪唑、吲哚、吡咯及二苯基二硫化物顺利地发生C—N和C—S偶联(Eq. 25).作者还将该催化体系用于抗肿瘤药尼洛替尼中间体的合成.
|
|
(25) |
在文献报道的Cu/L16催化体系中, 认为L16之所以具有较好催化效果除了因为其具有好的水溶性之外, 结构当中的NH2也起了非常重要的作用.为了进一步验证在催化循环中NH2是否参与了与铜配位进而催化反应, 2016年, 周少东等[35]通过实验和密度泛函理论计算(DFT)证实, 在氧化加成-还原消除的催化循环中, 是L16中C-3、C-4和C-6上的羟基发挥了重要的作用, 而并非之前人们所认为的NH2和C-1上的羟基在起作用.
2019年, 钱超等[36]设计合成了一类糖基表面活性剂L18, 可以促进水相中铜催化的Ullmann型C—N和C—S键的构建(Eq. 26).在反应中, L18中的亲水性乳糖部分负责螯合铜催化剂,充当配体作用; 而C14长链烷基部分负责捕获反应底物, 将其包裹在胶束中心促使偶联反应的发生.
|
|
(26) |
2019年, 葛新等[37]使用可生物降解的二糖基乙二胺类化合物L19作为配体, 建立了水溶液中铜催化芳香碘与多种含氮杂环的偶联反应方法(Eq. 27).该反应不需要额外添加相转移催化剂, L19可同时起到配体和增加底物溶解度的作用.
|
|
(27) |
糖类配体是近年来发展的一类可用于水相铜催化C—N偶联反应的配体, 该类配体的最大优点是来源广泛和水溶性好.因其结构中含有多个氧原子或氮原子, 使其具有较好的配位能力和可调节的配位位置.但配位位置的不确定性, 也是研究Cu-糖类催化体系的催化反应机理中的一个难点.
2009年, 周向葛课题组[38]首次使用双吡啶-N-氧化物(L20)作为CuSO4配体, 用于纯水中咪唑类化合物的N-芳基化反应(Eq. 28), 该催化体系简单高效, 能够以较高的产率获得相应的N-芳基化产物.配体L20具有较好的结构稳定性和水溶性是该体系具有较好催化效果的重要原因.
|
|
(28) |
2015年, 姚其正等[39]以草酰胺酸型吡啶-N-氧化物L21为配体, CuI为催化剂, 芳基碘代物、溴代物和活化氯代物与含氮杂环在水相中可以顺利地发生交叉偶联反应(Eq. 29).作者研究了吡啶环上不同位置的取代基对配体促进N-芳基化效果的影响, 结果表明当吡啶环4-位被硝基取代时, 催化活性最高.该反应还可以在DMSO中顺利发生, 可适用于一些对水敏感的底物.作者提出了该反应的机理, 推测反应中的活性配合物为吡啶-N-氧化物中的氧和酰胺中的羰基氧与Cu(Ⅰ)配位形成八元螯合物, 随后经历氧化加成和还原消除历程.
|
|
(29) |
同年, 该课题组[40]还发现, 在室温条件下, DMSO和水(10:1, V/V)的混合溶剂中, 该类配体还可促进CuI催化芳香碘代物与脂肪胺的偶联反应(Eq. 30), 环状仲胺、脂肪伯胺和氨基酸等均适应于该催化体系.对于非活化的芳香溴代物则需要将反应温度升高到50~80 ℃.他们还发现加入DMSO/H2O (V:V=10:1)比为10%的水, 对反应的催化效果有重要的促进作用.
|
|
(30) |
我们课题组[41]结合万一千等报道的酰肼类配体和周向葛等报道的吡啶-N-氧化物配体的结构特点, 设计合成了一类结构新颖的酰肼吡啶-N-氧化物类配体, 建立了Cu(0)/2-叔丁基酰肼吡啶-N-氧化物(L23)催化体系, 可催化纯水中芳香碘代物和溴代物与含氮杂环的偶联反应(Eq. 31).随后, 我们又合成了2, 6-双酰肼吡啶-N-氧化物和1-酰肼异喹啉-N-氧化物类配体, 以研究双配位基团和空间结构对活性的影响, 并分别建立了对应的Cu2O/2, 6-双(2-甲基酰肼)吡啶-N-氧化物(L24)[42] (Eq. 32)和Cu2O/1-(2-甲基酰肼)异喹啉-N-氧化物(L25)[43] (Eq. 33)两种催化体系.两种催化体系均能在纯水中实现芳香卤代物与氮杂环、脂肪胺和醇胺等的偶联, 并显示较好的化学选择性.
|
|
(31) |
|
|
(32) |
|
|
(33) |
在水相反应中, 为了增加底物的溶解性, 一般都要加入相转移催化剂.基于此, 我们又设计合成了N-长链烷基取代的酰肼吡啶-N-氧化物类配体, 以期在保持与铜配位的基团不变的基础上, 长链烷基能起到相转移催化剂的作用, 从而降低相转移催化剂的用量或完全取代相转移催化剂并保持较好的催化活性.烷基链长筛选结果显示C12效果最好, 建立的Cu2O/N-十二烷基酰肼吡啶-N-氧化物(L26)[44](Eq. 34)催化体系适应于芳香碘代物与咪唑类化合物的偶联.遗憾的是, 该配体并不能完全取代相转移催化剂四丁基溴化铵(TBAB), 但可以将它的用量由常规20 mol%量降低到5 mol%.
|
|
(34) |
文献报道吡啶-N-氧化物中的氧原子显负电性, 可被视为类似苯氧负离子的中性结构[45], 因此,我们将酰肼吡啶-N-氧化物配体结构中的吡啶-N-氧化物换成苯酚结构, 合成了系列2-羟基苯甲酰肼类化合物.正如我们所预期的, N-甲基-2羟基苯甲酰肼(L27)与CuSO4构成的催化体系具有很好的催化活性, 在水相中, 芳香卤代物可以很好地与各种含氮杂环发生偶联反应[46](Eq. 35).
|
|
(35) |
吡啶-N-氧化物类配体由于结构中的N-O基团具有较好的水溶性和适中的配位能力, 该类配体的催化效果较好, 特别是对于含氮杂环的N-芳基化, 底物适应性和反应产率都表现较优异.
2005年, Liu课题组[47]以8-羟基喹啉(L28)为配体, CuI为催化剂, 在DMF-水的混合溶剂中, 咪唑类杂环可顺利地发生N-芳基化反应(Eq. 36).在该反应中, 作者用了一种具有较好溶解性的碳酸二(四乙基铵)作为碱以代替C—N偶联反应中常用的K2CO3或Cs2CO3等.
|
|
(36) |
2012年, 韩世清课题组[48]也以L28为配体, Cu2O为催化剂, 在水相中100 ℃下实现了含氮杂环化合物与卤代芳烃的C—N偶联反应, 得到中等及优良的产率(Eq. 37).
|
|
(37) |
喹啉类配体与邻菲罗啉类配体类似, 具有较大的芳香骨架结构, 导致其水溶性较差, 同样在水相铜催化剂C—N偶联反应中应用较少.
2010年, 付华课题组[49]首次采用乙二醛二肟(L29)为配体, 协助CuCl或CuBr实现了水相中芳基碘、溴代物和氯代物与氮杂环化合物或脂肪胺的交叉偶联反应(Eq. 38).该催化体系不仅适用于吡咯、咪唑和吲哚, 还可与苯并咪唑及咔唑等较难反应的含氮杂环化合物发生反应.当3-硝基氯苯与咪唑偶联时, 只需加入KI, 反应就可以顺利进行.
|
|
(38) |
2011年, 吴香梅课题组[50]报道了一例采用丁二酮肟(L30)作为配体, 水相CuSO4催化的咪唑和苯并咪唑的N-芳基化方法(Eq. 39).位阻较大的咪唑类底物对该体系的催化效果影响较大, 如2-甲基咪唑与碘苯的反应仅得到66%的偶联产率.
|
|
(39) |
2012年, 王德平等[51]报道了CuI/6, 7-二氢喹啉-8-酮肟(L31)催化的唑类的水相N-芳基化反应(Eq. 40).该反应条件下, 各种唑类、芳基碘代物、溴代物以及含吸电子基团的氯代物均可以得到较理想的收率.该体系最大的优点是催化体系的用量少, 最低0.1 mol%的CuI和0.2 mol%的L31就可以高效地完成偶联反应. 2015年, 该课题组[52]又报道了室温下CuI/L31催化的水相二甲胺和甲胺的芳基化方法(Eqs. 41, 42).甲胺在该催化体系中具有较好的选择性, 能够以较高的产率得到唯一产物单芳基甲胺. 2017年, 该课题组[53]将该体系进一步扩展到芳香溴代物或碘代物与脂肪伯胺和仲胺的偶联, 该反应在室温下纯水中反应, 不需要额外添加相转移催化剂, 以最高95%的产率得到对应的偶联产物(Eq. 43).
|
|
(40) |
|
|
(41) |
|
|
(42) |
|
|
(43) |
肟类配体在水相铜催化C—N偶联反应中是一类非常重要的配体, 该类型配体结构简单, 制备容易, 催化效果良好.含氮杂环、芳香胺、脂肪胺、氨基酸和氨基醇等均能顺利的发生N-芳基化反应, 且产率普遍较高.
2016年, Schmitt等[54]用2, 2, 6, 6-四甲基-3, 5-庚二酮(L32)为配体, Cu(OTf)2为催化剂, 在温和条件下(25~50 ℃)实现了水介质中芳基溴代物和一级脂肪胺的偶联反应(Eq. 44).在反应中, D-葡萄糖作为还原剂, 原位还原Cu(Ⅱ)为Cu(Ⅰ); 质量分数为2%的TPGS-750-M作为相转移催化剂及反应溶剂, 它形成的胶束大大增加了反应底物的溶解性.在此条件下, 多种类型的一级脂肪胺均能高效转化, 含有CONH2, CN, COOtBu和CHO等基团的底物均适应于该体系.
|
|
(44) |
Jain等[55]以异丁酰环己酮(L33)为配体、CuI为催化剂、水为溶剂、报道了两性天然或非天然氨基酸在微波辅助加热条件下的N-芳基化反应(Eq. 45).该反应中, 对于邻位、间位或对位取代的各种溴代物均能与α-或β-氨基酸均能顺利的偶联, 50 min之内即可反应完全, 且手性HPLC检测偶联产物的构型保持良好, 为氨基酸的N-芳基化提供了一种新的思路和方法.
|
|
(45) |
二酮类配体在非水相体系中有广泛的应用且催化效果良好, 但在水相体系中报道较少.水体系所起的一个重要作用是对反应体系中的添加剂或反应底物具有好的溶解能力.
2018年, Vaccaro等[56]用生物质来源的糠醇(L34)作为配体, CuI为催化剂, 报道了芳香碘代物和含氮杂环的偶联(Eq. 46).该反应最大的亮点是使用糠醇-H2O的共沸物作为配体和溶剂, 反应完后, 又可以通过蒸馏共沸的方法近乎等量地回收糠醇-H2O的共沸物, 从而实现回收再用, 并最大限度的减少废弃物的排放.
|
|
(46) |
2018年, 马大为小组[57]研究发现, 在CuI催化剂中加入BMPO配体(L35)后, 在水相体系中, 即可实现卤代芳烃和水合肼的偶联, 产物经酸化处理, 芳香肼盐酸盐从萃取溶剂二氯甲烷中析出, 避免了柱层析分析(Eq. 47).该反应中, 催化剂用量少, 水为反应溶剂, 反应时间短和催化效率高, 为芳香肼的合成提供了一种非常温和和高效的方法.
|
|
(47) |
2019年, 陈国良小组[58]报道了肌醇(L36)促进的铜粉催化的水相胺的N-芳基化方法(Eq. 48).环状/链状仲胺、苯胺、醇胺和氨基酸均可在此体系下顺利发生反应.由于L36具有多个手性中心, 为手性胺类化合物的合成提供了一种新的可能.
|
|
(48) |
该类型的配体中, 草酰胺类配体(L35)是马大为课题组最近几年设计合成的一类非常有效的铜催化剂配体, 可顺利实现非水相中芳香氯代物与各种胺类的C—N偶联, 但用于该类型反应的水相体系报道较少.而另外两类配体是多元醇类, 羟基的存在可有效增加配体的水溶性和铜配合物的溶解性.
Salen配体是一类非常有效的助剂和金属螯合剂, 其与铜形成的配合物结构稳定、制备简单、催化活性高.基于此, 周向葛课题组以邻苯二胺、3-醛基-4-羟基苯磺酸和醋酸铜为原料, 在甲醇-水体系中以82%的收率一步合成Cu-salen配合物, 由于磺酸基的存在, 该化合物具有很好的水溶性.活性筛选时, 作者发现该配合物具有很好的水相催化C—N偶联反应效果.在较温和的条件下, 咪唑类含氮杂环(Eq. 49)[59]、氨水(Eq. 50)[60]、脂肪胺/氨基醇/氨基酸(Eq. 51)[61]等均能顺利的发生N-芳基化反应, 且显示较好的反应选择性.作者还发现, 该配合物可以在水相条件下, 催化邻氯/溴苯胺的分子间自偶联, 以最高85%的产率合成具有生物活性的吩嗪类衍生物[62](Eq. 52).
|
|
(49) |
|
|
(50) |
|
|
(51) |
|
|
(52) |
1993年, Pellón等[63]首次报道了无配体的水溶液中铜粉催化的C—N偶联反应, 但该体系底物仅适用于邻位带有羧酸的芳基卤代物和胺的反应(Eq. 53).
|
|
(53) |
2007年, Larhed课题组[64]在微波辅助下研究了CuI催化的溴苯与氨基酸及氨基酸酯之间的偶联反应(Eq. 54).该反应中KI的加入对该反应起到了促进作用, 多种氨基酸及氨基酸酯都可以在40 min内反应完全, 以较高产率得到对应的偶联物, 且仅有少于6%的消旋产物.在线ESI-MS和MS-MS检测发现, 在反应液中有氨基酸-铜配合物的存在, 因此在该反应中, 氨基酸既作反应底物又作金属配体.另外该反应的不足之处在于需要在高温下(185 ℃)进行.
|
|
(54) |
2010年, 冯乙巳等[65]以CuCl为催化剂, 在水相中实现了多种芳基碘或溴代物与苄胺、正己胺和吡咯的偶联反应(Eq. 55).质量分数为40%的四丁基氢氧化铵水溶液在该催化体系中既作溶剂、又充当碱和相转移催化剂的作用.
|
|
(55) |
同年, Mukhopadhyay等[66]报道了在PEG-水体系中, CuI催化的2-卤代重氮氨基苯类化合物的分子内偶联反应(Eq. 56).在这一反应中, 含有不同取代基的2-碘代、溴代、氯代重氮氨基苯均可以顺利地发生反应, 并以高的产率获得1-芳基苯并三唑类衍生物.值得说明的是, 当重氮氨基苯的2-位都含有卤素取代基时, 产物依然结构单一, 并没有出现重氮氨基苯的互变异构体.
|
|
(56) |
2011年, 冯乙巳等[67]又发现以纳米CuI粒子为催化剂, 质量分数为40%的n-Bu4NOH水溶液为碱和溶剂, 在不加配体和其它有机溶剂的条件下, 芳基卤代物可顺利地转化成对应的苯酚、苯胺和苯硫醇类化合物(Scheme 1).当使用芳基碘代物时, 催化剂用量少, 反应条件温和, 产率高.反应完成后, 经过简单的离心分离, 该催化剂重复利用三次后催化活性略有降低, 可能主要来自催化剂在分离中的损失.透射电镜(TEM)和X射线衍射(XRD)对反应前后催化剂的表征表明, 纳米CuI在反应前后的形态和晶型保持良好, 没有明显变化.
2011年, Teo课题组[68]报道了纯水相中, TBAB为相转移催化剂、Cu2O催化的芳基卤代物与吲哚、苯并吡唑和咪唑等杂环之间的N-芳基化反应(Eq. 57).该催化体系对于芳基碘代物有高的催化作用, 但对于溴代物不太理想.随后, 他们又发现该类似体系, 还可催化水相体系中吡咯烷酮、戊内酰胺、噻吩-2-酰胺和苯甲酰胺的N-芳基化反应, 但大部分反应的产率中等[69](Eq. 58).
|
|
(57) |
|
|
(58) |
2011年, 魏俊发课题组[70]报道以铜粉为催化剂, 无需碱和相转移催化剂的加入即可在水相中实现芳基碘、溴或氯代物与一级脂肪胺的N-芳基化反应(Eq. 59).该催化体系具有较广的底物适用范围, 各类一级脂肪胺都可以与芳基卤代物以优良的产率获得相应的偶联产物.在这一反应中, 少量空气的存在对该催化体系具有极大的促进作用, 在充入氮气或者氧气的情况下该反应则不能进行, 其原因主要是Cu(0)需要空气中的氧气氧化成Cu(Ⅰ), 而如果氧气太多, 则会把Cu(Ⅰ)进一步氧化成Cu(Ⅱ), 从而不能完成催化循环.另外, 二级脂肪胺和芳香胺不太适应该催化体系.
|
|
(59) |
2013年, 韩世清课题组[71]报道了CuI催化的芳香碘代物与醇胺的选择性N-芳基化反应(Eq. 60).该反应以水作为反应溶剂、无需额外的配体或相转移催化剂, 以64%~93%的产率选择性得到N-芳基化产物.
|
|
(60) |
2014年, Teo课题组[72]报道了水相Cu2O催化的芳香碘代物与甲磺酰胺的偶联反应(Eq. 61).该反应中, 催化剂用量仅需2 mol%, 大多数芳基碘代物都可以高效转化、但对于2-取代碘代物或溴代物活性较低, 需要增加催化剂的用量才能顺利发生反应.
|
|
(61) |
2015年, Heidarizadeh课题组[73]合成了一种“表面活性剂/铜基离子液体”, 并将其用于水相芳基碘或溴代物与含氮杂环之间的偶联反应(Eq. 62), 并显示较好的催化效果.作者推测可能是由于催化剂具有表面活性剂的性质, 在水中形成胶束颗粒, 从而增加了反应物在水中的溶解性, 进而促进偶联反应的发生.该催化体系循环使用四次后, 依然可保持着较高的催化活性.
|
|
(62) |
2016年, Sakhuja课题组[74]报道了一种反应条件温和、高效和环境友好的方法来合成具有荧光标记作用的N-香豆酰氨基酸方法(Eq. 63).该方法以CuI作为催化剂, 通过微波辅助催化4-氯香豆素与N-末端未保护的氨基酸在水中发生交叉偶联反应, 反应10 min后即可获得最高96%产率的偶联产物.该方法还被用于荧光标记物内吗啡-2的合成.
|
|
(63) |
同年, 钱超课题组[75]在中性的CuI水溶液中加入β-环糊精(β-CD), 经离心分离后制得负载型CuI/β-CD.随后以对甲氧基碘苯和咪唑的偶联为模型反应, 对影响反应的各条件进行了优化, 并将最优条件扩展到各种取代的卤代烃和胺, 均得到较好的产率(Eq. 64).热重分析显示该催化剂在290~350 ℃才会分解, 说明它具有较好的热稳定性.该催化剂循环使用五次后, 催化活性基本保持不变, TEM显示催化剂结构反应前后保持良好; 电感耦合等离子体原子发射光谱法(ICP-AES)测试发现反应后Cu的负载量为0.71 mmol/g, 与反应前的0.76 mmol/g相当.
|
|
(64) |
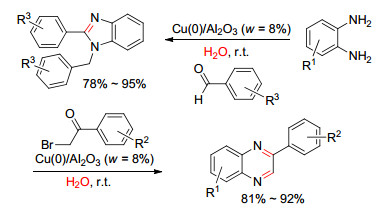
2017年, Likhar等[76]以Cu-Al水滑石为原料, 在氢气氛下热解还原制得负载型Cu(0)/Al2O3催化剂, 并将其作为非均相催化剂用于邻苯二胺与芳香醛或α-溴代苯乙酮的偶联, 构建1, 2-二取代苯并咪唑和喹喔啉类化合物(Scheme 2).该反应以水作为反应溶剂, 在室温下反应7 min~5 h即可.该催化剂循环使用5次后, 催化效率略有下降, 原子吸收光谱(AAS)检测溶液中Cu的溢出量基本可以忽略, 且反应后的催化剂通过氢气氛围下高温还原可以再生, 催化活性可恢复到原有水平.
2018年, Albadi等[77]用共沉淀法制得纳米CuO/ ZnO/Al2O3, 并用XRD、扫描电镜(SEM)、X射线能谱(EDS)和氮气吸附脱附法进行了结构表征.该化合物可催化芳香卤代物与氨水的反应得到相应的芳香胺, 反应在水溶液中回流温度下即可发生, 反应时间短.催化剂易于分离, 并可循环使用5次后, 催化活性没有明显下降(Eq. 65).
|
|
(65) |
同年, Kumar等[78]用六苯基苯衍生物作为纳米反应器和稳定剂, 利用自组装方式随后制得纳米HPB@ Cu2O-Fe2O3.该化合物可在水体系中, 光照条件下, 催化卤代芳烃和胺的C—N偶联.该反应在室温下即可发生, 一级或二级脂肪胺、芳香或杂环胺均能顺利发生N-芳基化, 催化剂还可循环使用(Eq. 67).
|
|
(66) |
无配体条件下的铜催化C—N偶联反应有较多的报道, 催化剂一般为Cu(0)、铜盐、铜氧化物、负载型的Cu盐或铜氧化物等.由于不需要外加配体, 使得反应操作更加简单.另外负载型催化剂在反应完后经简单过滤或离心即可与反应体系分离, 并经洗涤和烘干处理, 可实现催化剂的重复使用.然而, 该类催化体系的催化效率整体上比铜-配体体系要低一些, 并且应用范围要窄一些.
水作为绿色化学反应中的理想溶剂, 一直受到大家的关注, 水相Ullmann型C—N偶联反应也取得了很大的进展.从上述报道的文献可以看出, 此类反应的研究重点主要还是集中在开发新的催化剂配体上.这些配体一般具有较好的水溶性和稳定性及适当的螯合能力, 并能与铜形成6~8元环状配合物等.然而, 该反应还存在一些问题, 例如:水除了作为溶剂外的其它作用不明确, 纯水中Cu(Ⅰ)/Cu(Ⅲ)氧化加成和还原消除机理缺乏相应的证据、催化体系的催化活性不高、需要额外添加相转移催化剂而导致新的污染等问题.因此, 未来的铜-配体催化体系除了应有较高的产率和较宽的底物适应性外, 还应该强调贯彻绿色化学精神, 所用的配体本身应该安全毒性小或无毒, 对环境友好, 易于回收重复使用, 或配体易于分解处理等.与此同时, 进一步深入探索水相催化反应机理也是本领域未来需要重点关注的方向之一.
Evano, G.; Blanchard, N.; Toumi, M. Chem. Rev. 2008, 108, 3054. doi: 10.1021/cr8002505
(a) Hartwig, J. F. In Palladium-Catalyzed Amination of Aryl Halides and Related Reactions, Wiley, New York, 2002.
(b) Jiang, L.; Buchwald, S. L. In Palladium-catalyzed Aromatic carbon-nitrogen Bond Formation in Metal-catalyzed Cross- coupling Reactions, 2nd ed., Wiley-VCH, Weinheim, 2004.
(c) Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954.
(d) Beletskaya, I. P.; Cheprakov, A. V. Organometallics 2012, 31, 7753.
Kosugi, M.; Kameyama, M.; Migita, T. Chem. Lett. 1983, 927. https://www.researchgate.net/publication/301936653_ChemInform_Abstract_PALLADIUM-CATALYZED_AROMATIC_AMINATION_OF_ARYL_BROMIDES_WITH_NN-DIETHYLAMINOTRIBUTYLTIN?ev=auth_pub
(a) Ullmann, F.; Bielecki, J. Ber. Dtsch. Chem. Ges. 1901, 34, 2174.
(b) Ullmann, F. Ber. Dtsch. Chem. Ges. 1903, 36, 2382.
Wolter, M.; Nordamann, G.; Job, G. E.; Buchwald, S. L. Org. Lett. 2002, 4, 973. doi: 10.1021/ol025548k
Nordamann, G.; Buchwald, S. L. J. Am. Chem. Soc. 2003, 125, 4978 doi: 10.1021/ja034809y
(a) Dai, L. Prog. Chem. 2018, 30, 1257 (in Chinese).
(戴立信, 化学进展, 2018, 30, 1257.)
(b) Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525.
(c) Ribas, X.; Gueell, I. Pure Appl. Chem. 2014, 86, 345.
(d) Okano, K.; Tokuyama, H.; Fukuyama, T. Chem. Commun. 2014, 50, 13650.
(e) Li, Z.; Wu, Z.; Deng, H.; Zhou, X. Chin. J. Org. Chem. 2013, 33, 760 (in Chinese).
(李正凯, 吴之清, 邓杭, 周向葛, 有机化学, 2013, 33, 760.)
(f) Chen, Y.; Sun, L. Chin. J. Org. Chem. 2013, 33, 877 (in Chinese).
(成宜娟, 孙丽萍, 有机化学, 2013, 33, 877.)
(g) Bariwal, J.; Van der Eychen, E. Chem. Soc. Rev. 2013, 42, 9283.
(h) Wang, Y.; Zeng, J.; Cui, X. Chin. J. Org. Chem. 2010, 30, 181 (in Chinese).
(王晔峰, 曾京辉, 崔晓瑞, 有机化学, 2010, 30, 181.)
(i) Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954.
(j) Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2008, 47, 3096.
Poliakoff, M.; Fitzpatrick, J.-M.; Farren, T.-R.; Anastas, P. T. Science 2002, 297, 807. doi: 10.1126/science.297.5582.807
Li, C.-J.; Chan, T.-H. In Comprehensive Organic Reactions in Aqueous Media, 2nd ed., John Wiley & Sons, New Jersey, 2007.
Carril, M.; SanMartin, R.; Domínguez, E.; Tellitu, I. Green Chem. 2007, 9, 219. doi: 10.1039/B614218D
Barbero, N.; Carril, M.; SanMartin, R.; Domínguez, E. Tetrahedron 2008, 64, 7283. doi: 10.1016/j.tet.2008.05.072
Oshovsky, G. V.; Ouali, A.; Xia, N.; Zablocka, M.; Boeré, R. T.; Duhayon, C.; Taillefer, M.; Majoral, J. P. Organometallics 2008, 27, 5733. doi: 10.1021/om800728m
Swapna, K.; Murthy, S. N.; Nageswar, Y. V. D. Eur. J. Org. Chem. 2010, 2010, 6678. doi: 10.1002/ejoc.201000964
Peng, J.; Ye, M.; Zong, C.; Hu, F.; Feng, L.; Wang, X.; Wang, Y.; Chen, C. J. Org. Chem. 2011, 76, 716. doi: 10.1021/jo1021426
Malakar, C. C.; Baskakova, A.; Conrad, J.; Beifuss, U. Chem.-Eur. J. 2012, 18, 8882. doi: 10.1002/chem.201200583
Shang, X.; Zhao, S.; Chen, W.; Chen, C.; Qiu, H. Chem.-Eur. J. 2014, 20, 1825. doi: 10.1002/chem.201303712
Shao, B.; Du, H.; Hao, X.; Lu, R.; Luo, Y.; Zhang, S. Chin. J. Chem. Eng. 2016, 24, 1000. doi: 10.1016/j.cjche.2016.01.010
Bollenbach, M.; Aquino, P. G. V.; de Araújo-Júnior, J. X.; Bourguignon, J.-J.; Bihel, F.; Salomé, C.; Wagner, P.; Schmitt, M. Chem.- Eur. J. 2017, 23, 13676. doi: 10.1002/chem.201700832
Ding, X.; Bai, J.; Wang, H.; Zhao, B.; Li, J.; Ren, F. Tetrahedron 2017, 73, 172. doi: 10.1016/j.tet.2016.11.066
Zhu, X.; Ma, Y.; Su, L.; Song, H.; Chen, G.; Liang, D.; Wan, Y. Synthesis 2006, 3955. http://www.researchgate.net/publication/244568822_Biscyclohexanone_OxalyldihydrazoneCopperII_Oxide_-_A_Novel_and_Efficient_Catalytic_System_for_Ullmann-type_CN_Coupling_in_Pure_Water
Zhu, X. H.; Su, L.; Huang, L.; Chen, G.; Wang, J.; Song, H.; Wan, Y. Eur. J. Org. Chem. 2009, 2009, 635. doi: 10.1002/ejic.200801049
Xie, J. W.; Zhu, X. H.; Huang, M. N.; Chen, W.; Wan, Y. Eur. J. Org. Chem. 2010, 2010, 3219. doi: 10.1002/ejoc.201000361
Meng, F.; Zhu, X. H.; Li, Y.; Xie, J.; Wang, B.; Yao, J.; Wan, Y. Eur. J. Org. Chem. 2010, 2010, 6149. doi: 10.1002/ejoc.201001150
Li, Y.; Zhu, X. H.; Meng, F.; Wan, Y. Tetrahedron 2011, 67, 5450. doi: 10.1016/j.tet.2011.05.068
Huang, M.; Lin, X.; Zhu, X.; Peng, W.; Xie, J.; Wan, Y. Eur. J. Org. Chem. 2011, 2011, 4523. doi: 10.1002/ejoc.201100458
Kurandina, D. V.; Eliseenkov, E. V.; Khaibulova, T. S.; Petrov, A. A.; Boyarskiy, V. P. Tetrahedron 2015, 71, 7931. doi: 10.1016/j.tet.2015.07.071
Huang, L.; Yu, R.; Zhu, X.; Wan, Y. Tetrahedron 2013, 69, 8974. doi: 10.1016/j.tet.2013.07.036
Yang, B.; Mao, Z.; Zhu, X.; Wan, Y. Catal. Commun. 2015, 6, 92. http://www.sciencedirect.com/science/article/pii/S1566736714004671
Engel-Andreasen, J.; Shimpukade, B.; Ulven, T. Green Chem. 2013, 15, 336. doi: 10.1039/C2GC36589H
Xu, H.; Luo, C.; Li, Z.; Xiang, H.; Zhou, X. J. Heterocycl. Chem. 2016, 53, 1207. doi: 10.1002/jhet.2385
Thakur, K. G.; Ganapathy, D.; Sekar, G. Chem. Commun. 2011, 47, 5076. doi: 10.1039/c1cc10568j
Thakur, K. G.; Srinivas, K. S.; Chiranjeevi, K.; Sekar, G. Green Chem. 2011, 13, 2326. doi: 10.1039/c1gc15469a
Huang, M.; Wang, L.; Zhu, X.; Mao, Z.; Kuang, D.; Wan, Y. Eur. J. Org. Chem. 2012, 2012, 4897. doi: 10.1002/ejoc.201200787
Wen, M.; Shen, C.; Wang, L.; Zhang, P.; Jin, J. RSC Adv. 2015, 5, 1522. doi: 10.1039/C4RA11183D
Ge, X.; Chen, X.; Qian, C.; Zhou, S. RSC Adv. 2016, 6, 29638. doi: 10.1039/C6RA03015G
Ge, X.; Zhang, S.; Chen, X.; Liu, X.; Qian, C. Green Chem. 2019, 21, 2771. doi: 10.1039/C9GC00964G
Zhou, G.; Chen, W.; Zhang, S.; Liu, X.; Yang, Z.; Ge, X.; Fan, H.-J. Synlett 2019, 193.
Liang, L.; Li, Z. K.; Zhou, X. G. Org. Lett. 2009, 11, 3294. doi: 10.1021/ol9010773
Wang, Y.; Zhang, Y.; Yang, B.; Zhang, A.; Yao, Q. Org. Biomol. Chem. 2015, 13, 4101. doi: 10.1039/C5OB00045A
Wang, Y.; Ling, J.; Zhang, Y.; Zhang, A.; Yao, Q. Eur. J. Org. Chem. 2015, 2015, 4153.
Wu, F.-T.; Yan, N.-N.; Liu, P.; Xie, J.-W.; Liu, Y.; Dai, B. Tetrahedron Lett. 2014, 55, 3249. doi: 10.1016/j.tetlet.2014.04.039
Wang, X.; Meng, F.; Zhang, J.; Xie, J.; Dai, B. Catal. Lett. 2018, 148, 1142. doi: 10.1007/s10562-018-2321-8
Xie, J.-W.; Yao, Z.-B.; Wang, X.-C.; Zhang, J. Tetrahedron 2019, 75, 3788. doi: 10.1016/j.tet.2019.05.067
汪小创, 张洁, 谢建伟, 高等学校化学学报, 2017, 38, 1178. doi: 10.7503/cjcu20170157Wang, X.; Zhang, J.; Xie, J. Chem. J. Chin. Univ. 2017, 38, 1178 (in Chinese). doi: 10.7503/cjcu20170157
Karayannis, N. M.; Pytlewski, L. L.; Mikulski, C. M. Coord. Chem. Rev. 1973, 11, 93. doi: 10.1016/S0010-8545(00)82007-X
Yan, N.-N.; Wu, F.-T.; Zhang, J.; Wei, Q.-B.; Liu, P.; Xie, J.-W.; Dai, B. Asian J. Org. Chem. 2014, 3, 1159. doi: 10.1002/ajoc.201402136
Liu, L.; Frohn, M.; Xi, N.; Dominguez, C.; Hungate, R.; Reider, P. J. J. Org. Chem. 2005, 70, 10135. doi: 10.1021/jo051640t
张敬先, 殷慧清, 韩世清, 有机化学, 2012, 32, 1429. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341429.shtmlZhang, J. X.; Yin, H. Q.; Han, S. Q. Chin. J. Org. Chem. 2012, 32, 1429 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract341429.shtml
Li, X.; Yang, D.; Jiang, Y.; Fu, H. Green Chem. 2010, 12, 1097. doi: 10.1039/c002172e
Wu, X.; Hu, W. Chin. J. Chem. 2011, 29, 2124. doi: 10.1002/cjoc.201180368
Wang, D.; Zhang, F.; Kuang, D.; Yu, J.; Li, J. Green Chem. 2012, 14, 1268. doi: 10.1039/c2gc35077g
Wang, D.; Kuang, D.; Zhang, F.; Yang, C.; Zhu, X. Adv. Synth. Catal. 2015, 357, 714. doi: 10.1002/adsc.201400785
Wang, D.; Zheng, Y.; Yang, M.; Zhang, F.; Mao, F.; Yu, J.; Xia, X. Org. Biomol. Chem. 2017, 15, 8009. doi: 10.1039/C7OB02126G
Bollenbach, M.; Wagner, P.; Aquino, P. G. V.; Bourguignon, J.-J.; Bihel, F.; Salomé, C.; Schmitt, M. ChemSusChem 2016, 9, 3244. doi: 10.1002/cssc.201600801
Sharma, K. K.; Mandloi, M.; Rai, N.; Jain, R. RSC Adv. 2016, 6, 96762. doi: 10.1039/C6RA23364C
Ferlin, F.; Trombettoni, V.; Luciani, L.; Fusi, S.; Piermatti, O.; Santoro, S.; Vaccaro, L. Green Chem. 2018, 20, 1634. doi: 10.1039/C8GC00287H
Kumar, S. V.; Ma, D. Chin. J. Chem. 2018, 36, 1003. doi: 10.1002/cjoc.201800326
Zhou, Q.; Du, F.; Chen, Y.; Fu, Y.; Chen, G. Tetrahedron Lett. 2019, 60, 1938. doi: 10.1016/j.tetlet.2019.06.033
Wang, Y.; Wu, Z.; Wang, L.; Li, Z.; Zhou, X. Chem. Eur. J. 2009, 15, 8971. doi: 10.1002/chem.200901232
Wu, Z.; Jiang, Z.; Wu, D.; Xiang, H.; Zhou, X. Eur. J. Org. Chem. 2010, 2010, 1854. doi: 10.1002/ejoc.201000060
Wu, Z.; Zhou, L.; Jiang, Z.; Wu, D.; Li, Z.; Zhou, X. Eur. J. Org. Chem. 2010, 2010, 4971. doi: 10.1002/ejoc.201000840
Yu, L.; Zhou, X.; Wu, D.; Xiang, H. J. Organomet. Chem. 2012, 705, 75. doi: 10.1016/j.jorganchem.2011.12.030
Pellón, R. F.; Carrasco, R.; Rodés, L. Synth. Commun. 1993, 23, 1447. doi: 10.1080/00397919308011235
Röttger, S.; Sjöberg, P. J. R.; Larhed, M. J. Comb. Chem. 2007, 9, 204. doi: 10.1021/cc060150r
Xu, H.-J.; Zheng, F.-Y.; Liang, Y.-F.; Cai, Z.-Y.; Feng, Y.-S.; Che, D.-Q. Tetrahedron Lett. 2010, 51, 669. doi: 10.1016/j.tetlet.2009.11.104
Mukhopadhyay, C.; Tapaswi, P. K.; Butcher, R. J. Org. Biomol. Chem. 2010, 8, 4720. doi: 10.1039/c0ob00177e
Xu, H.-J.; Liang, Y.-F.; Cai, Z.-Y.; Qi, H.-X.; Yang, C.-Y.; Feng, Y.-S. J. Org. Chem. 2011, 76, 2296. doi: 10.1021/jo102506x
Yong, F.-F.; Teo, Y.-C.; Tay, S.-H.; Tan, B. Y.-H.; Lim, K.-H. Tetrahedron Lett. 2011, 52, 1161. doi: 10.1016/j.tetlet.2011.01.005
Yong, F.-F.; Teo, Y.-C.; Chua, G.-L.; Lim, G. S.; Lin, Y. Tetrahedron Lett. 2011, 52, 1169. doi: 10.1016/j.tetlet.2011.01.003
Jiao, J.; Zhang, X.-R.; Chang, N.-H.; Wang, J.; Wei, J.-F.; Shi, X.-Y.; Chen, Z.-G. J. Org. Chem. 2011, 76, 1180. doi: 10.1021/jo102169t
Jin, M.; Zhao, D.; He, G.; Tong, Y.; Han, S. Chin. J. Catal. 2013, 34, 1651. doi: 10.1016/S1872-2067(12)60623-8
Tan, B. Y.-H.; Teo, Y.-C.; Seow, A.-H. Eur. J. Org. Chem. 2014, 2014, 1541. doi: 10.1002/ejoc.201301561
Heidarizadeh, F.; Majdi-nasab, A. Tetrahedron Lett. 2015, 56, 6360. doi: 10.1016/j.tetlet.2015.09.128
Kumari, S.; Shakoor, S. M. A.; Bajaj, K.; Nanjegowda, S. H.; Mallu, P.; Sakhuja, R. Tetrahedron Lett. 2016, 57, 2732. doi: 10.1016/j.tetlet.2016.05.016
Ge, X.; Chen, X.; Qian, C.; Zhou, S. RSC Adv. 2016, 6, 58898. doi: 10.1039/C6RA13536F
Pogula, J.; Laha, S.; Likhar, P. R. Catal. Lett. 2017, 147, 2724. doi: 10.1007/s10562-017-2166-6
Albadi, J.; Jalali, M.; Samimi, H. A. Catal. Lett. 2018, 148, 3750. doi: 10.1007/s10562-018-2567-1
Singh, G.; Kumar, M.; Bhalla, V. Green Chem. 2018, 20, 5346. doi: 10.1039/C8GC02527D

图式 1 纳米铜催化合成苯酚、苯胺和苯硫酚
Scheme 1 CuI-nanoparticles-catalyzed synthesis of phenols, anilines and thiophenols

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:










 下载:
下载:
