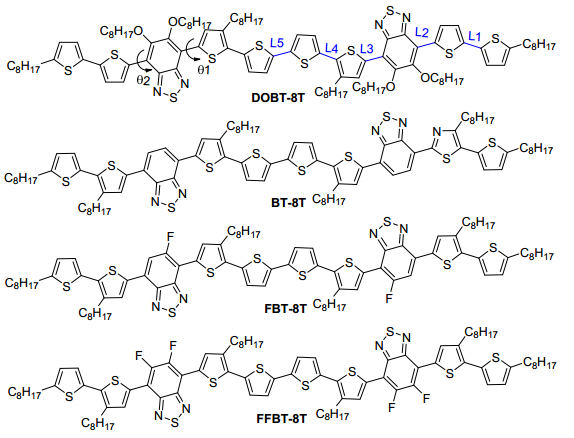
Figure 1.
Molecular structures of DOBT-8T,
BT-8T, FBT-8T and FFBT-8T

Organic solar cells (OSCs) have gained considerable attention during the past decade due to their advantages of low energy consumption, light weight, flexible and easy fabrication.[1] Using a blend of polymers donor materials and fullerene derivatives acceptor materials as the active layer, the polymer-based OSCs with bulk hetero-junction (BHJ) structure demonstrated high power conversion efficiency (PCE) up to 11% in single-junction devices.[2] Compared to the polymeric material, OSMs donor materials have also made considerable progress, due to their definite structure, high purity, better batch-to-batch reproduction and high charge carrier mobility. A high PCE of 11.53% was observed in the single BHJ OSCs based on OSMs: PC71BM blend film, which is abreast to that of polymer solar cells.[3] However, the overall performances of BHJ OSCs based on the OSMs as donors and fullerene or fullerene derivative as acceptors are still behind those of polymer counterparts. Therefore, OSMs donor materials require better material design toward high PCEs.
For the design of high performance OSMs donor materials, it is necessary to strike a delicate balance between a narrowed band gap (Eg) and a low highest occupied molecular orbital (HOMO) energy level.[4] A narrowed Eg is beneficial for getting wider absorption range, providing a larger short-circuit current density (Jsc) of OSCs, while a low HOMO energy level can increase the open-circuit voltage (Voc). The HOMO level and Eg value of OSMs can be well tuned by modifying donor unit, acceptor unit, substituent and side chains, because they are crucial in controlling energy levels.[5] Electron-withdrawing 2, 1, 3-ben-zothiadiazole (BT) is one of the most popular acceptor units used for organic photovoltaic (PV) donor materials, because it can be easy synthesized and purified.[6] OSMs comprised of BT as acceptor unit have shown relatively higher PCE over 6%.[7] In order to further tune the energy levels, crystallinity and solubility, different substituent and side chains could be attached to the BT unit. Introduction of octyloxy chains on BT unit has shown to be an effective way to improve crystallinity and solubility, the resulting polymers or molecules containing 5, 6-bis-(octyloxy)benzo[c]-[1, 2, 5]-thiadiazole (DOBT) exhibited good PV performance.[8] Adding the fluorine (F) atom on BT unit was found to successfully lower the HOMO level and promote intermolecular interactions. OSMs based on 5-fluorobenzo[c]-[1, 2, 5]thiadiazole (FBT) and 5, 6-difluoro-2, 1, 3-benzothia- diazole (FFBT) acceptor units have demonstrated lower HOMO energy levels and resulted in higher Voc in PV devices.[9] However, systematic investigation of the modified BT effects on the OSMs donor material wasseldom reported, which will be critical for deeply understanding the relationship between structure and property, thus providing useful information for the better molecular design in the OSMs PV donor materials. On the other hand, oligothiophenes (OTs) unit has been widely used in OSCs because of their excellent charge transport properties, high polarizability, tunable optical/electro-chemical properties and relatively simple synthesis, Chen and his coworkers have designed and synthesized a series of high PCE OSMs based on OTs unit.[10] However, high PCEs of D-A-D-A-D typed OTs-BT based OSMs OSCs were seldom reported.[11] Furthermore, The effects of different BT acceptor units on the PV properties of OTs based OSMs were never systematically investigated.
A theoretical analysis of the electronic structures of OSMs donor materials has an important influence on further understanding of the relationship between molecular structures and PV properties.[12] In order to seek the structure modification effects of BT acceptor unit, we theoretical designed a series of D-A-D-A-D structured OSMs containing tetrathiophene as the core donor unit, bithiophene as terminal donor units, different electron-withdrawing fragments DOBT, BT, FBT and FFBT as acceptor unit, respectively, named DOBT-8T, BT-8T, FBT-8T and FFBT-8T. The chemical structure of these designed OSMs is shown in Figure 1. In this work, we modified the BT moieties with octyloxy group and fluoruine (F) atom to construct DOBT, FBT and FFBT acceptor units in the OSMs donor materials, thus fully investigating their effects on tuning the energy levels and Eg. The extended of D-A conjugation mainly constructed by OTs is beneficial for intermolecular interactions, and consequently improves the device performances. Alky chains on the thiophene units were introduced to improve solubility of the four designed OSMs. A systematic theoretical investigation on the four designed OSMs was performed. The ground state geometries and electronic properties of the four designed OSMs were computed by density functional theory (DFT), and the optical properties were also simulated by time-dependent density functional theory (TD-DFT). At last, the PCEs of OSCs based on the four designed OSMs were predicted with Scharber model, which were discussed to evaluate our structural modification effects on the BT unit, providing insightful guidance for future judicious design of OSM donor materials based on OTs-BT backbone.
The optimizations of ground state geometry and simulation of optical absorption spectra of the designed OSMs in gas phase were calculated by DFT and TD-DFT with B3LYP functional[13] at the 6-31G(d) basis set levels[14] in the Gaussian 09 package, [15] respectively. B3LYP is arguably the most used functional within the DFT framework, and this level of theory and basis set combination has been shown to provide reliable electronic trends (when compared to available experimental data) for OSMs PV materials.[16] It has been reported that the side chain alkyl of the investigated molecules was merely beneficial for improving solubility without affecting electronic and optical properties.[17] To simplify the calculation, we replaced the alkyl chain by methyl groups. The frontier orbitals of HOMO and the lowest unoccupied molecular orbitals (LUMO) of the four designed OSMs were calculated by B3LYP/6-31G(d) on the basis of optimized structures at the ground states (S0). The band-gap was obtained from the energy difference between HOMO and LUMO of the OSMs. The electronic absorption band and excited-stated energy in vacuo were carried out using the TD-DFT method of B3LYP/6-31G(d) on the basis of optimize structures at S0. For PC61BM, HOMO was obtained at the DFT/B3LYP/6-31G(d) level, and LUMO was derived by adding the HOMO energy to the first singlet excitation energy at TD-B3LYP/6-31G(d) in gas phase; this approach was verified by Nelson and Musgrave et al.[18] In order to evaluate the influence of diffuse function for the excited state energies and absorption spectra, the four compounds were also computed by the TD-B3LYP/6-31+G(d) level in gas phase. Meanwhile, in Li and her coworkers' work, they calculated HOMO, LUMO and Eg for BDCTMBT, BDCTTMBT, BDCTFBT and BDCTTFBT at the B3LYP/6-31G(d) level in gas phase, and tested PV performances of these OSMs in experiment. The calculated and experimental data were shown in Table 1.[19] In compare with the experimental data, the calculated values were observed to have some deviations from the experimental data. However, the trends for the calculated values are normally the same with the experiments results, thus illustrating that the theoretically calculated data can give a guide line to molecular design in experiment.
 下载:
导出CSV
下载:
导出CSV
| Compd. | Theoretical | Experimental | Theoretical | Experimental | |||||
| HOMO/eV | Eg/eV | HOMO/eV | Eg/eV | Voc/V | Voc/V | ||||
| BDCTMBT | -4.91 | 2.30 | -5.30 | 2.02 | 0.94 | 1.05 | |||
| BDCTTMBT | -4.73 | 2.05 | -5.15 | 1.85 | 0.76 | 0.96 | |||
| BDCTFBT | -4.95 | 2.27 | -5.41 | 2.09 | 0.98 | 1.11 | |||
| BDCTTFBT | -4.77 | 2.03 | -5.25 | 1.89 | 0.80 | 1.01 | |||
In this paper, we are only interested in the change trend of introducing DOBT, BT, FBT and FFBT as acceptor unit for OSM OSCs, so the DFT/B3LYP method is sufficient for this study. The absorption spectra and the energy levels of all the compounds in CHCl3 solution and in the solid state were also computed to confirm that the calculated data in gas phase can provide reliable change trends for the designed OSMs (The calculation details were provided in the supporting information). The charge distribution and the electron-transfer mechanism of the four OSMs were analyzed based on the optimized structure of the ground state (S0) obtained at DFT/B3LYP/6-31G(d, p) level by the Natural Bond Orbital (NBO) analysis, [20] which has been obtained by the calculated orbital populations using NBO 3.1 program included in the Gaussian package program.
It is useful to analyze the dihedral angle (θ) and bond lengths (L) of the OSMs for understanding the structure modifying effects on the BT unit and molecular conjugation. Figure 2 illustrates the significant θ and L influenced by attaching the substituent on the BT unit. The values of L and θ for DOBT-8T, BT-8T, FBT-8T and FFBT-8T are listed in Table 2. It was observed that the structural properties of the four designed OSMs showed large dependency on the substituent attached to the BT unit. The θ1 and θ2 of BT-8T, FBT-8T and FFBT-8T are all less than 6°, which indicated the good planarity of the two OSMs. However, θ1 and θ2 of DOBT-8T are bigger than those of BT-8T, this is caused by the large steric hindrance of octyloxy chain substituent. In comparison with BT-8T, FBT-8T demonstrated a more smoothly smaller dihedral angles of θ1 and θ2, resulting from the improved planarity by introducing the F atom onto the BT unit, while the F/S interaction existed between BT and the adjacent thiophene unit.[21] FFBT-8T showed the smallest dihedral angle of the four designed OSMs, which may be assigned to strong F/S interaction for having two atom in the FFBT unit. It has been reported that the high planarization between adjacent aromatic units can make parallel p-orbital interactions to extend conjugation and facilitate delocalization.[22]
 下载:
导出CSV
下载:
导出CSV
| Compound | θ1 | θ2 | L1 | L2 | L3 | L4 |
| DOBT-8T | 9.29 | 13.74 | 0.1446 | 0.1456 | 0.1458 | 0.1444 |
| BT-8T | 4.54 | 5.07 | 0.1448 | 0.1452 | 0.1453 | 0.1443 |
| FBT-8T | 1.27 | 4.51 | 0.1441 | 0.1444 | 0.1452 | 0.1451 |
| FFBT-8T | 0.11 | 0.18 | 0.1442 | 0.1445 | 0.1453 | 0.1452 |
The lengths of all bridge bonds L1, L2, L3 and L4 are all within 0.1436~0.1453 nm, which are longer than a typical C=C bond (0.133 nm) and shorter than a typical C—C single bond (0.154 nm) at the same level of theory.[23] This is in part caused by the π-bonding interaction and resulted in partial double-bond properties on the bridge bond, thus strengthening and shortening the bridge bond.[24] The results demonstrated that the π-electrons has good delocalized over the whole molecular skeleton. The average bond lengths of L1, L2, L3 and L4 in FBT-8T and FFBT-8T are smaller than those in DOBT-8T and BT-8T, which is in agreement with their planarity and can be assigned to the extent of π conjugation of the OSMs. From the viewpoint of molecular geometrical structure, the attachment of F atom on BT acceptor unit will not only improve molecular planarity, but also strengthen the π-conjugation of the OSMs in some extent.
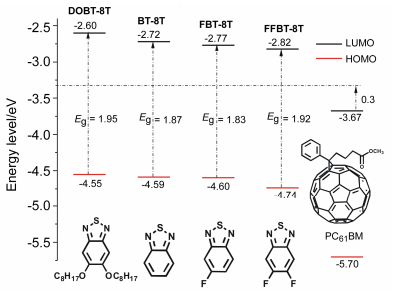
To gain insight into the effects of modified BT on the optoelectronic properties of the OSMs, the energy levels of HOMO and LUMO, the energetic driving force (ΔEL-L) and Voc for all the four designed OSMs were investigated. As we know, Voc is one of the critical parameters to evaluate the PCE of BHJ PV devices, [25] and which has a close relation with the energy difference between HOMO of the donor materials and LUMO of the acceptor materials, and it could be estimated by Eq. (1):
|
$ {V_{{\rm{oc}}}}{\rm{ = }}\left( {1/e} \right)(\mid {E_{{\rm{HOMO}}}}({\rm{D}})\mid - \mid {E_{{\rm{LUMO}}}}\left( {\rm{A}} \right)\mid ) - 0.3{\rm{ V}} $ |
(1) |
where e is the elementary charge, 0.3 V is the empirical value for efficient charge separation, EHOMO(D) is the HOMO energy level of OSMs donor material and ELUMO(A) is the LUMO energy level of acceptor material PC61BM.
A schematic energy diagram of OSMs:PC61BM system with HOMO, LUMO and the band-gap energy was shown in Figure 2. The calculated HOMO levels of DOBT-8T, BT-8T, FBT-8T and FFBT-8T are -4.55, -4.59, -4.60 and -4.74 eV, respectively. The HOMO energy level of DOBT-8T is higher than that of BT-8T, due to the electron-donating effect of octyloxy chain. Compared to BT-8T, FBT-8T and FFBT-8T got a relatively lower HOMO energy level due to substitution of the F atoms on the BT group, which increased the electron withdrawing effect of the acceptor unit.[26] FFBT-8T attained the lowest HOMO energy level of the four designed OSMs, because double F atoms are attached to BT group. This deeplying HOMO energy level of FFBT-8T is expected to promote the improvement of Voc in OSCs.[27] The LUMO energy levels of DOBT-8T, BT-8T, FBT-8T and FFBT-8T are -2.60, -2.72, -2.77 and -2.82 eV, respectively. The results indicated that attaching octyloxy chains on BT can up-shift the LUMO energy levels, while the incorporation of F atoms also led to a reduction of the LUMO energy levels. Their Eg values are in the order of FBT-8T < BT- 8T < FFBT-8T < DOBT-8T=1.95 eV, all the four designed OSMs have relatively narrowed Eg, which can be ascribed to the extended of D-A conjugation and strong D-A intramolecular interaction providing by the OTs.[28] The most narrowed Eg of FBT-8T benefited from its ELUMO decreasing more than EHOMO by attaching one F atom on BT unit. In order to evaluate the reliability of change trends of energy levels for the designed OSMs calculated in gas phase, DFT and TD-DFT calculation at the B3LYP/ 6-31G(d) level were also performed in solution and solid states, and the corresponding data are summarized in Table 3. The calculated results of HOMO and LUMO levels in solution and solid phase have some deviation from the values in gas phase, but change trend of HOMO and LUMO levels for the four OSMs calculated in solution and solid phase agrees well with the gas phase values.
下载:
导出CSV
| Compound | In gas state | In solution state | In solid state | |||||||||||
| HOMO | LUMO | Eg | Voc | ΔEL-L | Eb | HOMO | LUMO | Eg | HOMO | LUMO | Eg | |||
| DOBT-8T | -4.55 | -2.62 | 1.95 | 0.58 | 1.20 | 0.30 | -4.70 | -2.73 | 1.97 | -4.73 | -2.75 | 1.98 | ||
| BT-8T | -4.59 | -2.72 | 1.87 | 0.62 | 1.08 | 0.28 | -4.71 | -2.83 | 1.88 | -4.74 | -2.85 | 1.89 | ||
| FBT-8T | -4.60 | -2.77 | 1.83 | 0.63 | 1.03 | 0.27 | -4.73 | -2.87 | 1.86 | -4.78 | -2.89 | 1.89 | ||
| FFBT-8T | -4.74 | -2.82 | 1.92 | 0.77 | 0.98 | 0.29 | -4.83 | -2.89 | 1.94 | -4.85 | -2.90 | 1.95 | ||
| a ΔEL-L, HOMO, LUMO, Eg, Eb are in eV, Voc is in V. | ||||||||||||||
ΔEL-L values which defined as the difference between the LUMOs of the donor material and the acceptor material have always been considered as an important parameter connecting to the efficiency of charge dissociation.[29] It has been reported in many theoretical papers to estimate charge dissociation ability by comparing ΔEL-L with the exciton binding energy Eb, which was defined as the difference between Eg and Eopt.[30] If ΔEL-L is larger than Eb, the donor materials are considered to have efficient exciton split and charge dissociation at the donor/acceptor interface.[31] As shown in Table 3, the ΔEL-L values of DOBT-8T, BT-8T, FBT-8T and FFBT-8T are 1.2, 1.08, 1.03 and 0.98 eV, respectively, and the Eb values of DOBT-8T, BT-8T, FBT-8T andFFBT-8T are 0.30, 0.28, 0.27 and 0.29 eV, respectively. The results indicated that the four designed OSMs could provide sufficient driving force to conquer Eb and generate the charge separation. However, some recent studies reported that overlarge ΔEL-L would result in lower Voc and energetic loss of excess energy of exciton, thus limiting the further elevation of their device efficiencies.[32] FBT-8T and FFBT-8T with a relatively smaller ΔEL-L values than other two OSMs might give rise to a small energy loss and relatively higher PV device performance.
The calculated Voc values for the OSMs:PC61BM system are listed in Table 3. The Voc values of these OSMs are in the order of FFBT-8T > FBT-8T > BT-8T > DOBT-8T, the difference of the Voc values is ascribed to structural modification effects on the BT unit. FFBT-8T has the highest Voc of the four designed OSMs, suggesting that the substituent group of the two F atoms on FFBT unit could effectively increase Voc. Attaching octyloxy chains on BT unit made DOBT-8T achieve a higher HOMO level and provide a relatively smaller Voc. In contrast to BT-8T, FBT-8T exhibited a lower HOMO level as well as most narrowed Eg, benefiting from the introduction of FBT unit, thus tending to realize a delicate balance between large Voc and high Jsc. The above discussion indicates that FBT-8Tand FFBT-8T are also potential candidates for PV cells in the four designed OSMs. Interestingly, the changing trend of Voc for the four designed OSMs is just in agreement with their energy loss. Notably, we compare the energy levels of the four OSMs with non-fullerene acceptor INPIC-4F, and the investigated results indicated that the four OSMs donor has the potential for constructing all-small-molecule solar cell.
In order to further analyze the charge distribution and the electron-transfer mechanism in DOBT-8T, BT-8T, FBT- 8T and FFBT-8T, the Natural Bond Orbital (NBO) analysis has been performed. The acceptor units of the above OSMs are DOBT, BT, FBT and FFBT, respectively. The core donor unit is tetrathiophene, while the terminal donor unit is bithiophene. The calculated NPA charges that populated in the acceptor unit, tetrathiophene and bithiophene are listed in Table 4.
下载:
导出CSV
| Compound | Bithiophene (left) | BT unit (left) | Tetrathiophene | BT unit (right) | Bithiophene (right) |
| DOBT-8T | 0.053 | -0.105 | 0.104 | -0.105 | 0.053 |
| BT-8T | 0.061 | -0.110 | 0.097 | -0.110 | 0.061 |
| FBT-8T | 0.076 | -0.136 | 0.120 | -0.136 | 0.076 |
| FFBT-8T | 0.080 | -0.149 | 0.138 | -0.149 | 0.080 |
As seen in Table 4, the NPA charges of the different BT acceptor units are -0.105e, -0.110e, -0.136e and -0.149e, respectively. The most negative charge value of FFBT acceptor unit from FFBT-8T is attributed to the strong electronegativity of two F atom which be introduced into the BT moiety. FBT units from FBT-8T exhibited relatively bigger negative charge values compared to BT and DOBT acceptor units, which can be assigned to the attaching single F atom on the BT moiety. DOBT units from DOBT-8T demonstrated the smaller negative charge than the BT unit in BT-8T, because modifying the BT moiety with octyloxy group effectively lowered its electronegativity. Meanwhile, the core donor tetrathiophene and the terminal donor bithiophene all exhibited positive charge, and their charge values also changed with the different BT acceptor units, which resulted from the structural modification effects on the BT unit. The positive charges of the donor moiety and the negative charges of the acceptor unit for the four designed OSMs demonstrated that they were an effective electron pushing or pulling units, indicating an effective intramolecular charge transfer (ICT) transition from the donor group to the acceptor. During the photo-excitation, the electrons on donor unit are transferred to the acceptor unit, and the charge separation state was formed in OSMs. That is, upon PC61BM surface, the electrons could be successfully transferred from the donor unit to acceptor unit, finally injected into the LUMO of PC61BM surface.[33]At last, the calculated NPA charge shows that the modification of BT structure induces the variation of negative/positive charge values, which in agreement with electron-donating/ electron withdrawing character of the functional group for octyloxy and F atom.
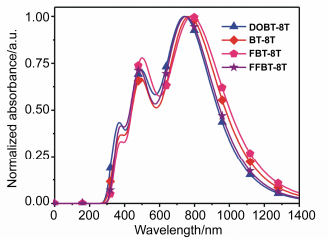
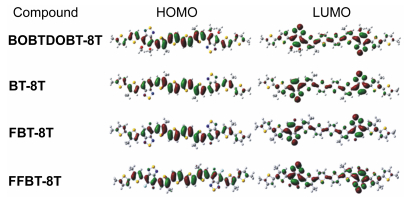
The OSMs donor material is the key component for sun light absorption in OSCs, because the absorption coverage of donor material within the solar spectrum crucially influences the Jsc of OSCs. Therefore, photo-excitation properties were discussed to better understand the underlying photo-physical processes involved in photo-current generation. For better understanding the nature of the absorption process, the electron density distributions of HOMO and LUMO for DOBT-8T, BT-8T, FBT-8T and FFBT-8T are depicted in Figure 3. The frontier molecular orbitals of the four designed OSMs have analogous distribution characteristics. All HOMOs show the typical aromatic features with electron delocalization for the whole conjugated molecules, and the LUMOs are mainly concentrated on electron-deficient BT unit.[34] The electronic excitation energy (E/eV), maximum absorption wavelengths (λmax/nm), oscillator strength (ƒ), and major configurations of main excited states for the four designed OSMs are listed in Table 5, and the simulated absorption spectra involved are gathered in Figure 4.
 下载:
导出CSV
下载:
导出CSV
| Compound | State | E/eV | λmax/nm | Oscillator | η | Main configuration |
| DOBT-8T | S1 | 1.65 | 750 | 2.07 | 0.9914 | HOMO → LUMO (69%) |
| S7 | 2.54 | 487 | 1.27 | HOMO → LUMO+2 (66%) | ||
| S12 | 2.69 | 404 | 0.19 | HOMO → LUMO+4 (68%) | ||
| BT-8T | S1 | 1.59 | 777 | 2.14 | 0.9927 | HOMO → LUMO (70%) |
| S7 | 2.55 | 485 | 1.25 | HOMO → LUMO+2 (66%) | ||
| S12 | 3.09 | 401 | 0.22 | HOMO → LUMO+4(68%) | ||
| FBT-8T | S1 | 1.56 | 793 | 2.15 | 0.9929 | HOMO → LUMO (70%) |
| S7 | 2.55 | 487 | 1.23 | HOMO → LUMO+2 (65%) | ||
| S12 | 3.01 | 411 | 0.10 | HOMO → LUMO+4 (68%) | ||
| FFBT-8T | S1 | 1.63 | 758 | 2.08 | 0.9917 | HOMO → LUMO (69%) |
| S7 | 2.57 | 482 | 1.38 | HOMO → LUMO+2 (68% | ||
| S12 | 3.15 | 393 | 0.21 | HOMO → LUMO+4(66%) |
As seen in Figure 4, the four designed OSMs all provide the similar absorption spectra that there are very strong absorption peaks in the low energy region (700~900 nm), relatively weak absorption in the moderate energy region (450~580 nm), and small shoulder peaks in the high energy region (300~450 nm). The maximum absorption peaks of all designed OSMs arose from the dominant excitation from state S0→S1 and mostly attribute to the transitions from HOMO to LUMO, which can deduce that the main ICT transitions occur at the low energy region.[33] The relatively weak absorption peaks in the moderate energy region resulted from the optical transitions of states S0→S7, which is correspondingly assigned to HOMO→ LUMO+2 transitions. The small shoulder peaks arose from the optical transitions of states S0→S12, and the main configuration is HOMO→LUMO+4 in the high energy region. The absorption band in the moderate and high energy region of the four designed OSMs can be ascribed to the π-π transitions.[17]
The designed four OSMs all have wide absorption spec- trum of 300~1400 nm, and the values of λmax were in the sequence of FBT-8T (793 nm) > BT-8T (777 nm) > FFBT- 8T (758 nm) > DOBT-8T (750 nm), which is consistent with their change trend of Eg. Compared to BT-8T, the maximum absorption peaks of DOBT-8T shows a blue shift of 27 nm, resulting from the introduction of octyloxy chains broadened its Eg. Obviously, FBT-8T obtained a reduced Eg, stronger and more red-shifted absorption of the maximum absorption peaks relative to BT-8T, which can be ascribed to the attaching one F atom on the BT unit. The light harvesting efficiency (η) of the OSMs can be calculated as η=1-10-f. According to the above equation, the higher the f is, the larger η will be.[35] The η values of the designed four OSMs are in the order of FBT-8T > FFBT-8T > BT-8T > DOBT-8T, which is in consistent in the trend of their Eg values. According the above discuss, we predicted that FBT-8T has better absorption ability to increase Jsc, because FBT-8T has higher value of λmax, larger f and η values than other three designed OSMs. Compared to FBT-8T, FFBT-8T achieved a relatively smaller maximum absorption peak, because two F atom attaching on the BT unit significantly decreased the HOMO and widened the Eg in some extent. However, FFBT-8T has the deeper HOMO level than other three molecules, which is beneficial for getting high Voc values in OSCs.
The calculated optical absorption profiles in solution and solid state are shown in the supporting information Figure S3. The simulated absorption spectra of the four OSMs all show a similar profile, which present two primary bands at short wavelength (450~580 nm) and long wavelength (700~900 nm), respectively, as well as a should peak in 300~450 nm. Notably, the calculated optical absorption spectra in solution and solid state are consistent with the gas phase absorption spectra. In addition, the excited state energies and absorption spectra of DOBT-8T, BT-8T, FBT-8T and FFBT-8T have been evaluated by diffuse function at TD-B3LYP/6-31+G(d) level. The calculated electronic excitation energy, the maximum absorption peak (λmax), oscillator strength (f > 0.5), η and main configuration of the four studied OSMs are gathered in Table S2 as well as the absorption spectra are depicted in Figure S4. The results indicated that excited state energies of the four studied OSMs from B3LYP/6-31+G(d) level calculation are identical with the data from B3LYP/6-31G(d) level calculation. Nevertheless, the simulated shape of absorption spectra and absorption region from B3LYP/6-31+G(d) level are similar to the one calculated from B3LYP/6-31G(d) level.
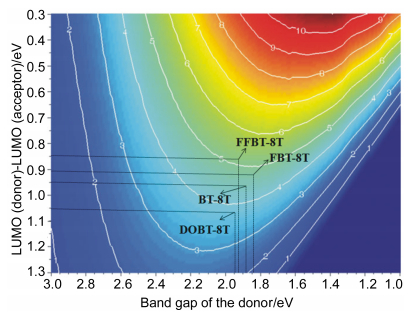
For the qualitative evaluation of the properties of the four designed OSMs, Scharber diagram can be used to estimate the PCE of the OSCs, which used the relationship between the Eg of donor materials as well as the energy difference of LUMO levels between donor materials and PC61BM.[36] This diagram has been widely used and accepted to evaluate the PCE of OSCs based on a blend of OSM/polymer donor materials and PC61BM acceptor materials. It is shown that the predicted PCEs are in consistent with the reported experimental values. According to the Scharber diagrams, the simulated PCEs of the designed OSMs/PC61BM are provided in Figure 5, the PCEs of DOBT-8T, BT-8T, FBT-8T and FFBT-8Tare ca. 3.8%, 4.4%, 4.7%, 5.2%, respectively. Compared to other OSMs, FBT-8T and FFBT-8T showed higher PCE than other two OSMs, which illustrated that the effect of introduction of F atom on BT acceptor is prominent. The FBT-8T is expected to have higher PCE by prediction, because they have deeper HOMO and the narrowest Eg, which illustrated that the introduction of F atom on BT unit can realized the balance between the deeper HOMO and narrowed Eg in some extent. FFBT- 8T get the highest predicted PCE of the four designed OSMs, because it has the lowest HOMO level and suitable Eg, which can be assigned to the introduction of two F atom on BT unit remarkably reducing the HOMO level, and the large conjugation framework mainly constructed by OTs units maintaining the good absorption ability. Among them, the performance of DOBT-8T is relatively bad because of introduction of octyloxy group on BT unit, not only enhanced the HOMO level but also widen the Eg. The results provided a positive evaluation on the modifying BT unit with different functional fragment to improve the PCEs of OSMs.
In this paper, OSMsDOBT-8T, BT-8T, FBT-8T and FFBT-8T have been theoretical designed, and the effects of different BT acceptor units on the PV properties of OTs based OSMs were firstly systematically investigated in this paper. Their geometrical structures, HOMOs, LUMOs, Eg, ΔEL-L and Eb have been calculated by using DFT- B3LYP/6-31G(d) method. Moreover, their absorption spectra were obtained by using TD-DFT-B3LYP/6-31G- (d) method. The results showed that the designed four OSMs can provide better matched energy levels with PC61BM, small Eb values and sufficient ΔEL-L of excitons for charge dissociation in the OSMs:PC61BM system. Notably, the results exhibited that attaching substituent on the BT acceptor unit could effectively affect molecular structures and tune photoelectric properties, which might further impact their PV performance. Among them, FBT-8T exhibited a most narrowed Eg than other three OSMs, and relatively higher Voc than BT-8T and DOBT-8T, which means that they could better balance large Voc and high Jsc in some extent. FFBT-8T showed a most deeplying HOMO level of the four designed OSMs, due to the introduction of two F atom on BT unit, which is beneficial for getting high Voc in OSCs. Furthermore, FFBT-8T also got a suitable Eg, which can be assigned to the large conjugation framework mainly constructed by OTs units, thus facilitating the improvement of the Jsc. According to the Scharber diagrams, the predicted PCEs of DOBT-8T, BT-8T, FBT-8T and FFBT-8T are ca. 3.8%, 4.4%, 4.7%, 5.2%, respectively, when they are used to combine PC61BM as an acceptor material. The results imply that FBT-8T and FFBT-8T could be potential high-efficiency PV donor materials of the D-A-D-A-D typed OTs-BT based OSMs. While some discrepancies in the calculated values may exist compared to the experimental data, we hope that the conclusion and predictions based from the obtained trends of the structured modification effects on BT unit can provide structural guidelines in the future designs of organic PV donor materials based on OTs-BT skeleton.
Supporting Information The absolute energies and atomic coordinates of all optimized structures, absorption spectra for the four studied OSMs in CHCl3 solution and in solid are provided. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn.
Cui C., Guo X., Min J., Guo B., Cheng X., Zhang M., Brabec C. J., Li Y.Adv. Mater. 2015, 27, 7469. doi: 10.1002/adma.201503815
Zhao J., Li Y., Yang G., Jiang K., Lin H., Ade H., Ma W., Yan H.Nat. Energy 2016, 1, 1. https://www.nature.com/articles/nenergy201527
Wan J., Xu X., Zhang G., Li Y., Peng Q.Energy Environ. Sci. 2017, 10, 1739. doi: 10.1039/C7EE00805H
Geng Y., Tang A., Tajima K., Zeng Q., Zhou E.J. Mater. Chem. A 2019, 7, 64. doi: 10.1039/C8TA09383K
Zhou Z., Xu S., Liu W., Zhang C., Hu Q., Liu F.J. Mater. Chem. A 2017, 5, 3425. doi: 10.1039/C6TA10559A
Du J., Biewer M. C., Stefan M. C.J. Mater. Chem. A 2016, 4, 15771. doi: 10.1039/C6TA06241E
Cheng M., Chen C., Yang X., Huang J., Zhang F., Xu B., Sun L.-C.Chem. Mater. 2015, 27, 1808. doi: 10.1021/acs.chemmater.5b00001
(a) Wang L.-H., Wang L.-J., Xie B., Ji C.-Y., Li Y.-Q.Dye Pigm. 2017, 140, 203.
(b) Wang L., Yin L., Ji C., Zhang Y., Gao H., Li Y.Org. Electron. 2014, 15, 1138.
Wang J.-L., Zhang H.-J., Liu S., Liu K.-K., Liu F., Wu H.-B., Cao Y.Sol. RRL 2018, 2, 1800. https://www.ncbi.nlm.nih.gov/pubmed/23965284
Ni W., Wan X., Li M., Wang Y., Chen Y.Chem. Commun. 2015, 51, 4936. doi: 10.1039/C4CC09758K
Cho A., Song C. E., Lee S. K., Shin W. S., Lim E.J. Mater. Sci. 2016, 51, 6770. doi: 10.1007/s10853-016-9964-x
(a) Chen R., Wang Y., Chen T., Li H., Zheng C., Yuan K., Wang Z., Tao Y., Zheng C., Huang W.J. Phys. Chem. B 2015, 119, 583.
(b) Franco F. C., Padama A. A. B.Polymer 2016, 97, 55.
(c) McCormick T. M., Bridges C. R., Carrera E. I., DiCarmine P. M., Gibson G.-L., Hollinger J., Kozycz L. M., Seferos D. S.Macromolecules 2013, 46, 3879.
(a) Becke A. D.J. Chem. Phys. 1993, 98, 1372.
(b) Lee C., Yang W., Parr R. G.Phys. Rev. B 1988, 37, 785.
(c) Becke A. D.Phys. Rev. A 1988, 38, 3098.
Lin L.-Y., Lu C.-W., Huang W.-C., Chen Y.-H., Lin H.-W., Wong K.-T.Org. Lett. 2011, 13, 4962. doi: 10.1021/ol2021077
Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B. G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A.Jr.; Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Kobayashi V. N. R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J.E; Cross J. B., Bakken V., Jaramillo C. J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R.L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J.Gaussian 09, Evision A.01, Gaussian, Inc., Wallingford CT, 2009.
(a) Rutledge L. R., McAfee S. M., Welch G. C.J. Phys. Chem. A 2014, 118, 7939.
(b) Sahu H., Panda A. N.J. Phys. Chem. C 2015, 119, 22855.
Wang Z., Song C., Li J., Li P., Zhang H.J. Phys. Chem. C 2018, 123, 1069. http://med.wanfangdata.com.cn/Paper/Detail/PeriodicalPaper_PM21303717
(a) Frost J. M., Faist M. A., Nelson J.Adv. Mater. 2010, 22, 4881.
(b) Zhang G., Musgrave C. B.J. Phys. Chem. A 2007, 111, 1554.
Wang L., Yin L., Ji C., Li Y.Dye Pigm. 2015, 118, 37. doi: 10.1016/j.dyepig.2015.02.017
Foster J. P., Weinhold F.J. Am. Chem. Soc. 1980, 102, 7211. doi: 10.1021/ja00544a007
Irfan M., Iqbal J., Sadaf S., Eliasson B., Rana U. A., Ud-din Khan S., Ayub K.Int. J. Quantum Chem. 2017, 117, 25363. doi: 10.1002/qua.25363
Cui Y., Li P., Song C., Zhang H.J. Phys. Chem. C 2016, 120, 28939. doi: 10.1021/acs.jpcc.6b09927
Zhang L., Shen W., He R., Tang X., Yang Y., Li M.Mater. Chem. Phys. 2016, 175, 13. doi: 10.1016/j.matchemphys.2016.01.062
Sun F., Jin R.Arab. J. Chem. 2017, 10, S2988. doi: 10.1016/j.arabjc.2013.11.037
Liu X., He R., Shen W., Li M.J. Power Sources 2014, 245, 217. doi: 10.1016/j.jpowsour.2013.06.137
Zhao Z. W., Pan Q. Q., Li S. B., Duan Y. A., Geng Y., Zhang M., Su Z. M.J. Mol. Graphics Modell. 2017, 77, 9. doi: 10.1016/j.jmgm.2017.07.027
Zhao Z.-W., Pan Q.-Q., Wu S.-X., Wu Y., Zhang M., Zhao L., Su Z. M.Theor. Chem. Acc. 2018, 137, 1. doi: 10.1007/s00214-017-2177-9
Wang J.-L., Liu K.-K., Yan J., Wu Z., Liu F., Xiao F., Chang Z.-F., Wu H.-B., Cao Y., Russell T. P.J. Am. Chem. Soc. 2016, 138, 7687. doi: 10.1021/jacs.6b03495
Pan Q.-Q., Li S.-B., Wu Y., Zhang J., Li H.-B., Geng Y., Su Z.-M.New J. Chem. 2013, 1, 1.
Bourass M., Touimi B. A., Hamidi M., Benzakour M., McHarfi M., Sfaira M.J. Saudi Chem. Soc. 2016, 20, S415. doi: 10.1016/j.jscs.2013.01.003
Wan J., Xu X., Zhang G., Li Y., Feng K., Peng Q.Energy Environ. Sci. 2017, 10, 1739. doi: 10.1039/C7EE00805H
Taouali W., Casida M. E., Darghouth A. A. M. H. M., Alimi K. Comput. Mater. Sci. 2018, 150, 54. doi: 10.1016/j.commatsci.2018.03.038
Bourass M., Touimi B. A., Benzakour M., McHarfi M., Jhilal F., Serein-Spirau F., Lère-Porte J.-P., Sotiropoulos J.-M., Bouzzine S. M., Bouachrine M.J. Saudi Chem. Soc. 2017, 21, 563. doi: 10.1016/j.jscs.2017.01.001
Zhang L., Shen W., He R., Liu X., Tang X., Yang Y., Li M.Org. Electron. 2016, 32, 134. doi: 10.1016/j.orgel.2016.01.023
Tai C.-K., Hsieh C.-A., Chang K.-H., Wang B.-C.Res. Chem. Intermed. 2016, 42, 6907. doi: 10.1007/s11164-016-2504-0
Dennler G., Scharber M. C.Brabec C. J.Adv. Mater. 2009, 21, 1323. doi: 10.1002/adma.200801283

Figure 2 Schematic energy diagram of OSMs:PC61BM system with HOMO, LUMO and Eg calculated in gas phase.

Figure 3 Electron density distribution of HOMO and LUMO for the four OSMs computed at B3LYP/6-31G(d) level

Figure 5 Scharber diagrams to estimate PCEs of the BHJ OSCs for OSMs DOBT-8T, BT-8T, FBT-8T and FFBT-8T
Table 1. Theoretical and experimental FMO energy levels of BDCTMBT, BDCTTMBT, BDCTFBT and BDCTTFBT
| Compd. | Theoretical | Experimental | Theoretical | Experimental | |||||
| HOMO/eV | Eg/eV | HOMO/eV | Eg/eV | Voc/V | Voc/V | ||||
| BDCTMBT | -4.91 | 2.30 | -5.30 | 2.02 | 0.94 | 1.05 | |||
| BDCTTMBT | -4.73 | 2.05 | -5.15 | 1.85 | 0.76 | 0.96 | |||
| BDCTFBT | -4.95 | 2.27 | -5.41 | 2.09 | 0.98 | 1.11 | |||
| BDCTTFBT | -4.77 | 2.03 | -5.25 | 1.89 | 0.80 | 1.01 | |||
 下载: 导出CSV
下载: 导出CSV
Table 2. Selected bond lengths (L, in nm) and dihedral angles (θ, in degree) of the four OSMs calculated at B3LYP/6-31G(d) level
| Compound | θ1 | θ2 | L1 | L2 | L3 | L4 |
| DOBT-8T | 9.29 | 13.74 | 0.1446 | 0.1456 | 0.1458 | 0.1444 |
| BT-8T | 4.54 | 5.07 | 0.1448 | 0.1452 | 0.1453 | 0.1443 |
| FBT-8T | 1.27 | 4.51 | 0.1441 | 0.1444 | 0.1452 | 0.1451 |
| FFBT-8T | 0.11 | 0.18 | 0.1442 | 0.1445 | 0.1453 | 0.1452 |
下载: 导出CSV
Table 3. Calculated ΔEL-L, HOMO, LUMO, Eg, Eb and Voc of the four OSMs obtained by B3LYP/6-31G(d) level in gas phase as well as the calculated HOMO, LUMO, and Eg values of the four OSMs in solution and solid states by B3LYP//6-31G(d) levela
| Compound | In gas state | In solution state | In solid state | |||||||||||
| HOMO | LUMO | Eg | Voc | ΔEL-L | Eb | HOMO | LUMO | Eg | HOMO | LUMO | Eg | |||
| DOBT-8T | -4.55 | -2.62 | 1.95 | 0.58 | 1.20 | 0.30 | -4.70 | -2.73 | 1.97 | -4.73 | -2.75 | 1.98 | ||
| BT-8T | -4.59 | -2.72 | 1.87 | 0.62 | 1.08 | 0.28 | -4.71 | -2.83 | 1.88 | -4.74 | -2.85 | 1.89 | ||
| FBT-8T | -4.60 | -2.77 | 1.83 | 0.63 | 1.03 | 0.27 | -4.73 | -2.87 | 1.86 | -4.78 | -2.89 | 1.89 | ||
| FFBT-8T | -4.74 | -2.82 | 1.92 | 0.77 | 0.98 | 0.29 | -4.83 | -2.89 | 1.94 | -4.85 | -2.90 | 1.95 | ||
| a ΔEL-L, HOMO, LUMO, Eg, Eb are in eV, Voc is in V. | ||||||||||||||
下载: 导出CSV
Table 4. Calculated NPA charge (e) of D and A units for all studied molecules
| Compound | Bithiophene (left) | BT unit (left) | Tetrathiophene | BT unit (right) | Bithiophene (right) |
| DOBT-8T | 0.053 | -0.105 | 0.104 | -0.105 | 0.053 |
| BT-8T | 0.061 | -0.110 | 0.097 | -0.110 | 0.061 |
| FBT-8T | 0.076 | -0.136 | 0.120 | -0.136 | 0.076 |
| FFBT-8T | 0.080 | -0.149 | 0.138 | -0.149 | 0.080 |
下载: 导出CSV
Table 5. Calculated electronic excitation energy, λmax, oscillator strength, η and main configuration of DOBT-8T, BT-8T, FBT-8T and FFBT-8T by TD-B3LYP/6-31G (d) level
| Compound | State | E/eV | λmax/nm | Oscillator | η | Main configuration |
| DOBT-8T | S1 | 1.65 | 750 | 2.07 | 0.9914 | HOMO → LUMO (69%) |
| S7 | 2.54 | 487 | 1.27 | HOMO → LUMO+2 (66%) | ||
| S12 | 2.69 | 404 | 0.19 | HOMO → LUMO+4 (68%) | ||
| BT-8T | S1 | 1.59 | 777 | 2.14 | 0.9927 | HOMO → LUMO (70%) |
| S7 | 2.55 | 485 | 1.25 | HOMO → LUMO+2 (66%) | ||
| S12 | 3.09 | 401 | 0.22 | HOMO → LUMO+4(68%) | ||
| FBT-8T | S1 | 1.56 | 793 | 2.15 | 0.9929 | HOMO → LUMO (70%) |
| S7 | 2.55 | 487 | 1.23 | HOMO → LUMO+2 (65%) | ||
| S12 | 3.01 | 411 | 0.10 | HOMO → LUMO+4 (68%) | ||
| FFBT-8T | S1 | 1.63 | 758 | 2.08 | 0.9917 | HOMO → LUMO (69%) |
| S7 | 2.57 | 482 | 1.38 | HOMO → LUMO+2 (68% | ||
| S12 | 3.15 | 393 | 0.21 | HOMO → LUMO+4(66%) |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们