
图式 1.
苯乙烯的2, 2, 2-三氟乙基化反应机理
Scheme 1.
Mechanism of 2, 2, 2-trifluoroethylation of styrene

20世纪下半叶以来, 随着全球经济的快速腾飞, 人类物质生活方面得到极大发展, 同时生态环境破坏问题却日趋严重.大量化学产物的肆意排放, 已经严重威胁到了人类的生存.因此, 减少化学过程中的有毒或污染废物的绿色化学得到了广泛的青睐[1].一般来说, 反应系统中的化学污染主要来源于有机溶剂和金属催化剂的大规模使用.因此, 环境友好型溶剂与新型催化剂的研究已成为化学研究中最具吸引力的领域之一[2].
水是地球生命的基础[3], 是生物体内酶催化反应的优良介质, 廉价、易得, 同时具有无毒、不致癌、不致突变和不易燃烧爆炸等绿色反应介质的所有特性[4].用水来代替有机溶剂, 符合当今绿色化学的主题[5].近年来, 以水为溶剂的金属催化[6]、生物催化[7]和胶束催化[8]等在加快反应速率中具有广阔的应用前景, 在水相中实现有机合成反应已经取得了重大进展[9].
光催化反应是在光和催化剂的同时作用下所进行的化学反应, 是光反应和催化反应的融合. 1972年, Fujishima和Honda[10]在n型半导体TiO2单晶电极上实现了水的光电催化分解, 自此光催化正式进入了人类的视野.光催化, 特别是可见光催化, 是当前有机化学的前沿研究领域, 近年来可见光介导的有机化学反应研究获得了突飞猛进的发展[11].可见光是一种清洁的、可再生的自然资源[12].与传统的金属催化、有机小分子催化以及酶催化相比较, 光催化剂具有用量少、反应条件温和、底物适用性好以及实验操作简单等优点[13].
目前为止, 绝大多数光催化反应都是在有机相中进行[14], 在纯水相中进行可见光介导的有机反应报道鲜少.在已有的水参与的光催化反应中, 水往往和有机溶剂一起作为双相溶剂[15]; 有时水还作为底物参与反应; 在其它反应中水虽然没有参与反应, 但是水对反应有显著的促进作用[16].对近年来水参与以及水作为反应介质的可见光催化的有机化学反应进行综述, 并对相应的机理进行讨论.
水具有的许多独特的物理和化学性质, 如极性和双亲性等, 均来源于水的独特结构——大量的氢键[17], 研究发现水有时还能够提高反应速率以及影响各种有机反应的选择性[6b, 18].
2015年, 陈庆云等[14]在水存在下, 氧气中, 可见光诱导苯乙烯与1, 1, 1-三氟-2-碘乙烷发生光氧化还原双官能化反应, 得到γ-三氟甲基醇(Eq. 1).在该自由基反应中,产物中的氧原子来自分子氧,其中水显示出对反应有促进作用。反应底物可适用于在芳环上带有给电子和卤素取代基的苯乙烯以及在双键的β-位具有取代基的苯乙烯.
|
|
(1) |
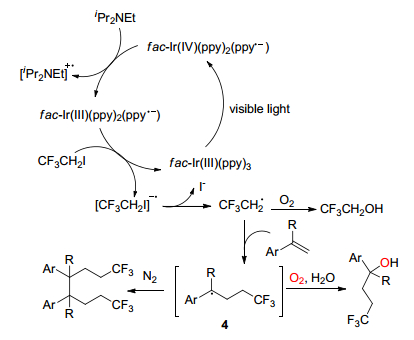
作者提出了该反应的反应机理见Scheme 1.首先, 可见光照射fac-Ir(Ⅲ)(ppy)3, 经金属-配体电荷转移(MLCT)产生氧化还原活性光激发态fac-Ir(Ⅳ)(ppy)2- (ppy•-), iPr2NEt作为还原猝灭剂, fac-Ir(Ⅳ)-(ppy)2(ppy•-)接受来自iPr2NEt的电子, 产生的fac-Ir(Ⅲ)(ppy)2(ppy•-)具有很高的还原能力, 通过fac-Ir(Ⅲ)(ppy)2(ppy•-)单电子还原CF3CH2I产生自由基阴离子, 然后产生2, 2, 2-三氟乙基(CF3CH2•), 如果反应在O2中进行, 则三氟乙基可以通过捕获分子氧生成CF3CH2OH.在另一种途径中, 三氟乙基可与苯乙烯反应得到苄基自由基4, 在分子氧存在下被水促进, 得到羟基产物; 如果反应在N2中进行, 则苄基自由基4与其自身偶联, 得到二聚产物.
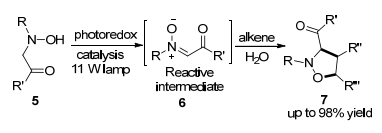
2014年, Rueping等[16]在可见光光氧还原催化下, 建立了N-取代的羟胺与烯烃的新的氧化[3+2]环加成反应(Scheme 2).这种新颖的策略对五元环异噁唑烷杂环化合物提供了简洁、快速、温和和高效的合成方法, 水的参与加速了反应速率.研究其机理发现, 在光氧还原循环后, 水对反应速率起着决定性的作用.
水不仅可以作为一种廉价、无毒、不易燃、高度稳定且易处理的“绿色”溶剂, 有时还可以参与反应过程.
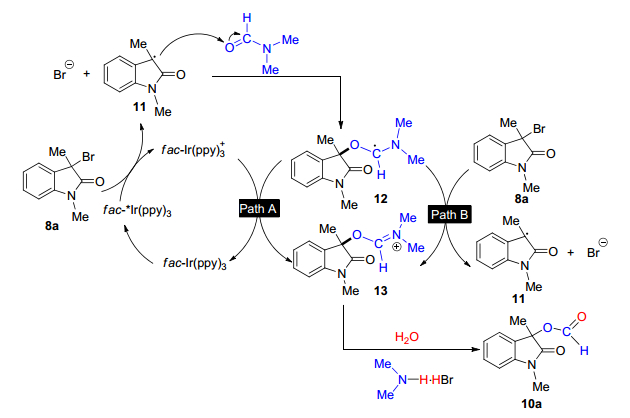
2014年, 肖文精和陈加荣等[19]在光氧化还原催化剂fac-Ir(ppy)3的存在和可见光照射下, 由3-溴甲氧基吲哚、N, N-二甲基甲酰胺(DMF)和水得到了3-甲酰氧基吲哚.这是一种新颖且有效的可见光诱导的自由基加成/氧化/水解级联反应(Eq. 2), 该转化可直接在羟吲哚的C-3位连接甲酰氧基.其中DMF作为“H, C, O”源, H2O作为“O”源.该反应适用范围广泛, 其中带有吸电子、中性电子和给电子取代基的化合物都以良好的产率得到相应的产物.
|
|
(2) |
该反应的机理见Scheme 3, 首先光催化剂fac-Ir(ppy)3通过可见光照射激发到高活性态fac-Ir*(ppy)3, 然后与烷基溴8a反应, 形成自由基中间体11, 通过氧化猝灭循环释放溴离子和fac-Ir(ppy)3+, 随后, 中间体11和DMF发生自由基加成反应, 得到自由基中间体12. 12被氧化成亚胺离子13, 可能通过两种途径: (1)中间体12被fac-Ir(ppy)3+氧化, 再生基态fac-Ir(ppy)3完成催化循环(路径A); (2)中间体12被另一个分子8a氧化, 通过链增长机制(路径B)得到亚胺离子13和自由基中间体11.最后, 13的水解得到末端产物10a, 同时生成副产物二甲基胺氢溴酸盐.
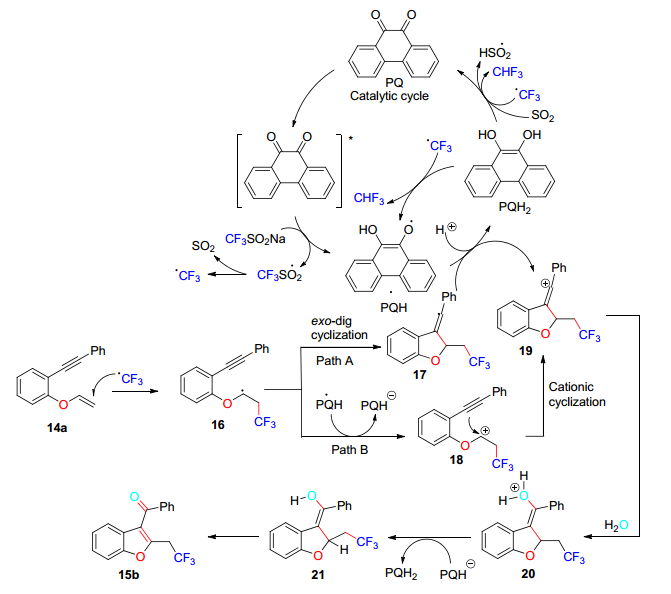
2017年, Kumar等[20]以Langlois试剂(CF3SO2Na)和9, 10-二酮菲蒽(PQ)作为光氧化还原催化剂, 水作为氧源, 实现了光诱导氧化、无金属串联自由基环化、三氟甲基化和1, 6-烯炔的脱氢氧化反应(Eq. 3).该方案可以得到各种含CF3的C(3)-芳酰基/酰化的苯并呋喃, 苯并噻吩和吲哚.
|
|
(3) |
其机理研究表明(Scheme 4), 在光诱导产生的PQ激发态作用下, CF3SO2Na产生CF3自由基, 将CF3自由基加到烯烃上, 经环化得到乙烯基自由基, 然后经电子转移生成乙烯基阳离子, 或者电子转移可以从加入CF3的烯烃部分发生, 形成碳阳离子, 再经历阳离子环化产生乙烯基碳阳离子, 随后向乙烯基阳离子中加入水, 除去氢气, 形成三氟甲基化的C(3)-芳酰基/酰化的杂环化合物.该方法能够使不稳定的1, 6-烯炔底物氧化, 同时在无氧化剂的条件下产生氢气, 且避免了过氧化产物的产生.
2018年, 魏伟等[21]在可见光诱导条件下, 由异氰化物、硫醇和水合成硫代氨基甲酸酯(Eq. 4).通过简单地使用廉价的玫瑰红作为光催化剂, 水作为反应试剂以及环境友好的助溶剂, 以良好的产率得到各种硫代氨基甲酸酯.机理研究发现反应是通过单电子转移过程形成自由基得到反应产物.
|
|
(4) |
2019年, 谢劲等[22]使用廉价的D2O作为理想的氘源, 在可见光与膦自由基协同作用下最终以86%的收率得到96%氘代的产物(Eq. 5).该反应对各种吸电子和给电子的邻、间及对位取代的芳香羧酸都具有很好的兼容性, 并以较优的产率得到高氘代化产物.反应底物含有的氨基、羟基、卤素、醛基、羰基、硼酸酯、末端烯烃及末端炔烃等敏感的官能团均不影响反应的正常进行.喹啉和吲哚等杂芳族酸以及脂肪羧酸也可以顺利进行脱氧氘化.在相同的条件下, 只需用H2O代替D2O, 可以在温和条件下选择性还原羧酸成醛, 并且具有良好的选择性和官能团兼容性.
|
|
(5) |
传统的有机合成在很大程度上依赖于有机溶剂, 包括溶解组分和促进化学反应, 因为许多试剂和活性物质与水不相溶或不混溶.与反应物相比, 有机溶剂大量被使用, 所以溶剂问题一直是环境问题的焦点.自1882年Baeyer和Drewsen在水中成功合成靛蓝后[2], 水被广泛用作有机合成反应研究的溶剂, 包括亲核取代反应[23]、氧化还原反应、Claisen重排反应[7]和Diels-Alder反应[24]等.近年来, 以水为溶剂的金属催化、生物催化、胶束催化等在加快反应速率中具有诱人的应用前景.与传统溶剂相比, 水是生态系统中最普遍和最友好的溶剂, 具有独特的优势.首先, 水具有安全、廉价、无毒和无污染等特点[18], 契合现代绿色化学的理念; 其次, 由于水自身能电离出H+与OH-, 对一些有机溶剂具有酸碱催化作用; 最后, 由于大量有机物在水中的溶解度较小, 当反应本身冷却到室温时, 反应产物可析出或分层, 后处理过程极为简便.
由于大多数底物不能完全溶解在水中, 然而溶解度一般是反应发生的先决条件, 溶解度低将大大影响反应的进程和结果, 因此在许多“水相反应”的例子中, 通常使用水混合有机溶剂以便提高有机反应物在水中的溶解度[25].
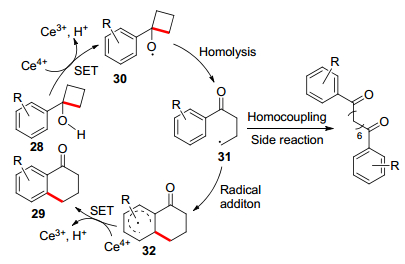
2018年, 龚行等[26]在水-乙腈混合物中, 用Ce4+介导串联氧化开环/环化反应合成了1-四氢萘酮(Eq. 6).该反应在0 ℃下开放式反应器中反应, 在30 s内快速完成, 且适用于各种环丁醇衍生物.
|
|
(6) |
该反应的反应机理如Scheme 5, 通过CAN介导的单电子转移(SET)过程形成氧自由基中间体30.然后以环张力释放为驱动力, 发生C—C均裂生成碳自由基31.随后, 进行自由基加成反应, 形成离域自由基中间体32.最后发生由Ce(NH4)2(NO3)6 (CAN)介导的另一个SET过程, 从而实现芳构化产物.
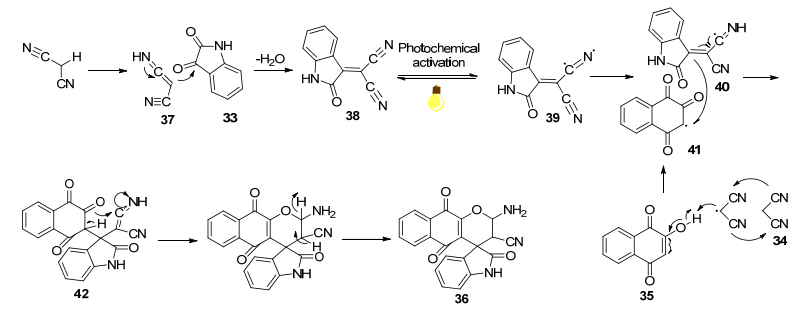
2017年, 吴银素等[27]报道了一种靛红、丙二腈和可烯醇化的C—H活化化合物(2-羟基萘1, 4-二酮、4-羟基香豆素和双甲酮)的一锅三组分反应(Eq. 7), 提供了一种可见光照射, 室温下在水-乳酸乙酯中, 合成螺噁吲哚-吡喃衍生物的高效环保的方案.
|
|
(7) |
其可能的自由基反应机理如Scheme 6.首先, 在共溶剂(EL/H2O)中用可见光照射, 通过丙二腈的互变异构形成中间体37, 37和靛红在溶液中经历Knoevenagel缩合, 得到氰基烯烃中间体38, 同时除去水.然后可见光活化38形成自由基中间体39, 39从丙二腈中夺取亚甲基氢, 产生丙二腈基自由基, 其反过来从2-羟基萘- 1, 4-二酮中提取氢得到中间体41.随后41进一步与40反应得到42, 最后进行分子内环化, 得到所需产物.该方法显示出显著的优点, 例如产率高, 反应条件温和清洁.清洁可见光作为能源, 不使用催化剂, 使用乳酸乙酯/水作为环境友好的溶剂, 且在室温下进行多组分反应, 无色谱分离, 适用于大规模合成.
2019年, 魏伟等[28]开发了一种以空气中的O2作为氧化剂, 在可见光驱动下, 使芳基二偶氮砜与硫醇氧化偶联, 构建不对称亚砜的反应(Eq. 8).该反应使用乙腈和水作混合溶剂, 可在室温下, 无需催化剂和添加剂的条件下进行, 由简单且容易获得的原料以良好的产率获得结构多样的亚砜结构.具有操作简单、选择性高、环境友好和无需催化剂等优点, 在有机合成中极具吸引力.
|
|
(8) |
除了使用混合溶剂以提高反应物在水中的溶解度以外, 引入极性官能团提高反应物的亲水性, 也能够提高化合物的水溶性[6a].人们还尝试通过添加外加配体[29]、表面活性剂[30]或采用超声波[31]等手段以解决有机底物在水相中溶解度低的问题, 从而实现纯水相中的有机反应.
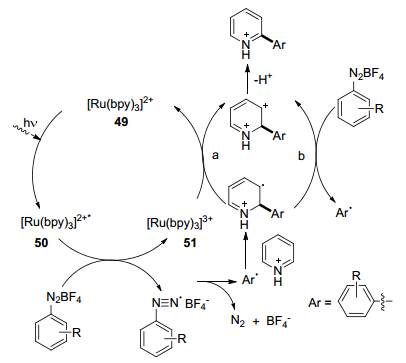
2014年, 薛东等[32]报道了一种可见光促进的, N-杂芳烃与芳基重氮盐在水中的“基团型”偶联反应, 在室温下用[Ru(bpy)3]Cl2•6H2O作为光敏剂, 家用灯泡作为光源进行反应(Eq. 9).该反应适用于各种取代的吡啶以及一系列的氧杂蒽、噻唑、吡嗪和哒嗪, 并且仅形成具有不同区域选择性的单取代产物.
|
|
(9) |
该偶联反应的反应机理见Scheme 7, 其中有三个关键步骤: (1)光照射催化剂49, 产生激发的[Ru(bpy)3]Ⅱ* 50; (2)通过单电子转移, 电子从50转移到芳基重氮盐, 释放出苯基, 同时催化剂被氧化成[Ru(bpy)3]Ⅲ (51); (3)苯基加成到吡啶盐酸盐上, 得到新的自由基中间体, 随后通过两种可能的途径将其转化为碳阳离子中间体:通过强氧化[Ru(bpy)3]Ⅲ(光催化芳基化, 途径a)或由芳基重氮盐氧化(途径b).考虑到两种氧化剂的还原电位以及避光芳基化的观察, 途径b不太可能发生自由基链转移反应.最后, 碳阳离子中间体的去质子化使芳族体系再生, 得到所需的偶联加合产物.
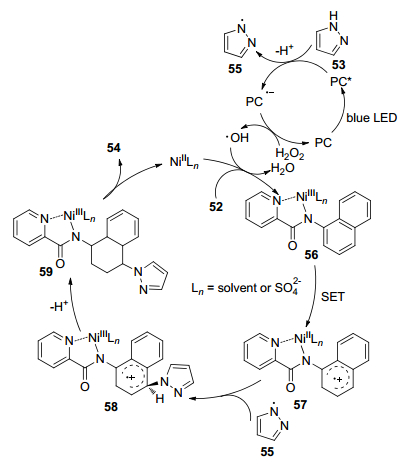
2018年, 夏成才等[33]报道了一种在温和条件下, 通过可见光介导, 镍(Ⅱ)催化, 芳基胺和吡唑之间的C—N交叉偶联, 区域选择性合成含吡唑化合物的绿色方法(Eq. 10).该反应底物适用范围广, 适用于含甲基、苯基、卤素、苯并三唑的芳基胺或其它萘酰胺的化合物的合成, 但带吸电子基团(CN和NO2)的吡唑衍生物不能进行该C—N交叉偶联反应.
|
|
(10) |
该反应单电子转移机制见Scheme 8.首先通过可见光照射, 激发光催化剂(PC)形成的激发物质PC*与吡唑(53)发生单电子转移(SET), 产生吡唑自由基55和PC•-.随后, PC•-被H2O2氧化产生羟基自由基和PC.同时, NiⅡLn与底物52结合, 氧化羟基自由基, 生成芳基-NiⅢLn络合物56和H2O.然后通过SET过程形成芳基-NiⅡLn络合物57.吡唑基55进攻芳基-NiⅡLn络合物56生成芳基-NiⅡLn络合物58, 58经去质子化形成芳基-NiⅡLn络合物59后, 通过金属离解过程获得目标产物54.该策略为含有吡唑的生物活性分子的绿色合成提供了强有力的方法.
目前已经开发了可回收的非均相催化剂代替单一金属催化剂, 在许多反应中表现出良好的催化活性.然而, 这些非均相催化剂仍然存在缺陷, 比如合成总是需要多个步骤, 结构确认难度大.因此, 在水中用简单且环保的催化剂进行反应仍然是比较好的选择.
2013年, 王心晨等[34]报道了一种绿色、无金属的光催化过程(Eq. 11).在介孔石墨氮化碳(mpg-CN)光催化剂介导下, 于水相中使用分子氧、水和可见光与4-甲氧基苄醇(MBA)发生选择性氧化反应.高稳定性的CN半导体可以通过改变不同的参数来调节光系统的催化性能, 例如和强酸/超强酸的协同催化.在该反应中, 水相中4-甲氧基苄醇可以在中性条件下, 60 ℃以89%的高选择性转化成相应的醛.水相中4-甲氧基苄醇可以在中性条件下, 60 ℃以89%的高选择性转化成相应的醛, 转化率为56%.当HCl存在时, 该体系催化苄醇转化为苄醛(49%), 具有93%的高选择性, 并且催化剂可回收, 而在强酸性溶液中不会失活.
|
|
(11) |
2018年, 蔡春课题组[8]报道了一种将光氧化还原催化与胶束催化相结合的胶束光催化体系, 用于催化原位亚硝化芳胺的芳基化反应(Eq. 12).该方法使用了Triton X-100作为外加表面活性剂, 有机染料曙红B作为光催化剂, 开发了不含过渡金属的水相光催化体系.该系统在室温下可以在没有任何有机溶剂和添加剂的情况下, 实现光催化芳基化反应.该反应具有广泛的底物适用范围, 适用于各种官能团取代的亚硝化芳胺化合物, 包括烷基、烷氧基、硝基、氰基、烷氧基和三氟甲基等, 在反应过程中, 通常带有吸电子和中性取代基的苯胺比带有给电子基团的苯胺获得更高的收率, 空间效应对这种转化没有显着影响.该方案操作简单, 催化系统中Triton X-100和曙红B都是市售且价格便宜; 水作为唯一的反应介质, 可见光作为能源, 两者都安全、清洁、丰富和廉价; 含有表面活性剂的含水反应介质可以循环数次, 具有清洁节能的优点.
|
|
(12) |
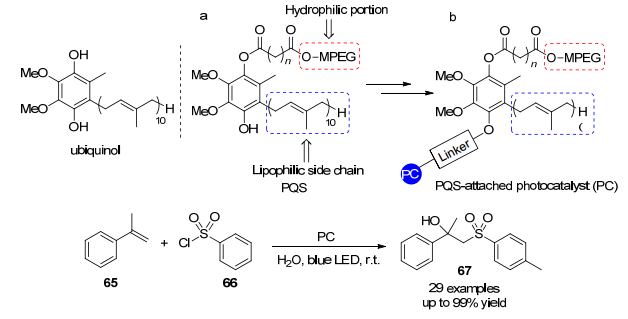
2018年, Lipshutz等[35]报道了一种新的催化体系(Scheme 9), 使用两性PQS附着的光催化剂, 在水中聚集成纳米胶束.这种共价结合的物质能够在不存在添加剂或共溶剂的情况下进行基于Ir的光催化氧化物催化.使用这种新的催化策略的反应, 实现了室温下在水中进行的光致还原催化, 无需加热或冷却, 整个含水反应混合物容易进行烧瓶内循环.在这种化学反应中, 水充当“绿色”反应介质, 可见光被用作安全和可再生的化学潜力来源.此外, 水介质和贵金属催化剂可以循环数次而无需从反应容器中除去, 这提供了一种可持续催化的策略.
地球上大部分植物和细菌通过光合作用完成对太阳能的吸收、捕获、转移和储存.在这个过程中, 来自阳光的能量被叶绿素分子捕获并转化为化学能.为了开发清洁和可持续的能源, 越来越多的科研团队研究如何模拟光合作用.
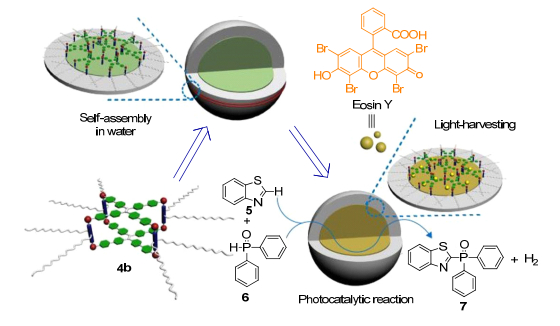
2019年, 张明明及其团队[36]以具有高荧光量子效率的金属铂配合物分子笼为供体, 以曙红Y为受体, 通过两种荧光分子的自组装在水溶液中制备了一种高效的光捕获系统, 其能量转移效率为45%.该光捕获系统被进一步作为光催化剂, 在可见光照射下, 铂分子笼作为天线传递光源来激发曙红Y产生可以催化反应的自由基, 使其表现出比单独的曙红Y更好的光催化活性.该系统组装形成的超分子胶束还可以减轻曙红Y的光漂白效应, 使其可以长时间保持较高的催化活性.该研究不仅提供了一种从金属铂配合物分子笼为供体的高效光捕获体系, 而且还利用其输出能量来催化析氢交叉偶联反应(Scheme 10), 实现了对叶绿体等光合作用载体结构和功能的系统模拟, 推动人们对光合作用的进一步研究.
2019年, 王磊和李品华等[37]道了一种使用N-烯丙基苯甲酰胺与CF3SO2Na (Langlois试剂)在可见光诱导光催化剂存在下水介质中一步合成三氟甲基化二氢异喹啉酮的策略(Eq. 13).该反应以高产率合成相应的三氟甲基化二氢异喹啉酮, 在温和条件下具有良好的官能团耐受性.
|
|
(13) |
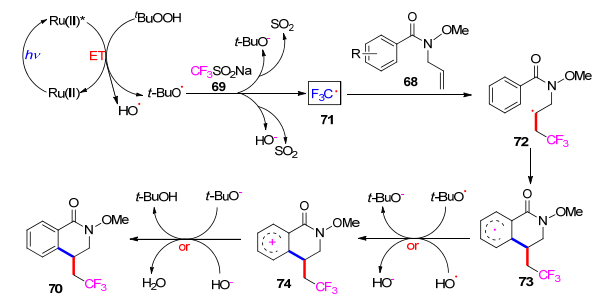
该反应的机理见Scheme 11.首先, [Ru(bpy)3]2+在可见光照射下被激活到其激发态[Ru(bpy)3]2+*.接下来, TBHP从激发的[Ru(bpy)3]2+*获得能量(能量转移过程), 通过过氧化物键的均裂解产生羟基和叔丁氧基.然后叔丁氧基或羟基与CF3SO2Na反应形成CF3自由基(71).随后, 获得的CF3自由基(71)被加入到N-烯丙基苯甲酰胺的双键中, 得到自由基中间体72, 然后进行分子内环化, 得到自由基中间体73. 73被叔丁氧基或羟基氧化, 通过SET过程产生阳离子中间体74.最后, 74失去质子产生所需产物, 同时形成水或叔丁醇.
可见光照射下的水相反应在有机合成中应用较多, 但是在生命体内的研究还非常的罕见.若将其应用到活细胞中的细胞质中, 实时调控细胞生命活动, 对研究细胞的生理功能具有重要价值.
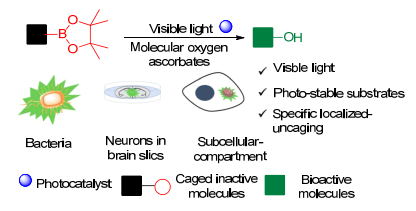
2019年, 陈以昀等[38]报道了将常用于荧光成像的有机荧光分子作为生物相容的光催化剂, 在细胞内利用氧气及抗氧化剂通过光催化氧化还原反应产生过氧化氢, 从而进行脱硼羟基化反应去笼, 释放生物活性分子.使用家用CFL灯或低能量蓝色LED灯作为光源, 该反应在中性水溶液中可以对含有酚、羟基、氨基基团的生物分子实现高效的光去笼释放(Scheme 12).该方法可以通过光释放异丙基-β-d-硫代半乳糖苷(IPTG)分子有效光调控大肠杆菌中的蛋白表达, 并且可以通过光释放巴氯芬药物有效调控γ-氨酪酸(GABAB)受体功能, 从而对小鼠脑片中神经细胞的膜电位进行高时空分辨率的光调控.最后使用线粒体定位荧光染料作为具有定位效应的光催化剂, 借助于过氧化氢在细胞内的有限自由扩散, 实现了亚细胞定位的可见光去笼.该研究展示了光催化氧化还原反应应用于细胞及生命体系研究的广阔前景.
可见光和水都是丰富的自然资源, 在生命中起着重要作用, 水是生化反应的介质, 光是生化反应的能源, 因此, 研究温和条件下水相中的光照反应具有重要的意义.水参与的可见光催化反应近年来获得了显著的进展, 但是在纯水相中进行的可见光催化反应研究相对较少, 反应类型种类还不够多, 在纯水相中反应的规律还没有充分揭示.随着近年来人们对纯水相中的光催化反应的日益关注, 探索新型的有机反应, 发展新型光催化剂成为化学家的研究热点.随着适用于水相反应的新的光催化剂(如新型的表面活性剂型光敏剂)的出现, 为发展纯水相光催化反应提供了新的策略.开发出新型基于水相反应的光敏剂, 将光敏剂设计成表面活性剂型催化剂, 将胶束效应、疏水活化和可见光催化有机结合起协同活化惰性底物, 从而在温和条件下实现传统光敏剂在水相很难实现的催化反应.这个挑战性的课题对促进绿色化学的发展具有重要的意义, 同样对于揭示生命体内的生化反应规律也具有重要的意义.因此, 纯水相中的可见光催化反应还有广阔的发展空间.
Anastas, P.; Eghbali, N. Chem. Soc. Rev. 2010, 39, 301. doi: 10.1039/B918763B
Nicewicz, D. A.; MacMillan, D. W. C. Science 2008, 322, 77. doi: 10.1126/science.1161976
Stillinger, F. H. Nature (London) 1999, 401, 850. doi: 10.1038/44698
(a) Simon, M.-O.; Li, C.-J. Chem. Soc. Rev. 2012, 41, 1415.
(b) Zhou, Z.; Duan; J.; Mu, X.; Xiao, S. Chin. J. Org. Chem. 2018, 38, 585(in Chinese).
(周曌, 段建凤, 穆小静, 肖尚友, 有机化学, 2018, 38, 585.)
Anastas, P. T. Chem. Rev. 2007, 107, 2167. doi: 10.1021/cr0783784
(a) Itami, K.; Yoshida, J.-I. Chem. Rec. 2002, 2, 213.
(b) Li, C.-J.; Meng, Y.; Yi, X.-H.; Ma, J.; Chan, T.-H. J. Org. Chem. 1997, 62, 8632.
Nicolaou, K. C.; Xu, H.; Wartmann, M. Angew. Chem., Int. Ed. 2005, 44, 756. doi: 10.1002/anie.200462211
Bu, M.-J.; Lu, G.-P.; Jiang, J.; Cai, C. Catal. Sci. Technol. 2018, 8, 3728. doi: 10.1039/C8CY01221K
黄依铃, 魏文廷, 化学进展, 2018, 30, 1819. http://www.cqvip.com/QK/98085X/201812/6100174196.htmlHuang, Y.; Wei, W. Prog. Chem. 2018, 30, 1819(in Chinese). http://www.cqvip.com/QK/98085X/201812/6100174196.html
Fujishima, A.; Honda, K. Nature (London) 1972, 238, 37. doi: 10.1038/238037a0
(a) Fan, X.-Z.; Rong, J.-W.; Wu, H.-L.; Zhou, Q.; Deng, H.-P.; Tan, J. D.; Xue, C.-W.; Wu, L.-Z.; Tao, H.-R.; Wu, J. Angew. Chem., Int. Ed. 2018, 57, 8514.
(b) Jiang, X.; Zhang, M.-M.; Xiong, W.; Lu, L.-Q.; Xiao, W.-J. Angew. Chem., Int. Ed. 2019, 58, 2402.
(c) Ye, S.; Li, X.; Xie, W.; Wu, J. Eur. J. Org. Chem. 2019 (DOI:10.1002/ejoc.201900396).
(d) Cai, B.-G.; Xuan, J.; Xiao, W.-J. Sci. Bull. 2019, 64, 337.
(e) Chen, Y.; Lu, L.-Q.; Yu, D.-G.; Zhu, C.-J.; Xiao, W.-J. Sci. China, Chem. 2018, 62, 24.
(f) Chen, J.-R.; Yan, D.-M.; Wei, Q.; Xiao, W.-J. ChemPhotoChem 2017, 1, 148.
(g) Ren, L.; Ran, M.; He, J.; Qian, Y.; Yao, Q. Chin. J. Org. Chem. 2019, 39, 1583(in Chinese).
(任林静, 冉茂刚, 何佳芯, 钱燕, 姚秋丽, 有机化学, 2019, 39, 1583.)
(h) Shang, T.-Y.; Lu, L.-H.; Cao, Z.; Liu, Y.; He, W.-M.; Yu, B. Chem. Commun. 2019, 55, 5408.
(i) Wang, L.; Bao, P.; Liu, W.; Liu, S.; Hu, C.; Yue, H.; Yang, D.; Wei, W. Chin. J. Org. Chem. 2018, 38, 3189(in Chinese).
(王雷雷, 鲍鹏丽, 刘维维, 刘思彤, 胡昌松, 岳会兰, 杨道山, 魏伟, 有机化学, 2018, 38, 3189.)
Zhang, W.-M.; Dai, J.-J.; Xu, J.; Xu, H.-J. J. Org. Chem. 2017, 82, 2059. doi: 10.1021/acs.joc.6b02891
(a) Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044.
(b) Hopkinson, M. N.; Sahoo, B.; Li, J.-L.; Glorius, F. Chem.-Eur. J. 2014, 20, 3874.
(c) Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322.
(d) Qiu, G.; Li, Y.; Wu, J. Org. Chem. Front. 2016, 3, 1011.
(e) Reckenthaeler, M.; Griesbeck, A. G. Adv. Synth. Catal. 2013, 355, 2727.
(f) Xuan, J.; Xiao, W.-J. Angew. Chem., Int. Ed. 2012, 51, 6828.
Li, L.; Huang, M.; Liu, C.; Xiao, J. C.; Chen, Q. Y.; Guo, Y.; Zhao, Z. G. Org. Lett. 2015, 17, 4714. doi: 10.1021/acs.orglett.5b02177
Zhang, M.; Ruzi, R.; Li, N.; Xie, J.; Zhu, C. Org. Chem. Front. 2018, 5, 749. doi: 10.1039/C7QO00795G
Hou, H.; Zhu, S.; Pan, F.; Rueping, M. Org. Lett. 2014, 16, 2872. doi: 10.1021/ol500893g
(a) Allavena, M. Mol. Eng. 1995, 5, 403. (b) Head-Gordon, T.; Hura, G. Chem. Rev. 2002, 102, 2651.
Jayaraman, M.; Batista, M. T.; Manhas, M. S.; Bose, A. K. Heterocycles 1998, 48, 1100.
Zou, Y.-Q.; Guo, W.; Liu, F.-L.; Lu, L.-Q.; Chen, J.-R..; Xiao, W.-J. Green Chem. 2014, 16, 3787. doi: 10.1039/C4GC00647J
Jana, S.; Verma, A.; Kadu, R.; Kumar, S. Chem. Sci. 2017, 8, 6633. doi: 10.1039/C7SC02556D
Wei, W.; Bao, P.; Yue, H.; Liu, S.; Wang, L.; Li, Y.; Yang, D. Org. Lett. 2018, 20, 5291. doi: 10.1021/acs.orglett.8b02231
Zhang, M.; Yuan, X. A.; Zhu, C.; Xie, J. Angew. Chem., Int. Ed. 2019, 58, 312. doi: 10.1002/anie.201811522
(a) Converso, A.; Burow, K.; Marzinzik, A.; Sharpless, K. B.; Finn, M. G. J. Org. Chem. 2001, 66, 4386.
(b) Converso, A.; Burow, K.; Marzinzik, A.; Sharpless, B.; Finn, M. G. J. Org. Chem. 2004, 69, 7336.
(c) Yudin, A. Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, p. 492.
(a) Breslow, R.; Maitra, U.; Rideout, D. Tetrahedron Lett. 1983, 24, 1901.
(b) Grieco, P. A.; Garner, P.; He, Z.-M. Tetrahedron Lett. 1983, 24, 1897.
(c) Grieco, P. A.; Yoshida, K.; Garner, P. J. Org. Chem. 1983, 48, 3137.
(d) Rideout, D. C.; Breslow, R. J. Am. Chem. Soc. 1980, 102, 7816.
Lindstróm, U. M. Chem. Rev. 2002, 102, 2751. doi: 10.1021/cr010122p
Fang, J.; Li, L.; Yang, C.; Chen, J.; Deng, G.-J.; Gong, H. Org. Lett. 2018, 20, 7308. doi: 10.1021/acs.orglett.8b03246
Zhang, M.; Fu, Q.-Y.; Gao, G.; He, H.-Y.; Zhang, Y.; Wu, Y.-S.; Zhang, Z.-H. ACS Sustainable Chem. Eng. 2017, 5, 6175. doi: 10.1021/acssuschemeng.7b01102
Liu, Q.; Wang, L.; Yue, H.; Li, J.-S.; Luo, Z.; Wei, W. Green Chem. 2019, 21, 1609. doi: 10.1039/C9GC00222G
(a) Cobo, I.; Matheu, M. I.; Castillór, S.; Boutureira, O.; Davis, B. G. Org. Lett. 2012, 14, 1728.
(b) Ganesamoorthy, S.; Shanmugasundaram, K.; Karvembu, R. J. Mol. Catal. A: Chem. 2013, 371, 118.
(c) Wallow, T. I.; Novak, B. M. J. Am. Chem. Soc. 1991, 113, 7411.
(a) Lysen, M.; Koehler, K. Synthesis 2006, 692.
(b) Soomro, S. S.; Roehlich, C.; Koehler, K. Adv. Synth. Catal. 2011, 353, 767.
(c) Xie, L.-Y.; Jiang, L.-L.; Tan, J.-X.; Wang, Y.; Xu, X.-Q.; Zhang, B.; Cao, Z.; He, W.-M. ACS Sustainable Chem. Eng. 2019.
(a) Solodenko, W.; Schoen, U.; Messinger, J.; Glinschert, A.; Kirschning, A. Synlett 2004, 1699.
(b) Zhang, J.; Yang, F.; Ren, G.; Mak, T. C. W.; Song, M.; Wu, Y. Ultrason. Sonochem. 2007, 15, 115.
(c) Wu, C.; Lu, L.-H.; Peng, A.-Z.; Jia, G.-K.; Peng, C.; Cao, Z.; Tang, Z.; He, W.-M.; Xu, X. Green Chem. 2018, 20, 3683.
Xue, D.; Jia, Z. H.; Zhao, C. J.; Zhang, Y. Y.; Wang, C.; Xiao, J. Chem.-Eur. J. 2014, 20, 2960. doi: 10.1002/chem.201304120
You, G.; Wang, K.; Wang, X.; Wang, G.; Sun, J.; Duan, G.; Xia, C. Org. Lett. 2018, 20, 4005. doi: 10.1021/acs.orglett.8b01395
Long, B.; Ding, Z.; Wang, X. ChemSusChem 2013, 6, 2074. doi: 10.1002/cssc.201300360
Bu, M.-J.; Cai, C.; Gallou, F.; Lipshutz, B. H. Green Chem. 2018, 20, 1233. doi: 10.1039/C7GC03866F
Zhang, Z.; Zhao, Z.; Hou, Y.; Wang, H.; Li, X.; He, G.; Zhang, M. Angew. Chem., Int. Ed. 2019, 58, 8862. doi: 10.1002/anie.201904407
Zou, L.; Li, P.; Wang, B.; Wang, L. Green Chem. 2019, 21, 3362. doi: 10.1039/C9GC00938H
Wang, H.; Li, W. G.; Zeng, K.; Wu, Y. J.; Zhang, Y.; Xu, T. L.; Chen, Y. Angew. Chem., Int. Ed. 2019, 58, 561. doi: 10.1002/anie.201811261

图式 1 苯乙烯的2, 2, 2-三氟乙基化反应机理
Scheme 1 Mechanism of 2, 2, 2-trifluoroethylation of styrene

图式 2 N-取代的羟胺与烯烃的[3+2]环加成反应
Scheme 2 [3+2] cycloaddition reaction of N-substituted hydroxylamine with olefins

图式 3 可见光诱导的3-溴甲基吲哚光催化甲酰氧基化反应的机理
Scheme 3 Mechanism of photocatalytic oxidation of 3-bromomethylhydrazine photocatalytic reaction induced by visible light

图式 4 在水中可见光诱导氧化剂和无金属脱氢级联三氟甲基化和1, 6-烯炔的氧化的机理
Scheme 4 Mechanism of visible light-induced oxidant and metal-free dehydrogenation cascade trifluoromethylation and oxidation of 1, 6-enyne in water

图式 5 水-乙腈中二次串联氧化开环/环化反应合成1-四氢萘酮的机理
Scheme 5 Mechanism of synthesis of 1-tetralone by double-stage oxidative ring-opening/cyclization reaction in water- acetonitrile

图式 6 在乳酸乙酯水溶液中无催化剂可见光促进一锅法合成螺内吲哚-吡喃衍生物
Scheme 6 Catalyst-free, visible-light promoted one-pot synthesis of spirooxindole-pyran derivatives in aqueous ethyl lactate

图式 7 在水中光催化N-杂芳烃与芳基重氮盐的直接芳基化反应的机理
Scheme 7 Mechanism of photocatalytic direct arylation of N-heteroarene with aryl diazonium salt in water

图式 8 可见光介导镍(Ⅱ)催化的水中C—N交叉偶联反应的机理
Scheme 8 Mechanism of visible light mediated nickel(Ⅱ) catalyzed C—N cross-coupling reaction in water

图式 9 两亲的PQS连接的铱光敏剂催化的光致还原反应
Scheme 9 Photoreduction reaction catalyzed by amphiphilic PQS-linked iridium

图式 10 基于水溶性荧光金属铂分子笼的光捕获系统用于光催化交叉偶联析氢反应
Scheme 10 Queous platinum(Ⅱ) cage-based light-harvesting system for photocatalytic cross-coupling hydrogen evolution Reaction

图式 11 室温下可见光催化N-烯丙基苯甲酰胺与CF3SO2Na在水相中合成三氟甲基化二氢异喹啉酮
Scheme 11 Visible-light-induced radical cyclization of N-allylbenzamide with CF3SO2Na to trifluoromethylated dihydroisoquinolinones in water at room temperature

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:












 下载:
下载:
