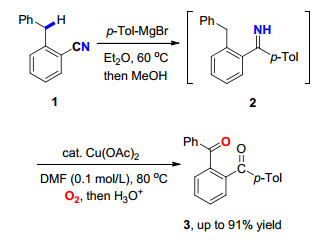
图式 1.
亚胺导向苄基参与的氧插入反应
Scheme 1.
Imine-directed benzyl-participated oxygenation

含氧化合物普遍存在于天然产物和生物活性分子中, 也是有机合成中重要的合成原料.因此, 含氧化合物的合成和制备已经成为当前有机合成研究的热点, 同时受到了有机合成化学家们的广泛关注.近十几年来, 很多新型合成含氧化合物的催化方法相继被报道[1~3].其中, 过渡金属催化氧气氧插入反应是一类新型而又高效构建C—C键和C—X (X=O, N等)的方法[4].该类反应在过渡金属催化下, 可以实现氧原子的高选择性插入, 从而高效地合成含氧杂原子的化合物.同时, 由于氧气的廉价易得及环境友好的特性, 使得该类反应具有操作简便、原子经济、绿色环保、无需隔绝空气和水等特点.该类反应的高效、绿色构建含氧化合物的优点使得过渡金属催化氧插入反应得以迅速发展, 尤其是铜催化和钯催化的氧插入反应研究得到了极大的发展.该类反应在合成含氧化合物的应用也非常广泛, 研究对象主要包括基本的酮类、醛类、芳烃类和炔烃类等, 涉及到的反应类型包括羟基化反应、羰基化反应、酯化反应和成环反应等.对该类反应机理研究显示反应主要通过活化C—H键和C—C键断裂两种基本方式实现氧气的插入过程.本文在前人研究报告的基础上[5], 同时结合本课题组的工作, 不仅综述了过渡金属催化氧插入反应在醛酮和烯炔烃的研究新进展, 同时还综述了该反应在去芳构化和构建吲哚类衍生物等领域上的新应用.
过渡金属催化氧化C—H键的氧插入反应主要包括C(sp3)—H键、C(sp2)—H键和C(sp)—H键参与的氧插入反应, 涉及到的研究对象包括烷烃类、烯烃类、芳烃类和炔烃类等.在过渡金属的催化氧化下, 氧气容易与金属结合形成金属-氧自由基复合物, 从而进行自由基历程的C—H键参与的氧插入反应, 实现C—H键向C—O键的转化.
过渡金属催化氧化C(sp3)—H键的氧插入反应可以将甲基、亚甲基或次甲基转换成含羰基类或含氧杂环类等化合物. 2011年, Chiba课题组[6]通过以苄基苯甲腈(1)为原料, 以分子内的N—H亚胺基团导向经1, 5-氢迁移的过程, 实现了铜催化苄位亚甲基C(sp3)—H键参与的氧插入反应, 最后通过酸化水解可以高效地得到了邻苯二甲酰基取代的芳香类化合物3 (Scheme 1).
2012年, Chiba课题组[7]通过溴化亚铜-双吡啶体系催化, 采用同样的亚胺基团导向和1, 5-氢迁移等策略进一步实现了次甲基参与的铜催化氧插入反应, 一步构建了含二氢噁唑环结构的化合物5 (Eq. 1).反应机理研究显示反应先生成叔碳自由基中间体, 随后进行铜氧条件下的氧插入反应过程, 最后环化水解得目标产物.
|
|
(1) |
2011年, 焦宁课题组[8]通过溴化亚铜-吡啶催化芳基乙醛与苯胺之间的氧插入反应, 实现了对α-芳基酮酰胺类化合物8的一步构建(Eq. 2).相比于传统合成α-芳基酮酰胺类化合物方法, 该反应过程涉及到两个C(sp3)—H键、一个C(sp2)—H键、一个N—H键和O—O键的断裂, 同时表现出很好的底物适用性范围.反应机理研究显示底物芳基乙醛与苯胺首先进行脱水缩合反应生成含亚胺结构的中间体, 随后进行过渡金属催化的氧插入反应.
|
|
(2) |
2012年, 焦宁课题组[9]进一步通过溴化亚铜-吡啶催化芳基乙醛和苯胺之间的氧插入反应, 在不加入分子筛和降低温度的条件下, 实现了对2-乙酰基脒类化合物10的合成.反应机理研究显示, 反应过程经过氨基自由基正离子加成反应, 生成含脒结构的自由基中间体; 随后经过活化C(sp3)—H键的氧插入反应.整个反应过程脱去了6个氢(Eq. 3).
|
|
(3) |
同年, 焦宁课题组[10]以具有α-H的伯胺和芳基乙醛为原料, 在氧气的环境中, 进一步通过溴化铜-吡啶催化体系催化的氧插入反应, 一步合成了2, 4-双取代的噁唑类化合物12 (Eq. 4).该反应涉及到六个氢原子的消除.相比传统合成噁唑类的方法, 该反应的底物廉价易得, 更适合于工业应用.
|
|
(4) |
2015年, 焦宁课题组[11]通过溴化亚铜-哌啶体系催化芳基乙酮与伯醇之间氧插入反应, 实现了一价铜催化甲基参与的氧插入反应.该反应过程通过哌啶活化C(α)—H键, 氧化氧气插入生成α-酮醛中间体.该中间体随后与伯醇进行亲核加成反应, 最终氧化脱氢得α-酮酸酯类化合物15 (Eq. 5).
|
|
(5) |
2016年, Kumar课题组[12]通过二价铜-吡啶体系催化苄位氢的氧化氧插入反应, 首次实现了从单底物的芳基亚胺酸酯向芳基α-酮酸酯的转化.该反应方法具有操作简单及底物范围广等特点.同时, 研究该反应机理时显示产物中其中一个氧原子来自反应中的水(Eq. 6).
|
|
(6) |
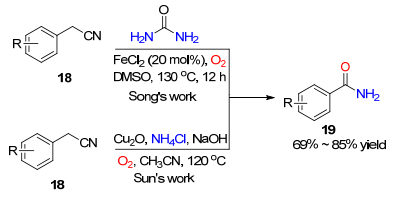
过渡金属催化活化C(sp3)—H键参与的氧插入反应还可以被用于合成芳酰伯胺类化合物. 2017年, 宋秋玲课题组[13]发展了一条以苯乙腈和尿素为底物, 二氯化亚铁催化活化苄位C(sp3)—H键的氧插入反应, 实现了对芳基甲酰胺类化合物的合成, 同时伴随着良好的产率(Scheme 2).反应机理研究显示尿素为氨基的来源.同年, 孙绍发课题组[14]通过氧化亚铜催化, 在强碱条件下实现了苯乙腈与氯化铵之间的氧插入反应, 合成了芳酰伯胺类化合物(Scheme 2). 18O同位素标记实验显示产物中的氧全部来自于氧气.
黄德光课题组[15]利用三齿配体的双吡啶类化合物与三氟甲磺酸铜配位, 得到金属复合物.该金属复合物在强碱叔丁醇钾的作用下, 可以催化室温下的2-吡啶甲基芳胺为底物的氧插入反应, 一步构建了N-苯基-2-吡啶甲酰胺类化合物21 (Eq. 7).该反应条件温和是配体辅助催化的一大特点, 然而, 该方法的产物专一性较差, 同时会伴随着吡啶酸和亚胺等副产物的生成.
|
|
(7) |
过渡金属催化氧化C(sp2)—H键的氧插入反应主要包括端烯、内部烯烃和芳环上C—H键参与的氧插入反应.该类反应通常先经过自由基的加成反应, 生成新的自由基中间体, 随后进行C(sp3)—H的氧气插入反应. 2013年, 焦宁课题组[16]通过三氟甲磺酸铜和硝酸铁共同催化, 实现了芳基肼与取代的苯乙烯之间的氧插入反应, 一步合成了1, 2-二芳基取代的乙二酮类化合物24.进一步的反应底物拓展结果显示, 该反应适用范围较窄, 仅限于端烯类化合物(Eq. 8).
|
|
(8) |
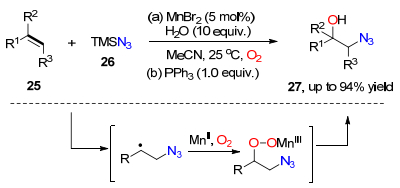
2015年, 焦宁课题组[17]通过过渡金属锰催化烯烃与三甲基叠氮硅烷之间的氧插入反应, 实现了对β-叠氮醇类化合物27的高效合成.该反应不仅可以在室温条件下进行, 而且具有很好的底物适用性, 端烯和内烯都可以参与反应.密度泛函理论(DFT)结果显示, β-叠氮取代的自由基中间体和三价锰中间体的生成是该反应成功转化的关键步骤(Scheme 3).
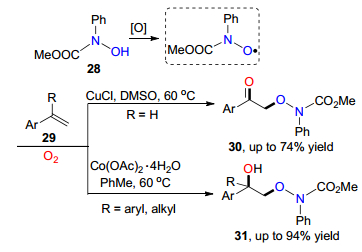
同年, 雷爱文课题组[18]通过氯化亚铜和醋酸钴分别催化异羟肟酸酯与端烯和1, 1-二取代端烯之间的氧插入反应, 实现了对α-氧基酮30和α-氧三级醇类化合物31的合成.该方法具有较高的化学选择性且不需要任何配体或碱等辅助添加剂等特点.反应的机理研究显示反应先生成异羟肟自由基, 随后进行自由基的加成反应, 最后实现氧自由基插入反应(Scheme 4).
砜类化合物具有广泛的生物或药物活性, 一直是有机合成化学家的研究内容之一[19]. 2013年, 王桦课题组[20]通过醋酸酮催化, 氧气活化内烯烃与磺酰肼之间的氧插入反应, 实现了对β-羰基砜类化合物33的合成.机理研究显示反应首先经过磺酰肼的氧化, 生成磺酰自由基中间体. 2014年, 王桦课题组[21]进一步通过四水氯化亚铁催化的亚磺酸与内烯烃之间的氧插入反应, 实现了对β-羰基砜类化合物的合成, 产率良好(Scheme 5).
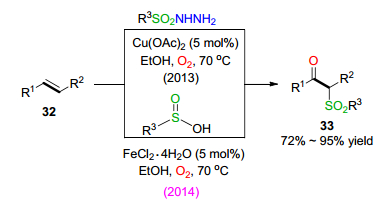
2018年, 魏文龙课题组[22]通过五水硫酸铜催化氧气氧化, 实现了芳基端烯、磺酸纳盐、芳基重氮盐及氧气等四组分之间的氧插入反应, 一锅法合成了α-芳肼基-β-酮砜类化合物36.同时, 该反应方法表现出很好的底物适用性.反应的机理研究显示反应过程首先经过砜自由基的加成, 随后进行氧气插入反应.该反应同时涉及到对C—O键、C—S键和C—N键一锅法的构建(Eq. 9).
|
|
(9) |
利用分子内活性基团与活性基团之间的反应是合成环状化合物常用的方法.而利用分子内环化反应也可以实现分子参与的环化-插氧串联反应. 2011年, Chiba课题组[23]通过二价的醋酸铜催化实现了分子内的环化氧插入反应.该反应以同时含端烯基团与缺电子烯胺基团的仲胺类化合物为底物, 在铜氧催化体系下, 一步构建了二取代3-吡咯甲醛类化合物38, 伴随着良好的产率和底物适用性(Eq. 10).
|
|
(10) |
过渡金属催化芳基参与的氧插入反应主要通过活化芳环上的C(sp2)—H键实现氧的插入.该部分涉及到的对象为芳烃类和杂环芳烃类, 涉及到的反应包括C(sp2)—H键的羟基化反应和芳基参与的去芳构化反应[24]. 2009年, 余金权教授课题组[25]通过醋酸钯催化活化苯环上的C(sp2)—H键, 实现了苯甲酸类化合物的氧插入反应, 合成了邻羟基苯甲酸类化合物(Eq. 11).反应机理研究显示产物中的氧来自于氧气, 同时该反应表现出很好的底物适用性.
|
|
(11) |
无独有偶, 2013年, 焦宁课题组[26]通过二价的氯化钯催化、N-羟基苯邻二甲酰胺辅助催化氧化C(sp2)—H键, 实现了对2-芳基吡啶环的C—H键的羟基化反应, 并伴随着良好的产率.反应机理研究显示, 底物吡啶环与钯配位, 生成双配位的金属络合物中间体, 在氮杂原子的孤对电子诱导效应下活化苯环上邻位C—H键(Eq. 12).
|
|
(12) |
2010年, Chiba等[27]以α-叠氮-N-苯基酰胺为底物, 发展了一条由二价铜催化的分子内环化-氧插入反应, 合成了一系列螺环醌类化合物, 实现了对苯环的去芳构化.推测的反应机理显示, 反应涉及到苯胺环上对位C(sp2)—H键的活化和氧插入过程(Eq. 13).
|
|
(13) |
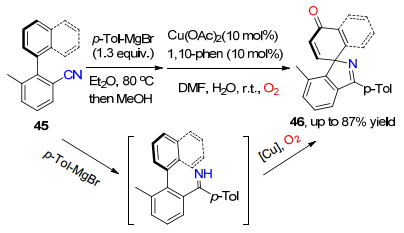
2012年, Chiba课题组[28]又发展了一种基于亚胺中间体活化苯环和萘环上C(sp2)—H键的氧插入反应.该方法以3-甲基-2芳基苯甲腈为底物, 先与格氏试剂对甲苯基溴化镁进行亲核加成反应生成亚胺中间体, 随后铜催化苯环和萘环的1, 4-氨基氧插入反应过程, 一锅法实现了对苯环和萘环的氧插入和氨基插入去芳构化的螺环化反应(Scheme 6).
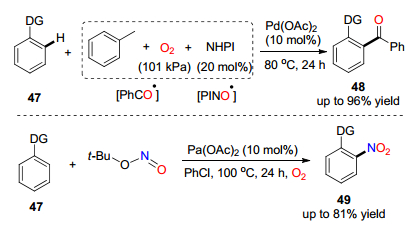
2015年, 焦宁课题组[29]发展了一条钯催化氧气活化苯环上C—H硝基化和酰基化反应(Scheme 7).该反应以叔丁基亚硝酸盐(TBN)和甲苯作为自由基前体, 通过氧气作为氧化剂用于激发自由基前体, 同时作为氧源进行氧气插入.对于苯环的酰基化反应, 加入自由基引发剂N-羟基苯邻二甲酰胺辅助催化促使苯甲酰基自由基的生成.对于苯环硝基化反应, 控制实验显示硝基中的氧有可能来自于氧气, 也有可能来自于底物本身.
2015年, 焦宁课题组[30]以芳基乙醛和芳基伯胺为底物, 在氧气环境中, 以三氟乙酸铜为催化剂, 实现了两者之间的氧插入反应, 合成了N-芳基取代吡啶酮类化合物(Eq. 14).反应机理推测可能先生成吡啶环, 而后经过活化脱苄基得吡啶正离子中间体, 最后铜催化氧插入得到目标产物51. 18O同位素标记试验显示产物中的氧同时来源于水或氧气.
|
|
(14) |
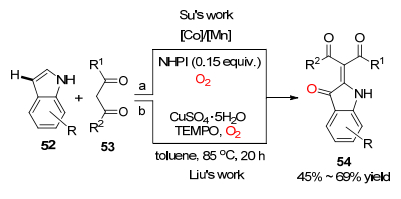
值得注意的是, 除了苯环上的C(sp2)—H参与的氧插入反应, 利用吲哚参与的氧插入反应合成吲哚类化合物也是有机合成研究的热点, 近几年对吲哚参与的氧插入反应也得到了迅速的发展.由于吲哚结构的特点, 其3-位C(sp2)—H键化学性质活泼, 易发生C—H功能化反应. 2011年, 苏伟平课题组[31]报道了通过醋酸钴和醋酸锰共同催化, N-羟基苯邻二甲酰胺辅助催化, 氧气氧化的吲哚与β-酮酸酯的氧化交叉偶联反应, 得到了3-位酮基化-2-烯化的吲哚酮类化合物54.与之类似的工作由刘良先课题组[32]报道, 以2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO)为氧化剂, 氧气条件下, 硫酸铜为催化剂, 实现了吲哚与乙酰乙酸乙酯之间的氧气插入反应, 完成了对吲哚酮类化合物的合成(Scheme 8).
2013年, 王彦广课题组[33]报道了在氧气环境中, 碘化亚铜催化吲哚与靛红之间的氧插入环化反应, 实现了对色胺酮类化合物的合成.反应的机理研究显示吲哚在铜氧体系催化条件下, 可以生成中间体靛红, 随后经过脱羧基偶联, 分子内的亲核加成反应, 最后氧化芳构化得到最终色胺酮类产物56 (Scheme 9).
2016年, 邓国军课题组[34]发展了一条由铜催化氧气活化的吲哚3-位C—H参与的氧插入环化反应.该反应在氯化铜为催化剂的条件下, 通过氧气氧化2-取代吲哚与苯乙酮肟之间的氧气插入环化过程, 一锅法实现了对吲哚的多功能化反应(Eq. 15).反应的机理研究显示, 该反应通过一个自由基的反应机理, 苯乙酮肟作为体内氧化引发剂激发氧气氧化插入反应.
|
|
(15) |
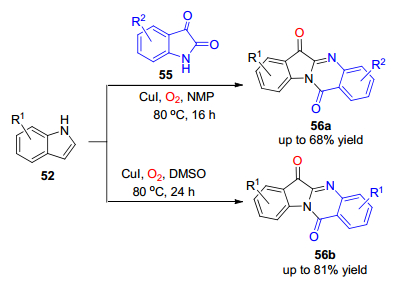
2018年, 范学森课题组[35]发展了溴化亚铜催化邻苯甲酰胺-2-取代吲哚的氧插入反应.该反应在空气环境中, 以N, N-二甲基甲酰胺(DMF)为溶剂, 可以实现对吲哚-3-酮类骨架的构建(Scheme 10).此外, 该反应在1, 4-二氧六环为溶剂时, 生成的是吲哚并喹啉结构类化合物.
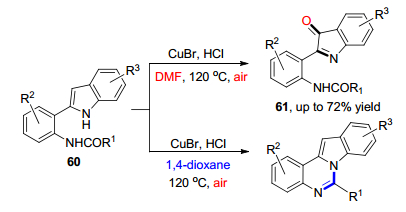
2019年, Borban等[36]发展了一种由氯化亚铜-吡啶体系催化的3H-吲哚与叔丁氧基羟胺之间的氧插入反应, 实现了对2-叔丁氧酰胺基取代的吲哚-3-酮类化合物64的合成(Eq. 16).然而, 该反应具有一定的底物局限性.在该条件下, 当3-取代吲哚参与反应时, 并不能生成吲哚酮类化合物, 而是进一步实现对2-位甲基的功能化的产物.
|
|
(16) |
2018年, 本课题组[37, 38]发展了一条铜催化3-取代吲哚的碳碳双键的氧插入环化反应(Eq. 17).在碱式碳酸铜和醋酸铜的催化作用下, 通过氧化吲哚C=C双键生成双氧桥环结构的中间体, 随后进一步氧化消去得到氧杂三元环结构的二氢吲哚中间体.该中间体通过与偶氮甲碱亚胺类的环化反应得到含氧杂环的二氢吲哚类衍生物67, 并且伴随着非常高的非立体选择性, 同时含有多个手性中心.对该反应的机理研究显示反应过程中的空间位阻和π电子作用是该反应具有高非立体选择性的主要原因.
|
|
(17) |
过渡金属催化C(sp)—H键参与的氧插入反应主要研究对象为端炔烃类.与烯烃类参与的氧插入反应类似, 炔烃首先进行金属催化的加成反应, 而后进行氧化活化C(sp2)—H或C(sp3)—H键的氧气插入反应过程. 2010年, 焦宁课题组[39]通过溴化亚铜-吡啶体系催化, 加入自由基捕捉剂四甲基哌啶氧化物(TEMPO)辅助催化, 实现了芳基伯胺与炔烃之间的氧插入反应, 合成了α-酮酰胺类化合物69.可能的反应机理显示该反应过程先经过脱质子氨基化反应过程, 得到烯胺类中间体, 然后经C(sp2)—H键的氧插入反应得到关键过氧四元环氮自由基中间体. 18O标记实验显示产物中的氧全部来自于氧气, 从而显示反应过程需要断裂O—O键(Eq. 18).
|
|
(18) |
2011年, 王彦广课题组[40]报道了以氧气为氧源, 溴化亚铜催化炔烃、磺酰叠氮和3H-吲哚三组分之间的氧插入反应, 合成了3-位含羰基的吲哚类化合物72.反应机理的研究显示, 反应过程首先经叠氮与炔烃在铜催化下反应原位形成的烯亚胺过渡态, 随后与吲哚发生亲电加成反应得到关键的烯胺中间体.最后, 经过铜氧催化氧化C(sp2)—H键的氧插入反应得到最终产物(Eq. 19).
|
|
(19) |
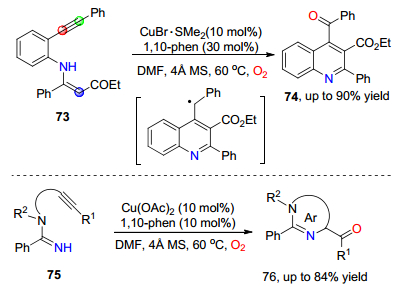
内部炔烃也可以作为底物实现过渡金属催化的氧插入反应, 反应过程不仅仅只涉及到对C(sp3)—H键的氧化.继分子内烯烃参与的氧插入反应之后, Chiba课题组[41]在2012年进一步通过溴化亚铜和1, 10-邻二氮杂菲联合催化, 实现了内炔与烯胺基团之间的分子内环化-氧插入反应.该反应首先通过活化烯胺的C(sp2)—H键与炔基进行分子内环化反应和电子重排, 生成苄自由基中间体; 再经过C(sp3)—H键参与铜催化氧化氧气插入过程, 高效合成了芳酰基取代的喹啉类化合物74 (Scheme 11).在该反应条件中, 通过二价的醋酸铜催化, 进一步实现了内炔与亚胺之间的氧插入反应, 伴随着对含羰基取代的咪唑和嘧啶类衍生物76的合成(Scheme 11).
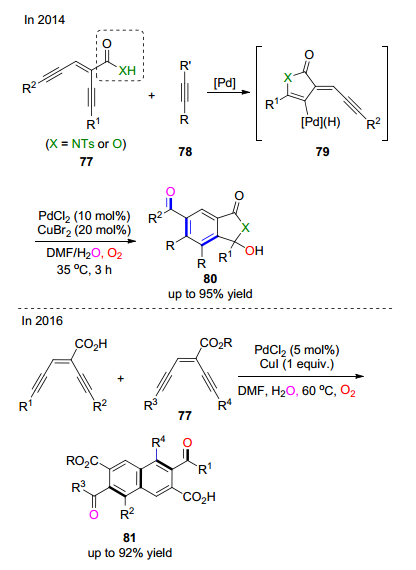
2014年, 马成课题组[42]通过氯化钯和溴化铜联合催化以烯二炔烃-酰亚胺类或羧酸类化合物77和内部炔烃为底物的氧插入反应, 经过串联的偶联-铜催化氧插入反应过程, 实现了对异吲哚啉酮类结构或邻酰基苯甲酸类化合物80的构建(Scheme 12).整个反应过程涉及到一个C(sp2)—H键的断裂.邻酰基苯甲酸类结构可以进一步经过脱羧反应得到1, 4-二酰基苯类化合物.该反应的机理研究显示反应通过[4+2]组合方式实现产物的芳构化. 2016年, 马成课题组[43]在前期工作的基础上进一步通过过渡金属盐氯化钯和碘化亚铜联合催化, 在氧气环境中, 实现了烯二炔类底物自身的二聚-氧插入串联反应, 高效地合成了二酰基取代的萘甲酸类化合物81 (Scheme 12).同位素标记实验显示产物中新加入的氧原子分别来自于水和氧气.值得一提的是, 该类反应具有非常高的原子经济性, 没有任何的副产物产生.
2017年, 马成等[44]通过二价的钯/铜联合催化体系, 以类似的交叉环化-氧插入串联反应方式, 进一步实现了烯二炔与炔基苯胺类化合物之间的氧插入反应, 高效地合成了酰基取代的菲啶类衍生物83.控制实验显示反应为自由基反应历程, 且产物新生成羰基中的氧全部来自于氧气.反应机理研究显示, 反应涉及到叔碳自由基中间体的生成及C(sp3)—H的氧插入反应过程(Eq. 20).
|
|
(20) |
2018年, 焦宁课题组[45]进一步以N-碘代琥珀酰亚胺(NIS)作为辅助添加剂, 通过溴化铜-吡啶催化体系催化, 实现了芳基伯胺与吸电子取代的炔烃之间的氧化插入反应, 一步合成了2, 4, 5-三取代的噁唑类化合物86 (Eq. 21).反应过程中会生成烯胺中间体, 同时涉及到一个C(sp2)—H键的断裂.对照试验结果显示, N-碘代琥珀酰亚胺可以促进不饱和碳氢键断裂, 表现出辅助消去氢自由基的作用.
|
|
(21) |
通过过渡金属催化氧化C—H键的氧插入反应涉及到氢原子的消除, 所以通常会伴随着水分子或氨气等副产物的产生. C—H键参与的氧插入反应应用范围广泛, 可以构建不同类型的醇酚类、酮酯类、酰胺类、含氧杂环类以及吲哚酮类化合物.除了活化C—H键之外, 过渡金属还可以通过催化断裂C—C键的方式进行氧气插入反应.
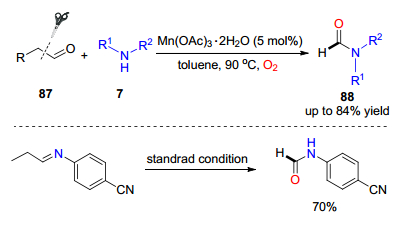
过渡金属催化断裂C—C键的氧插入反应过程中往往会生产过氧环中间体, 涉及到碳碳键和氧氧键的同时断裂. 2012年, 焦宁课题组[46]进一步以二水醋酸锰催化, 通过断裂C—C键和O—O键的反应过程, 实现了醛与仲胺之间的氧插入反应, 合成了对甲酰胺类化合物88.该反应条件也可以适用于烷基取代的醛, 并以更高的产率得到目标产物.在该反应条件下, 直接以芳基亚胺为底物时, 反应也可以进行, 说明该反应过程首先生成亚胺中间体(Scheme 13).
2013年, 焦宁教题组[47]通过溴化亚铜-吡啶体系催化1, 3-二酮类化合物和一级醇之间的氧插入反应, 在氧气环境中, 实现了芳基α-酮酸酯类90的合成.反应机理研究显示反应过程涉及到C—C键的断裂, 同时伴随着副产物芳基甲酸酯91的产生(Eq. 22).
|
|
(22) |
2014年, 焦宁课题组[48]通过溴化亚铜-吡啶体系进一步催化芳基烷基酮类与一级醇之间的氧插入反应, 合成了芳基酸酯类化合物93.同位素标记实验显示产物羰基中的氧约三分之二来自于酮类原料92, 只有三分之一的氧来自于空气中的氧.机理显示反应同时涉及到了C—C键和O—O键的断裂, 伴随着副产物醛的生成(Eq. 23).
|
|
(23) |
2017年, 郭凯课题组[49]报道了芳基酮与饱和的仲胺之间的氧插入反应(Eq. 24).该反应在异硫氰酸铜的催化及4-N, N-二甲基吡啶(DMAP)的碱性条件下, 通过活化C(α)—H键进行氧气插入反应.该反应经过芳基酮的C(α)—C(β)键断裂、氧-氧键的断裂以及仲胺的亲核加成, 最后铜氧气氧化得到α-酮酰胺类化合物.该反应条件下产物专一性较差, 同时会伴随着多个副产物的生成, 例如醛类、酰胺类等副产物.
|
|
(24) |
过渡金属催化断裂C—C的氧插入反应目前只应用在合成酯和酰胺类化合物, 是过渡金属催化氧气氧插入反应的另一重要部分.然而, 目前该类反应只涉及到C—C单键的断裂, 还未涉及到断裂C=C键或C≡C键的氧插入反应.因此, 通过过渡金属催化断裂不饱和碳碳键的氧插入反应是这部分未来的发展方向.
相对于传统合成含氧化合物的方法, 过渡金属催化氧气氧插入反应具有简单、高效、绿色和经济等特点.该类反应近几年的发展已经实现了对甲基、亚甲基、次甲基、烯烃、炔烃、苯环、吲哚环等参与的氧插入反应, 并合成了结构多样的含羟基、羰基或含氧杂环类化合物.目前, 过渡金属催化氧气氧插入反应已经成为有机合成或工业上合成含氧化合物的最重要的方法之一.然而, 该类反应发展虽然非常迅速, 但随着绿色化学和可持续发展化学的新要求, 该类反应仍然存在未解决的问题和挑战.比如说该类反应需要多种金属参与, 且反应选择性还不够好, 反应的条件比较苛刻, 同时通常需要加入催化辅助剂, 底物局限性和反应立体选择性未解决等.这些不足都限制了这类反应的广泛应用和快速发展.近年来, 光催化氧气氧插入反应引起了合成化学家的极大兴趣.通过光催化诱导自由基历程的氧插入反应避免了过渡金属的使用, 使得反应更加绿色环保.因此, 如何实现温和条件下的氧气插入反应, 如何根据该类催化体系活性中间体设计新型自由基参与的氧插入反应及其过渡金属催化立体选择性氧插入反应是该领域今后重点发展的方向.
尹志平, 王泽超, 吴小锋, 有机化学, 2019, 39, 573. doi: 10.6023/cjoc201809004Yin, Z.; Wang, Z.; Wu, X. F. Chin. J. Org. Chem. 2019, 39, 573(in Chinese). doi: 10.6023/cjoc201809004
Zhang, Y.; Riemer, D.; Schilling, W.; Kollmann, J.; Das, S. ACS Catal. 2018, 8, 6659. doi: 10.1021/acscatal.8b01897
Thatikonda, T.; Deepake, S. K.; Das, U. Org. Lett. 2019, 21, 2523. doi: 10.1021/acs.orglett.9b00115
袁斯甜, 王艳华, 邱观音生, 刘晋彪, 有机化学, 2017, 37, 566. http://journal15.magtechjournal.com/Jwk3_hxs/hxxb/CN/abstract/566Yuan, S.; Wang, Y.; Qiu, G.; Liu, J. B. Chin. J. Org. Chem. 2017, 37, 566(in Chinese). http://journal15.magtechjournal.com/Jwk3_hxs/hxxb/CN/abstract/566
Liang, Y. F.; Jiao, N. Acc. Chem. Res. 2017, 50, 1064. http://www.ncbi.nlm.nih.gov/pubmed/28967737
Zhang, L.; Ang, G. Y.; Chiba, S. Org. Lett. 2011, 13, 1622. doi: 10.1021/ol200425c
Wang, Y. F.; Chen, H.; Zhu, X.; Chiba, S. J. Am. Chem. Soc. 2012, 134, 11980. doi: 10.1021/ja305833a
Zhang, C.; Xu, Z.; Zhang, L.; Jiao, N. Angew. Chem., Int. Ed. 2011, 50, 11088. doi: 10.1002/anie.201105285
Zhang, C.; Zhang, L.; Jiao, N. Adv. Synth. Catal. 2012, 354, 1293. doi: 10.1002/adsc.201100892
Xu, Z.; Zhang, C.; Jiao, N. Angew. Chem., Int. Ed. 2012, 51, 11367. doi: 10.1002/anie.201206382
Huang, X.; Li, X.; Zou, M.; Pan, J.; Jiao, N. Org. Chem. Front. 2015, 2, 354. doi: 10.1039/C5QO00028A
Kumar, Y.; Jaiswal, Y.; Kumar, A. J. Org. Chem. 2016, 81, 12247. doi: 10.1021/acs.joc.6b02176
Xu, X.; Li, B.; Zhao, Y.; Song, Q. Org. Chem. Front. 2017, 4, 331. doi: 10.1039/C6QO00635C
Chen, X. L.; Peng, Y. H.; Li, Y.; Wu, M. H.; Guo, H. B.; Wang, J.; Sun, S. F. RSC Adv. 2017, 7, 18588. doi: 10.1039/C7RA02207G
Wang, Y.; Liu, H. X.; Zhang, X. F.; Zhang, Z. L.; Huang, D. G. Org. Biomol. Chem. 2017, 15, 9164. doi: 10.1039/C7OB02192E
Su, Y.; Sun, X.; Wu, G.; Jiao, N. Angew. Chem., Int. Ed. 2013, 52, 9808. doi: 10.1002/anie.201303917
Sun, X.; Li, X.; Song, S.; Zhu, Y.; Liang, Y.-F.; Jiao, N. J. Am. Chem. Soc. 2015, 137, 6059. doi: 10.1021/jacs.5b02347
Lu, Q. Q.; Liu, Z. L.; Luo, Y.; Zhang, G. H.; Huang, Z. Y.; Wang, H. M.; Liu, C.; Miller, J. T.; Lei, A. W. Org. Lett. 2015, 17, 3402. doi: 10.1021/acs.orglett.5b01223
周豪, 陈知远, 有机化学, 2018, 38, 719. http://journal15.magtechjournal.com/Jwk3_hxs/hxxb/CN/abstract/abstract346410.shtmlZhou, H.; Chen, Z. Y. Chin. J. Org. Chem. 2018, 38, 719(in Chinese). http://journal15.magtechjournal.com/Jwk3_hxs/hxxb/CN/abstract/abstract346410.shtml
Wei, W.; Liu, C. L.; Yang, D. S.; Wen, J. W.; You, J. M.; Suo, Y. R.; Wang, H. Chem. Commun. 2013, 49, 10239. doi: 10.1039/c3cc45803b
Wei, W.; Wen, J. W.; Yang, D. S.; Wu, M.; You, J. M.; Wang, H. Org. Biomol. Chem. 2014, 12, 7678. doi: 10.1039/C4OB01369G
Gao, W. C.; Cheng, Y. F.; Shang, Y. Z.; Chang, H. H.; Li, X.; Zhou, R.; Qiao, Y.; Wei, W. L. J. Org. Chem. 2018, 83, 11956. doi: 10.1021/acs.joc.8b01843
Toh, K. K.; Wang, Y.-F.; Ng, E. P. J.; Chiba, S. J. Am. Chem. Soc. 2011, 133, 13942. doi: 10.1021/ja206580j
钱少平, 马尧睿, 高珊珊, 骆钧飞, 有机化学, 2018, 38, 1930. http://journal15.magtechjournal.com/Jwk3_hxs/hxxb/CN/abstract/abstract346568.shtmlQian, S. P.; Ma, Y. R.; Gao, S. S.; Luo, J. F. Chin. J. Org. Chem. 2018, 38, 1930(in Chinese). http://journal15.magtechjournal.com/Jwk3_hxs/hxxb/CN/abstract/abstract346568.shtml
Zhang, Y. H.; Yu, J. Q. J. Am. Chem. Soc. 2009, 131, 14654. doi: 10.1021/ja907198n
Yan, Y.; Feng, P.; Zheng, Q.-Z.; Liang, Y.-F.; Lu, J.; Cui, Y.; Jiao, N. Angew. Chem., Int. Ed. 2013, 52, 5827. doi: 10.1002/anie.201300957
Chiba, S.; Zhang, L.; Lee, J.-Y. J. Am. Chem. Soc. 2010, 132, 7266. doi: 10.1021/ja1027327
Tnay, Y. L.; Chen, C.; Chua, Y. Y.; Zhang, L.; Chiba, S. Org. Lett. 2012, 14, 3550. doi: 10.1021/ol301583y
Liang, Y.-F.; Li, X.; Wang, X.; Yan, Y.; Feng, P.; Jiao, N. ACS Catal. 2015, 5, 1956. doi: 10.1021/cs502126n
Li, Z.; Huang, X.; Chen, F.; Zhang, C.; Wang, X.; Jiao, N. Org. Lett. 2015, 17, 584. doi: 10.1021/ol5035996
Wu, W.; Xu, J.; Huang, S.; Su, W. Chem. Commun. 2011, 47, 9660. doi: 10.1039/c1cc10545k
Bao, Y.-H.; Zhu, J.-Y.; Qin, W.-B.; Kong, Y.-B.; Chen, Z.-W.; Tang, S.-B.; Liu, L.-X. Org. Biomol. Chem. 2013, 11, 7938. doi: 10.1039/c3ob41589a
Wang, C.; Zhang, L. P.; Ran, A.; Lu, P.; Wang, Y. G. Org. Lett. 2013, 15, 2982. doi: 10.1021/ol401144m
Huang, H.; Cai, J.; Ji, X.; Xiao, F.; Chen, Y.; Deng, G. J. Angew. Chem. 2016, 128, 315. doi: 10.1002/ange.201508076
Guo, S. H.; Wang, F.; Tao, L.; Zhang, X. Y.; Fan, X. S. J. Org. Chem. 2018, 83, 3889. doi: 10.1021/acs.joc.8b00231
Zhang, J.; Kohlbouni, S. T.; Borban, B. Org. Lett. 2019, 21, 14. doi: 10.1021/acs.orglett.8b03185
Yu, L. M.; Zhong, Y.; Yu, J. C.; Gan, L.; Cai, Z. J.; Wang, R.; Jiang, X. X. Chem. Commun. 2018, 54, 2353. doi: 10.1039/C7CC09640B
余乐茂, 博士论文, 中山大学, 广州, 2018.Yu, L. M. Ph. D. Dissertation, Sun Yat-Sen University, Guangzhou, 2018 (in Chinese).
Zhang, C.; Jiao, N. J. Am. Chem. Soc. 2010, 132, 28. doi: 10.1021/ja908911n
Wang, J.; Wang, J.; Zhu, Y.; Lu, P.; Wang, Y. Chem. Commun. 2011, 47, 3275. doi: 10.1039/c0cc04922k
Toh, K. K.; Sanjaya, S.; Sahnoun, S.; Chong, S. Y.; Chiba, S. Org. Lett. 2012, 9, 2290. http://www.ncbi.nlm.nih.gov/pubmed/22490011/
Ling, F.; Li, Z. X.; Zhang, C. G.; Liu, X.; Ma, C. J. Am. Chem. Soc. 2014, 136, 10914. doi: 10.1021/ja506795u
Ling, F.; Wan, Y.; Wang, D.; Ma, C. J. Org. Chem. 2016, 81, 2770. doi: 10.1021/acs.joc.5b02870
Liu, X.; Mao, R. J.; Ma, C. Org. Lett. 2017, 19, 6704. doi: 10.1021/acs.orglett.7b03427
Pan, J.; Li, X. Y.; Qiu, X.; Luo, X.; Jiao, N. Org. Lett. 2018, 20, 2762. doi: 10.1021/acs.orglett.8b00992
Zhang, C.; Xu, Z.; Shen, T.; Wu, G.; Zhang, L.; Jiao, N. Org. Lett. 2012, 14, 2362. doi: 10.1021/ol300781s
Zhang, C.; Feng, P.; Jiao, N. J. Am. Chem. Soc. 2013, 135, 15257. doi: 10.1021/ja4085463
Huang, X.; Li, X.; Zou, M.; Song, S.; Tang, C.; Yuan, Y.; Jiao, N. J. Am. Chem. Soc. 2014, 136, 14858. doi: 10.1021/ja5073004
Liu, C.; Yang, Z.; Zeng, Y.; Fang, Z.; Guo, K. Org. Chem. Front. 2017, 4, 2375. doi: 10.1039/C7QO00690J

图式 3 锰催化氧插入反应合成β-叠氮醇类化合物
Scheme 3 Synthesis of β-azidol compounds by Mn2+ catalyzed oxygenation

图式 4 铜/钴催化氧肟酸与端烯之间的氧插入反应
Scheme 4 Copper/cobalt catalyzed oxygen insertion reaction between oxime acid and terpene

图式 5 铜/铁催化的氧插入反应合成β-羰基砜类化合物
Scheme 5 Copper/iron catalyzed oxygenation for the synthesis of β-carbonyl sulfones

图式 6 铜催化芳环的氧插入螺环反应
Scheme 6 Copper catalyzed spiral cyclization of aromatic rings with oxygenation

图式 7 钯催化氧插入的酰基化和硝基化反应
Scheme 7 Palladium-catalyzed acylation and nitrogenation of Aromatic rings with oxygenation

图式 8 过渡金属催化吲哚与1, 3-二酮之间的氧插入反应
Scheme 8 Transition metal catalyzed oxygenation between indole and 1, 3-diketone

图式 9 铜氧催化氧插入反应合成色胺酮类化合物
Scheme 9 Copper catalyzed oxygenation for the synthesis of tryptamine compounds

图式 10 铜催化氧插入反应合成吲哚3-酮类化合物
Scheme 10 Copper catalyzed oxygenation for the synthesis of indole 3-ketones

图式 11 铜催化炔烃参与的分子内环化-氧插入反应
Scheme 11 Copper catalyzed intramolecular cyclization- oxygenation involved by internal alkynes

图式 12 钯/铜催化的炔烃之间的偶联-氧插入串联反应
Scheme 12 Palladium/copper catalyzed coupling-oxygenation cascade between alkynes

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

























 下载:
下载:

