图 1.
含有胺基基团化合物
Figure 1.
Amine group compound

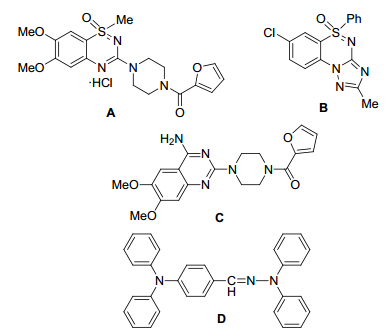
胺基基团作为重要的结构骨架[1], 广泛存在于各种生物活性分子、药物分子和材料分子中. 图 1列举了部分胺基类或含氮类化合物, 例如官能化的噻二嗪-1-氧化物A, 在动物试验中表现出降血压活性, 其功能同α-肾上腺素受体阻滞剂相似[2]; 三环稠合亚砜亚胺Gö4962 B是一种苯二氮䓬受体激动剂, 具有良好的抗焦虑和抗惊厥活性[3]; 哌唑嗪C能降低心脏的前、后负荷, 也可以治疗心功能不全[4]; 芳基醛腙D作为一类新的无定形分子材料, 可以制作成更稳定的空穴传输分子材料[5].因此, 高效且经济地合成胺基类化合物在药物化学、材料化学及有机化学领域受到广泛关注.
过渡金属催化C—N键胺化反应一般包括两种方法(Scheme 1): (ⅰ)卤代烷烃或卤代杂芳烃与胺化试剂交叉偶联反应[6], 这类反应具有反应收率高, 选择性好, 底物普适性高且应用广泛等优点, 但是需要对底物进行预官能团化, 并且不可避免地产生定量的卤代物副产物. (ⅱ)碳氢化合物与胺化试剂直接C—H胺化反应, 大量的碳氢化合物可以作为反应底物, 此反应不需要预先安装功能性官能团, 具有步骤及原子经济性高等优点[7].
过渡金属催化C—H官能化反应, 主要利用昂贵的第五周期(钌、铑、钯、银)或第六周期(铱、金)过渡金属作为催化剂[8].然而, 在地球上具有丰富储量的第四周期过渡金属中, 钴因低成本、低毒性和独特的催化反应性受到了越来越多的关注[9].钴元素符号为Co, 位于元素周期表第四周期、第VⅢ族, 原子序数为27, 外层电子排布3d7 4s2, 常见化合价为+2和+3.钴是一种银白色铁磁性金属, 比较硬而脆, 在常温下不和水反应, 在潮湿的空气中也很稳定.
廉价、低毒的钴作为催化剂, 可以实现C(sp2)—H胺化[10]和C(sp3)—H胺化[11]反应.本文将围绕这两种反应类型综述钴催化C—H胺化反应的研究进展及其反应机理, 并对该领域所面临的挑战及发展前景做了总结和展望.
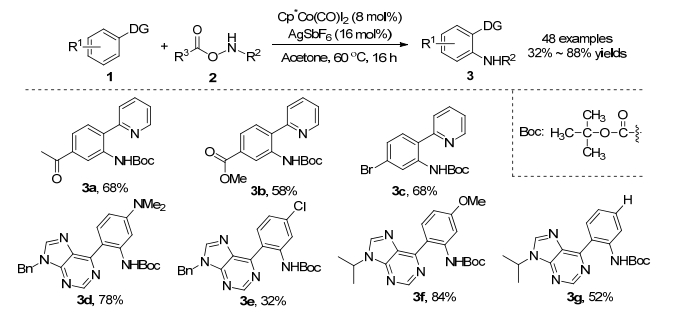
2015年, Chang课题组[12]报道了钴催化芳烃1与氨基甲酸酯C—H胺化反应(Scheme 2).通过对氨基甲酸酯筛选, 作者发现乙酰氧基氨基甲酸叔丁酯是最佳的胺化试剂, 用其作为底物时, 产物分离收率为88%, 并且产生的副产物乙酸很容易去除.该反应以8 mol% CoCp*(CO)I2作为最佳的催化剂, 16 mol% AgSbF6作为银添加剂, 不需要外源氧化剂和碱, 在60 ℃温度下, 以32%~88%的收率得到胺化产物3.该反应具有良好的官能团耐受性, 可以耐受乙酰基、酯基、氯基和溴基等取代基.同时, 作者利用具有生物活性的嘌呤作为导向基时, 该反应仍具有良好的兼容性(甲氧基、二甲胺基和氯基).
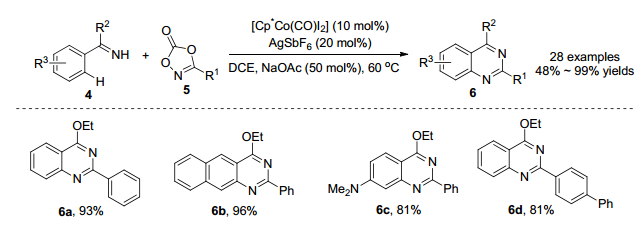
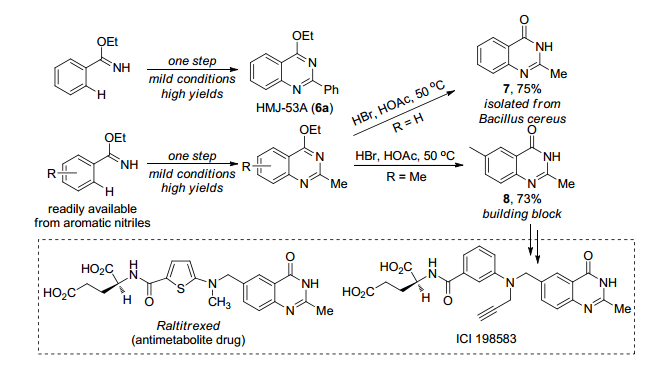
2016年, Glorius课题组[13]报道了利用钴催化亚胺导向芳烃4与二噁唑5生成喹唑啉化合物6 (Scheme 3).通过对[Cp*IrCl2]2、[Cp*RhCl2]2和[Cp*Co(CO)I2]三种催化剂的筛选, 发现钴催化剂在该反应中催化效果最佳, 这是因为钴催化剂具有强路易斯酸性和对空间位阻的高敏感性.该反应条件温和, 一步直接合成喹唑啉化合物, 分离收率高达99%.这一合成方法具有良好的底物普适性, 对于各种芳环、杂环以及脂肪烃都可得到较好的结果.利用该策略生成的产物6a可以抑制花生四烯酸诱导的血小板聚集(Scheme 4).喹唑啉化合物6通过水解可以合成具有生物和药物活性的喹唑啉酮衍生物.产物6b、6c和6d在pH比色和pH发光传感器方面具有潜在应用.证明了该合成方法具有一定的应用价值.
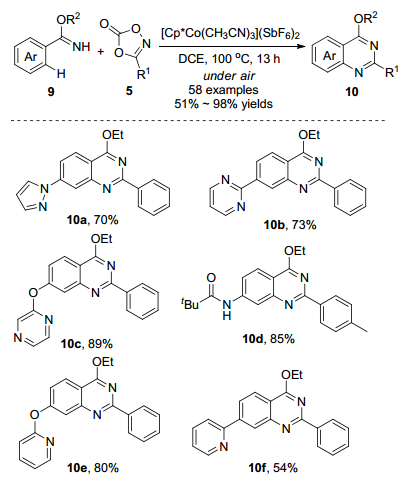
2016年, Ackermann课题组[14]报道了钴催化亚胺酸酯导向苯甲亚胺类化合物9和二噁唑C—H胺化反应(Scheme 5).该反应不需要添加剂, 利用5.0 mol%的[Cp*Co(CH3CN)3](SbF6)2作为催化剂, 1, 2-二氯乙烷(DCE)作为溶剂, 在100 ℃的条件下高效地合成喹唑啉化合物10.该反应具有高官能团耐受性和优异化学选择性, 当化合物9的苯环上取代基为吸电子官能团(氟基、氯基、酯基、羰基和硝基)或供电子官能团(甲氧基)时, 收率中等至优秀; 对于具有间位取代基的底物, 反应选择性地发生在空间位阻较小的C—H键上.若化合物9的苯环上取代基为强配位的N-杂环(吡唑、嘧啶、吡嗪和吡啶)时, 钴(Ⅲ)催化剂完全耐受这些具有导向作用的N-杂环, 只生成喹唑啉化合物10.作者进行了分子间竞争实验, 首次确定了钴(Ⅲ)催化C—H官能团化中不同N-杂环导向基团的配位能力, 依次为亚氨酸酯≥吡啶≈吡唑>噁唑啉>嘧啶.
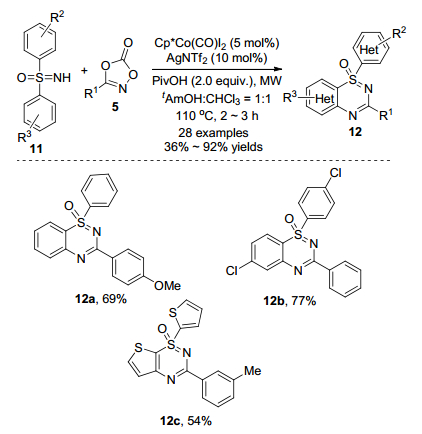
在2017年, 陈知远课题组[15]使用NH-亚磺酰亚胺11作为原料, 二噁唑5作为胺化试剂, 在Cp*Co(CO)I2催化和微波辅助下一锅一步生成噻二嗪-1-氧化物12(Scheme 6).该反应易于处理, 不需要外源氧化剂, 对空气中的水分或氧气不敏感.反应产生的副产物为二氧化碳和水, 符合绿色化学的发展理念.当反应规模放大到1 mmol时, 以78%的分离收率得到噻二嗪1-氧化物.在底物拓展过程中, 无论底物上的取代基是吸电子基团(氟基、溴基和碘基)还是供电子基团(甲基和甲氧基), 都能以中等至优秀的收率得到目标产物.这一新方法可以简单、高效地合成具有生物活性的噻二嗪-1-氧化物, 具有较好的实际应用价值.
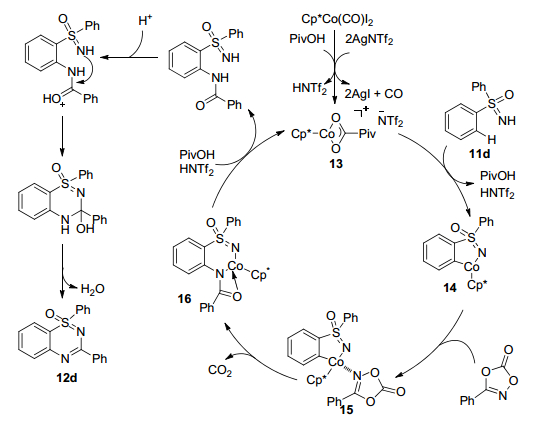
作者提出如下反应机理(Scheme 7):首先, 在Piv-OH存在下, 中性络合物Cp*Co(CO)I2与AgNTf2反应产生阳离子物种13; 然后, 物种13与亚磺酰亚胺11d进行亲电金属化得到五元钴化合物14, 同时释放PivOH和HNTf2.胺化试剂与钴化合物14配位产生物种15, 然后经历插入、脱水和环化反应生成产物12d.
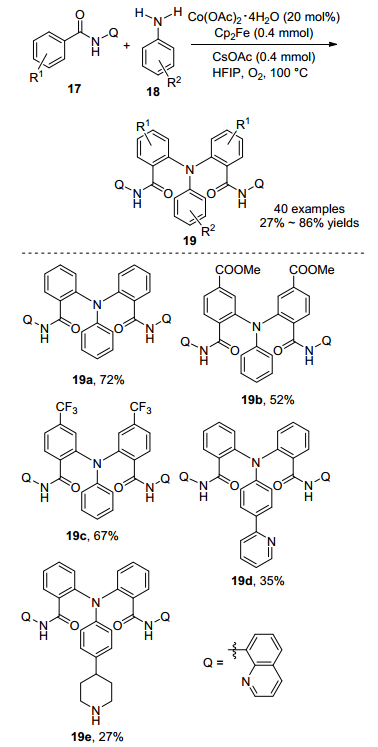
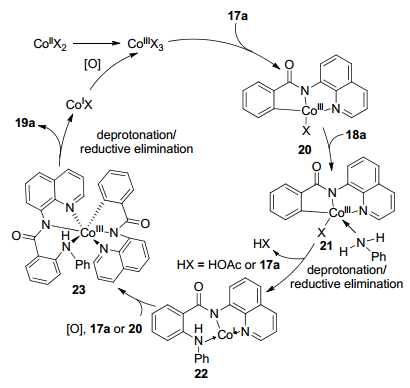
在2017年, 宋毛平课题组[16]利用廉价的Co(OAc)2• 4H2O催化8-氨基喹啉导向未预活化的芳烃17和简单的苯胺C—H/N—H交叉偶联反应(Scheme 8).使用氧气作为氧化剂, 二茂铁(Cp2Fe)作为助氧化剂, 六氟异丙醇作为促进剂, 在100 ℃条件下高化学选择性地生成三芳基胺19, 分离收率高达86%.对底物进行拓展, 发现其可以兼容许多官能团, 譬如烷基、卤素、苯基、酯基和强配位基团(芳基吡啶和哌啶).该方法可进行克级规模反应, 当芳烃17投加量为4.0 mmol时, 以67%的收率获得三芳基胺化合物.利用NaOH水解芳烃19便可除去8-氨基喹啉导向基团, 回收率高达96%, 显示出该合成方法的应用潜力.
反应可能的机理如Scheme 9所示:首先在Cp2Fe和O2存在下, 钴(Ⅱ)盐被氧化成钴(Ⅲ)物种, 其活化苯甲酰胺邻位C—H键形成三价钴络合物20; 再与苯胺18a配位、去质子化得到中间体22; 中间体22被氧化成钴(Ⅲ)物种, 然后通过C—H键活化与苯甲酰胺配位, 生成中间体23, 或者中间体22与络合物20之间配体交换生成中间体23; 最后, 通过去质子化、还原消除获得胺化产物19a.
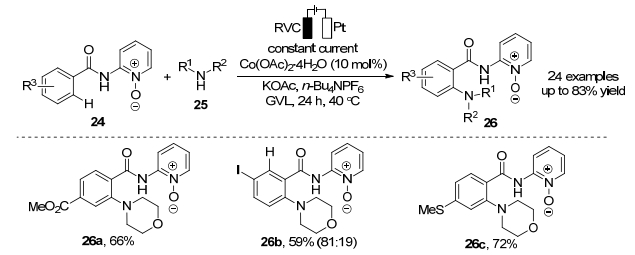
2018年, Ackermann课题组[17]报道了钴催化苯甲酰胺类化合物24和烷基胺C—H胺化反应(Scheme 10). Co(OAc)2•4H2O作为最佳催化剂, 利用廉价的电作为氧化剂[18], 可再生的γ-戊内酯(GVL)作为反应溶剂, KOAc作为添加剂, 在40 ℃反应条件下生成芳胺化合物26.该反应不添加任何金属氧化剂, 避免产生化学计量的金属副产物, 生成的副产物为H2, 符合原子经济性要求.该策略具有高官能团耐受性, 能够耐受各种吸电子官能团(硫醚基、酯基和碘基)和给电子官能团(甲基和叔丁基), 分离收率最高达83%.
在2014年, Kanai课题组[19]利用Cp*Co(CO)I2配合物作为阳离子Co(Ⅲ)活性催化剂的前体, 嘧啶作为导向基, 定向催化吲哚27和磺酰叠氮化合物28胺化反应(Scheme 11). AgSbF6作为银盐, KOAc作为添加剂, 在DCE溶剂中反应12 h生成胺化产物, 反应副产物为N2气体. Cp*Co(CO)I2配合物在空气环境中非常稳定且容易获得, 在大于10 g规模下收率为86%[20].当催化剂用量降低至1.25 mol%时, 仍具有良好的催化效率(TON=68).该方法具有较好的底物普适性, 对于吸电子基团(酯基、氯基和溴基等)和供电子基团(甲氧基和甲基), 反应收率都在85%以上.
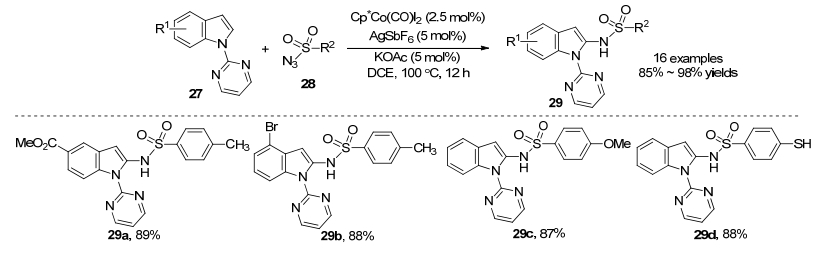
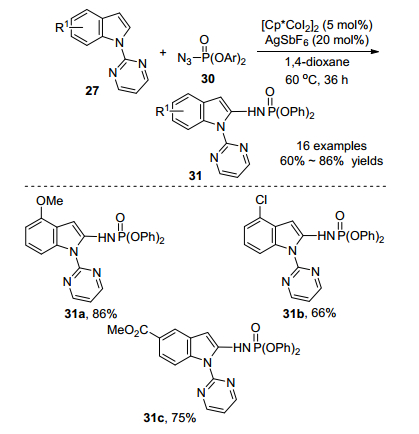
Cp*Co(CO)I2配合物作为催化剂, 在反应过程中会释放出有毒的一氧化碳气体.为了避免这个安全问题, 2015年, Kanai课题组[21]利用[Cp*CoI2]2配合物作为阳离子Co(Ⅲ)活性催化剂的前体, 催化磷酰基叠氮化物30和吲哚27的C-2选择性胺化反应(Scheme 12).嘧啶作为导向基团, AgSbF6作为银盐, 1, 4-二噁烷作为溶剂, 以86%的收率得到氨基磷酸酯31.该反应具有良好的官能团耐受性, 可以兼容烷基、酯基、醚和卤素等官能团.该反应条件温和, 收率中等至优秀, 由于氨基磷酸酯衍生物是许多生物活性化合物中的重要结构骨架, 该方法具有潜在的应用价值.
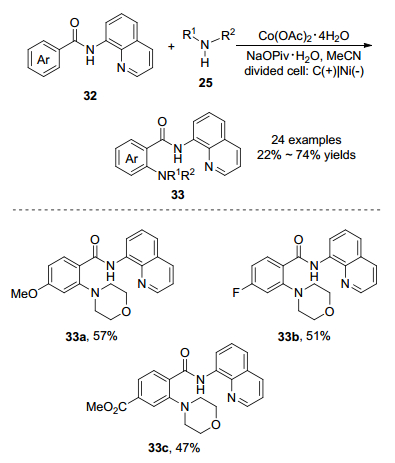
2018年, 雷爱文课题组[22]报道了钴(Ⅱ)催化喹啉酰胺双齿导向芳烃32与烷基胺电氧化C—H胺化反应(Scheme 13).该反应Co(OAc)2•4H2O作为催化剂, 在阳极使用NaOPiv•H2O作为添加剂, 乙腈作为溶剂, 在阴极用甲醇作为溶剂, 10 mA恒流电解3 h就可得到胺化产物.在分隔池中, 阳极碳布替代外源氧化剂, 避免使用金属氧化剂, 符合原子经济学的要求.对底物进行拓展, 发现可以兼容多种官能团, 譬如氟、酯、醚和杂环(哌啶、硫代吗啉和1, 2, 3, 4-四氢异喹啉), 产物的收率最高达到74%.作者以5.0 mmol规模进行钴催化电氧化C-H胺化反应, 收率为59%.
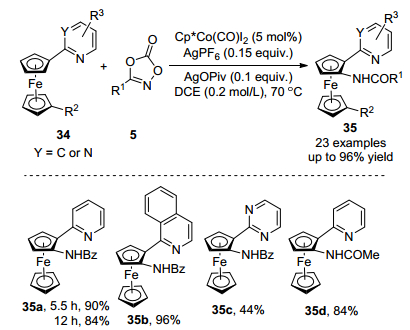
2018年, 游书力课题组[23]报道了钴催化吡啶导向二茂铁34和二噁唑C—H胺化反应(Scheme 14). 5 mol% Cp*Co(CO)I2作为催化剂, AgPF6作为银盐, AgOPiv作为添加剂, 在温和的反应条件下生成N-二茂铁酰胺衍生物35, 分离收率高达96%.在该反应中异喹啉和嘧啶可以作为导向基, 分别生成35b和35c.当二噁唑5上取代基为芳基, 杂芳基和烷基时, 具有中等至优秀的分离收率.该反应可以扩大到克级规模, 当以3 mmol的34a作为底物时, 反应12 h, 分离收率为84%.生成的N, N-二齿二茂铁产物可作为配体或催化剂的中间体, 说明该反应具有一定实用价值.
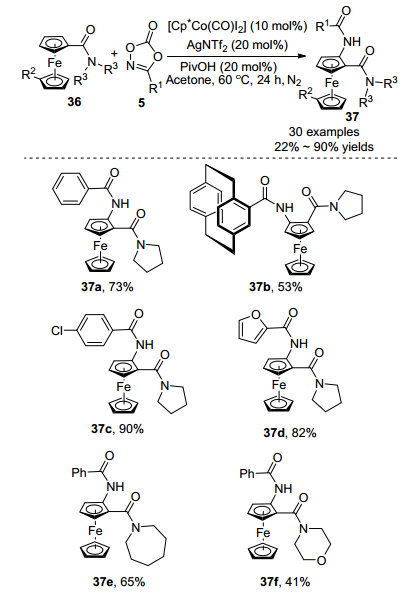
2019年, 史炳锋课题组[24]使用稳定的1, 4, 2-二噁唑-5-酮作为胺化试剂, 在氮气中使用Cp*Co(CO)I2催化酰胺导向二茂铁化合物36胺化反应(Scheme 15).丙酮作为反应溶剂, AgNTf2作为银盐, PivOH为添加剂, 在60℃温度条件下反应24 h生成胺化产物37.产物37a的结构得到X射线单晶体衍射确认, 产物37b是一种广泛应用于有机合成和材料科学的重要结构骨架.该反应具有良好的官能团耐受性, 1, 4, 2-二噁唑-5-酮作为底物时, 可以兼容吸电子取代基(氟基、溴基和氯基等)或给电子取代基(甲基、甲氧基和叔丁基), 吸电子取代的二噁唑酮反应效果更好, 生成的胺化产物分离收率高达90%.二茂铁的另一个Cp环可以兼容异丙基、1-(呋喃-2-基)乙基和苯乙基.该反应可扩大到4 mmol规模, 催化剂用量由原来的10 mol%降低至5 mol%, 以70%的收率得到胺化产物, 增强了该方法的应用性.
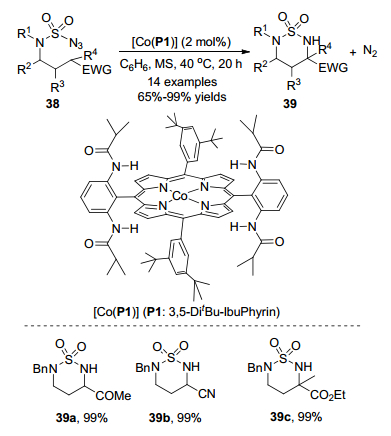
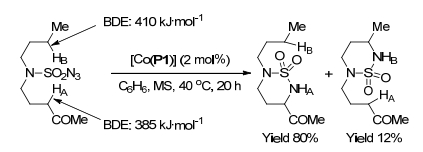
2012年, 张小祥课题组[25]报道了钴(Ⅱ)金属自由基催化磺酰叠氮化物38分子内C—H胺化反应(Scheme 16).在该反应中, 2 mol%的[Co(P1)]作为缺电子C—H键的分子内胺化的最佳催化剂, 苯作为反应溶剂, 在40 ℃下反应20 h直接生成α-氨基酸衍生物39, 收率高达99%.当底物为酰胺、酮和腈时, 具有优秀的分离收率.当底物为β-取代叠氮化物和γ-取代的叠氮酯时, 不受取代基的影响, 分子内α-C—H胺化反应, 具有高的区域选择性和非对映选择性.环状氨基酸衍生物39在4-二甲氨基吡啶(DMAP)存在下用Cl3CCH2OH处理时, 可以高效地转化为α, γ-二氨基酸酯, 收率为88%.由于α-和γ-胺基团被其他基团保护, 进一步转化可生成各种α, γ-二氨基酸衍生物.为了阐明钴(Ⅱ)对C—H胺化的催化能力, 作者进行了分子间竞争实验(Scheme 17).该实验结果表明缺电子C—HA的胺化速度明显快于供电子的C—HB.因为C—HA键(385 kJ•mol-1)键能低于C—HB键(410 kJ•mol-1)[26], 所以钴(Ⅲ)-硝基自由基中间体更容易脱除HA原子.
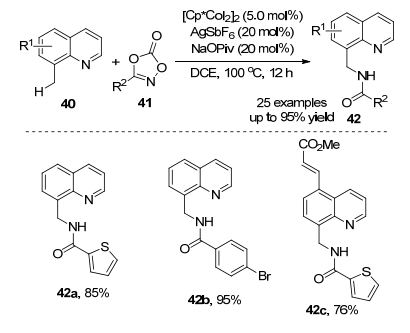
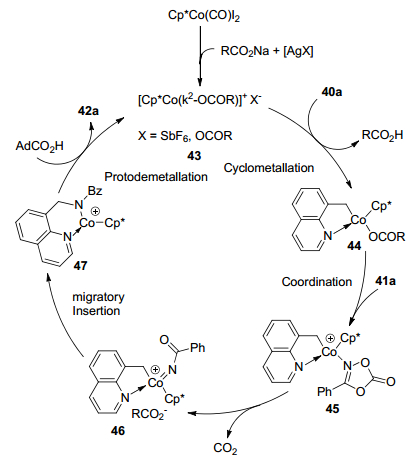
2016年, Sundararaju课题组[27]报道了钴催化喹啉40和噁唑酮C(sp3)—H胺化反应(Scheme 18).该反应以5 mol%的[Cp*CoI2]2为催化剂, 催化量的AgSbF6和NaOPiv作为添加剂, 不需要外源氧化剂, 在1, 2-二氯乙烷中成功实现了C—H胺化反应.产物42a的结构得到X射线单晶体衍射的确认.该反应底物普适性广, 当取代基在喹啉40的C-5 (甲基、4-茴香基、1-萘基)、C-6(甲基、苯基)和C-7(甲基)位置时, 具有良好至优秀的收率.噁唑酮41能兼容吸电子官能团(溴基、氟基)或给电子官能团(甲氧基、叔丁基和甲基), 产物收率最高为95%.
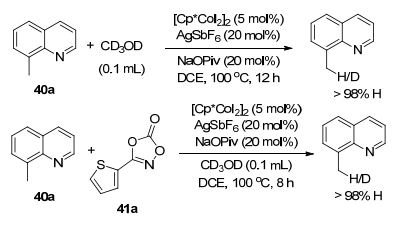
作者对去质子化途径进行探究(Scheme 19), 在优化条件下没有观察到8-甲基喹啉与CD3OD的氘交换, 表明该反应是不可逆的.作者提出了一种合理的反应机理(Scheme 20).阳离子络合物43在过量的NaOPiv辅助下活化喹啉化合物40a的C—H键生成物种44.之后与噁唑酮41a反应得到物种45, 在羧酸盐辅助下开环生成钴-氮烯中间体46和CO2.中间体46经历迁移插入和金属化生成42a.
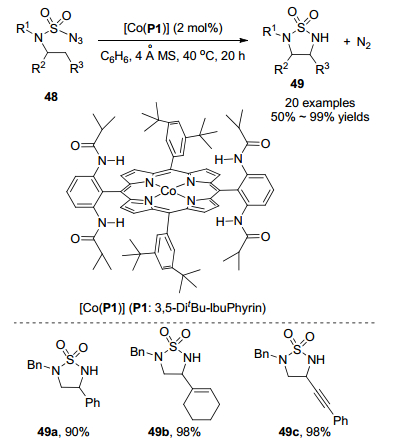
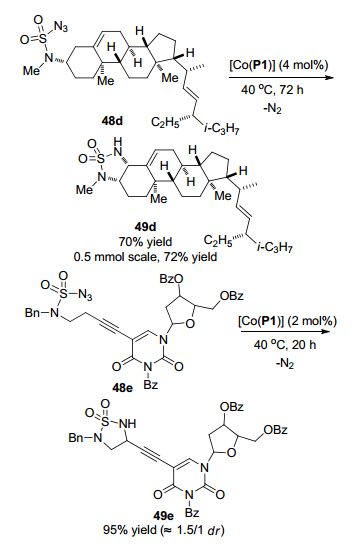
2016年, 张小祥课题组[28]报道了钴(Ⅱ)金属自由基在中性和非氧化条件下催化氨磺酰叠氮化物48分子内1, 5-C(sp3)—H胺化生成5-元环状磺酰胺化合物49 (Scheme 21). 2 mol%的[Co(P1)]作为催化剂, 不需要任何外源添加剂, 以高产率直接一步得到胺化产物49, 氮气是唯一的副产物.该策略具有优异的化学选择性和高官能团耐受性, 可以高效地胺化杂芳环(呋喃和噻吩)、非苄型底物、酯、烯烃和炔烃, 分离收率中等至优秀.豆甾醇叠氮化物48d作为底物时, 以70%的收率生成稠合的多环磺酰胺49d, 都是顺式立体异构体(Scheme 22).当该底物的量放大到0.5 mmol时, 反应收率为72%. [Co(P1)]可以催化脱氧尿苷底物48e C-H胺化生成脱氧尿苷衍生的5-元环磺酰胺49e.利用该策略对具有生物活性的复杂有机化合物进行官能化, 可以对生成的5-元环状磺酰胺化合物进行一些生物活性研究.
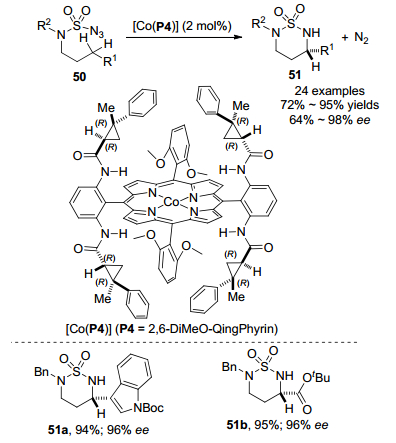
2018年, 张小祥课题组[29]利用钴(Ⅱ)金属自由基催化氨磺酰叠氮化物50的分子内1, 6-C(sp3)—H胺化反应(Scheme 23). 2 mmol%的[Co(P4)]作为催化剂, 甲基叔丁基醚(MTBE)作为最佳反应溶剂, 利用氮气保护在室温条件下以高达98%收率生成六元手性杂环磺酰胺51, 同时表现出优异的对映选择性.钴(Ⅱ)金属自由基催化胺化反应表现出高官能团耐受性和对映选择性, 可以耐受烯烃、炔烃、醇、酮、酯和酰胺等, 具有良好至优秀的分离收率.光学活性的磺酰胺51b在不损失对映体纯度情况下可以容易地转化为1, 3-二胺衍生物, 再通过进一步转化生成光学活性的α, γ-二氨基酸衍生物.
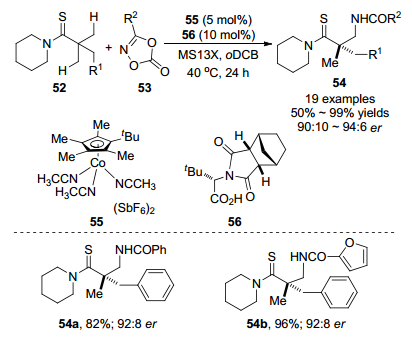
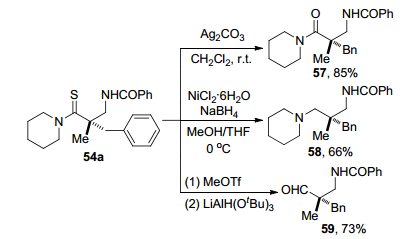
2019年, Matsunaga课题组[30]报道了使用非手性钴(Ⅲ)/手性羧酸杂化体系催化硫代酰胺52和二噁唑酮53 C(sp3)—H胺化反应(Scheme 24).在该反应中55作为最佳催化剂, (S)-H2-BHTL 56为手性羧酸, MS13X作为添加剂, 邻二氯苯(oDCB)是最佳溶剂, 在温和条件下C(sp3)—H胺化直接合成β-氨基硫代羰基化合物54.该反应具有良好的官能团耐受性, 可以很好地耐受吸电子基团(氯基和溴基)和给电子基团(甲氧基和甲基), 具有中等至优秀的分离收率和良好的对映选择性.以4 mmol规模进行放大反应, 产物收率为92%和93:7 er. β-氨基硫代羰基化合物54a通过甲基化、还原生成手性β-氨基醛59 (Scheme 25), 说明对映选择性酰胺化产物具有一定的应用性.
综述了近年来Co催化C—H胺化反应, 反应具有原子经济性、步骤经济性的优点, 为胺基类化合物的合成提供了简便绿色的合成方法.然而钴催化C—H胺化反应领域仍然存在一些挑战.首先, 目前使用的钴催化剂的种类有限, 催化剂的转化效率和稳定性有待提高, 发展高选择性和高效的催化剂是重要的研究方向.其次, 目前芳香C—H胺化反应主要局限在导向基团邻位, 如何实现导向基团间位、对位C—H键胺化将是一个巨大的挑战.最后, 在钴催化胺化反应中, 导向基的引入提高了反应的选择性, 但是导向基团的难去除也限制了产物的进一步衍生化, 如何更好地利用导向基团是一个重要的研究方向.虽然钴催化C—H胺化反应具有这些挑战, 但在该研究领域里充满活力, 相信在接下来的时间里, 该反应将会更加经济、高效和实用.
(a) Ning, Z.; Tian, H. Chem. Commun. 2009, 5483.
(b) Michael, J. P. Nat. Prod. Rep. 2008, 25, 166.
(c) Liu, D.; Zhang, Z.; Zhang, H.; Wang, Y. Chem. Commun. 2013, 49, 10001.
(d) Mphahlele, M. J.; Paumo, H. K.; El-Nahas, A. M.; ElHendawy, M. M. Molecules 2014, 19, 795.
(e) Lücking, U. Angew. Chem., Int. Ed. 2013, 52, 9399.
(f) Liu, L.-T.; Chen, Y.-Y.; Zhang, A.-A.; Liu, X.; Zhang, L.; Bai, J.-R.; Li, H.; Mao, G.-L. Chin. J. Org. Chem. 2019, 39, 475(in Chinese).
(刘澜涛, 陈莹莹, 张安安, 刘雪, 张丽, 白静茹, 李恒, 毛国梁, 有机化学, 2019, 39, 475.)
(a) Dillard, R.; Yen, T.; Stark, P.; Pavey, D. J. Med. Chem. 1980, 23, 717.
(b) Zhou, H.; Chen, Z.-Y. Chin. J. Org. Chem. 2018, 38, 719(in Chinese).
(周豪, 陈知远, 有机化学, 2018, 38, 719.)
Bartoszyk, G.; Dooley, D.; Barth, H.; Hartenstein, J.; Satzinger, G. J. Pharm. Pharmacol. 1987, 39, 407. doi: 10.1111/j.2042-7158.1987.tb03410.x
(a) Achelle, S.; Rodríguez-Lopez, J.; Guen, F. R. J. Org. Chem. 2014, 79, 7564.
(b) Witt, A.; Bergman, J. Curr. Org. Chem. 2003, 7, 659.
(a) Strohriegl, P.; Grazulevicius, J. V. Adv. Mater. 2002, 14, 1439.
(b) Nishimura, K.; Kobota, T.; Inada, H.; Shirota, Y. J. Mater. Chem. 1991, 1, 897
(a) Wolfe, J. P.; Wagaw, S.; Marcoux, J.-F.; Buchwald, S. L. Acc. Chem. Res. 1998, 31, 805.
(b) Yang, B. H.; Buchwald, S. L. J. Organomet. Chem. 1999, 576, 125.
(c) Hartwig, J. F. Angew. Chem., Int. Ed. 1998, 37, 2046.
(d) Hartwig, J. F. Nature 2008, 455, 314.
(a) Park, Y.; Kim, Y.; Chang, S. Chem. Rev. 2017, 117, 9247.
(b) Moselage, M.; Li, J.; Ackermann, L. ACS Catal. 2016, 6, 498.
(c) Zhao, F.-Q.; Yang, Q.; Zhang, J.-J.; Shi, W.-M.; Hu, H.-H.; Liang, F.; Wei, W.; Zhou, S.-L. Org. Lett. 2018, 20, 7753.
(d) Gao, F.; Han, X.; Li, C.-P.; Liu, L.-J.; Cong, Z.-Q.; Liu, H. RSC Adv. 2018, 8, 32659.
(e) Yetra, S. R.; Shen, Z.-G.; Wang, H.; Ackermann, L. Beilstein J. Org. Chem. 2018, 14, 1546.
(f) Wang, F.; Jin, L.; Kong, L.-H.; Li, X.-W. Org. Lett. 2017, 19, 1812.
(g) Mei, R.-H.; Loup, J.; Ackermann, L. ACS Catal. 2016, 6, 793.
(h) Zhang, L.-B.; Zhang, S.-K.; Wei, D.-H.; Zhu, X.-J.; Hao, X.-Q.; Su, J.-H.; Niu, J.-L.; Song, M.-P. Org. Lett. 2016, 18, 1318.
(i) Sun, J.-S.; Liu, M.; Zhang, J.; Dong, L. J. Org. Chem. 2018, 83, 10555.
(a) Kuhl, N.; Schröder, N.; Glorius, F. Adv. Synth. Catal. 2014. 356 1443.
(b) Song, G.; Li, X. Acc. Chem. Res. 2015. 48 1007.
(c) Newton, C. G.; Wang, S.-G.; Oliveira, C. C.; Cramer, N. Chem. Rev. 2017. 117, 8908.
(d) Hummel, J. R.; Boerth, J. A.; Ellman, J. A. Chem. Rev. 2017. 117, 9163.
(a) Gandeepan, P.; Cheng, C. H. Acc. Chem. Res. 2015, 48, 1194.
(b) Grigorjeva, L.; Daugulis, O. Org. Lett. 2014, 16, 4684.
(c) Prakash, S.; Muralirajan, K.; Cheng, C. H. Angew. Chem., Int. Ed. 2016, 55, 1844.
(d) Zhang, Z.-Z.; Liu, B.; Xu, J.-W.; Yan, S.-Y.; Shi, B.-F. Org. Lett. 2016, 18, 1776.
(e) Wei, D.-H.; Zhu, X.-J.; Niu, J.-L.; Song, M.-P. ChemCatChem 2016, 8, 1242.
(f) Shin, K.; Kim, H.; Chang, S. Acc. Chem. Res. 2015, 48, 1040.
(g) Kim, H.; Chang, S. ACS Catal. 2016, 6, 2341.
(h) Gu, Z.-Y.; Ji, S.-J. Acta Chim. Sinica 2018, 76, 347(in Chinese).
(顾正洋, 纪顺俊, 化学学报, 2018, 76, 347.)
(i) Zhang, J.-H.; Hao, X.-Q.; Wang, Z.-L.; Ren, C.-J.; Niu, J.-L.; Song, M.-P. Chin. J. Org. Chem. 2017, 37, 1237(in Chinese).
(张家恒, 郝新奇, 王正龙, 任常久, 牛俊龙, 宋毛平, 有机化学, 2017, 37, 1237.)
(j) Cheng, B.; Lu, P.; Zhao, J.-J.; Lu, Z. Chin. J. Org. Chem. 2019, 39, 1704(in Chinese).
(程彪, 陆鹏, 赵家金, 陆展, 有机化学, 2019, 39, 1704.)
(a) Park, J.; Chang, S. Angew. Chem., Int. Ed. 2015, 54, 14103.
(b) Liang, Y.-J.; Liang, Y.-F.; Tang, C.-H.; Yuan, Y.-Z.; Jiao, N. Chem. Eur. J. 2015, 21, 16395.
(d) Wu, F.-F.; Zhao, Y.; Chen, W.-Z. Tetrahedron 2016, 72, 8004.
(e) Wang, F.; Wang, H.; Wang, Q.; Yu, S.-J.; Li, X.-W. Org. Lett. 2016, 18, 1306.
(g) Figg, T. M.; Park, S.; Park, J.; Chang, S.; Musaev, D. G. Organometallics 2014, 33, 4076.
(h) Liu, Y.; Xie, F.; Jia, A.-Q.; Li, X.-W. Chem. Commun. 2018, 54, 4345.
(i) Bera, S. S.; Sk, M. R.; Maji, M. S. Chem. Eur. J. 2019, 25, 1806.
(j) Yan, Q.-Q.; Xiao, T.-X.; Liu, Z.-X.; Zhang, Y.-H. Adv. Synth. Catal. 2016, 358, 2707.
(k) Cheng, H.-C.; Hernández, J. G.; Bolm, C. Adv. Synth. Catal. 2018, 360, 1.
(l) Borah, G.; Borah, P.; Patel, P. Org. Biomol. Chem. 2017, 15, 3854.
(m) Yu, X.-L.; Ma, Q.; Lv, S.-Y.; Li, J.; Zhang, C.; Hai, L.; Wang, Q.-T.; Wu, Y. Org. Chem. Front. 2017, 4, 2184.
(n) Shi, P.-F.; Wang, L.-L.; Chen, K.-H.; Wang, J.; Zhu, J. Org. Lett. 2017, 19, 2418.
(a) Villanueva, O.; Weldy, N. M.; Blakey, S. B.; MacBeth, C. E. Chem. Sci. 2015, 6, 6672.
(b) Harden, J. D.; Ruppel, J. V.; Gao, G.-Y.; Zhang, X. P. Chem. Commun. 2007, 4644.
(c) Pang, S.-F.; Yuan, H.-K.; Wu, Y.-J.; Shi, F. J. Mol. Catal. 2017, 31, 105(in Chinese).
(庞少峰, 袁航空, 吴亚娟, 石峰, 分子催化, 2017, 31, 105.)
(d) Wu, X.-S.; Yang, K.; Zhao, Y.; Sun, H.; Li, G.-G.; Ge, H.-B. Nat. Commun. 2015, 6, 6462.
(e) Lu, H.-J.; Li, C.-Q.; Jiang, H.-L.; Lizardi, C. L.; Zhang, X. P. Angew. Chem., Int. Ed. 2014, 53, 7028.
(f) Lu, H.-J.; Zhang, X. P. Chem. Soc. Rev. 2011, 40, 1899.
(g) Ragaini, F.; Penoni, A.; Gallo, E.; Tollari, S.; Gotti, C. L.; Lapadula, M.; Mangioni, E.; Cenini, S. Chem. Eur. J. 2003, 9, 249.
(h) Cenini, S.; Tollari, S.; Penoni, A.; Cereda C. J. Mol. Catal. 1999, 137, 135.
(i) Ruppel, J. V.; Kamble, R. M.; Zhang, X. P. Org. Lett. 2007, 9, 4889.
(j) Lu, H.-J.; Tao, J.; Jones, J. E.; Wojtas, L.; Zhang, X. P. Org. Lett. 2010, 12, 1248.
(k) Lu, H.-J.; Subbarayan, V.; Tao, J.; Zhang, X. P. Organometallics 2010, 29, 389.
(l) Cenini, S.; Gallo, E.; Penoni, A.; Ragainia, F.; Tollari, S. Chem. Commun. 2000, 2265.
(m) Tan, P.-W.; Mak, A. M.; Sullivan, M. B.; Dixon, D. J.; Seayad, J. Angew. Chem., Int. Ed. 2017, 56, 16550.
(n) Lu, H.-J.; Jiang, H.-L.; Wojtas, L.; Zhang, X. P. Angew. Chem., Int. Ed. 2010, 49, 10192.
Patel, P.; Chang, S. ACS Catal. 2015, 5, 853. doi: 10.1021/cs501860b
Wang, X.-M.; Lerchen, A.; Glorius, F. Org. Lett. 2016, 18, 2090. doi: 10.1021/acs.orglett.6b00716
Wang, H.; Lorion, M.-M.; Ackermann, L. Angew. Chem., Int. Ed. 2016, 55, 10386. doi: 10.1002/anie.201603260
Huang, J.-p.; Huang, Y.-F.; Wang, T.; Huang, Q.; Wang, Z.-H.; Chen, Z.-Y. Org. Lett. 2017, 19, 1128. doi: 10.1021/acs.orglett.7b00120
Du, C.; Li, P.-X.; Zhu, X.-J.; Han, J.-N.; Niu, J.-L.; Song, M.-P. ACS Catal. 2017, 7, 2810. doi: 10.1021/acscatal.7b00262
Sauermann, N.; Mei, R.-H.; Ackermann, L. Angew. Chem., Int. Ed. 2018, 57, 5090. doi: 10.1002/anie.201802206
(a) Parry, J. B.; Fu, N.; Lin, S. Synlett 2018, 29, 257.
(b) Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230.
(c) Feng, R.; Smith, J. A.; Moeller, K. D. Acc. Chem. Res. 2017, 50, 2346.
(d) Jiao, K.-J.; Zhao, C.-Q.; Fang, P.; Mei, T.-S. Tetrahedron Lett. 2017, 58, 797.
(e) Hou, Z.-W.; Mao, Z.-Y.; Xu, H.-C. Synlett 2017, 28, 1867.
Sun, B.; Yoshino, T.; Matsunaga, S.; Kanai, M. Adv. Synth. Catal. 2014, 356, 1491. doi: 10.1002/adsc.201301110
Li, W.; Weng, L.; Jin, G. Inorg. Chem. Commun. 2004, 7, 1174. doi: 10.1016/j.inoche.2004.09.005
Sun, B.; Yoshino, T.; Matsunaga, S.; Kanai, M. Chem. Commun. 2015, 51, 4659. doi: 10.1039/C4CC10284C
Gao, X.-L.; Wang, P.; Zeng, L.; Tang, S.; Lei, A.-W. J. Am. Chem. Soc. 2018, 140, 4195. doi: 10.1021/jacs.7b13049
Wang, S.-B.; Gu, Q.; You, S.-L. J. Catal. 2018, 361, 393. doi: 10.1016/j.jcat.2018.03.007
Huang, D.-Y.; Yao, Q.-J.; Zhang, S.; Xu, X.-T.; Zhang, K.; Shi, B.-F. Org. Lett. 2019, 21, 951. doi: 10.1021/acs.orglett.8b03938
Lu, H.-J.; Hu, Y.; Jiang, H.-L.; Wojtas, L.; Zhang, X. P. Org. Lett. 2012, 14, 5158. doi: 10.1021/ol302511f
Luo, Y.-R. In Comprehensive Handbook of Chemical Bond Energies, Vol. 3, Ed.: Luo, Y.-R., Taylor & Francis Group, Boca Raton, 2007, p. 19.
Barsu, N.; Rahman, M. A.; Sen, M.; Sundararaju B. Chem. Eur. J. 2016, 22, 9135. doi: 10.1002/chem.201601597
Lu, H.-J.; Lang, K.; Jiang, H.-L.; Wojtas, L.; Zhang, X. P. Chem. Sci. 2016, 7, 6934. doi: 10.1039/C6SC02231F
Li, C.-Q.; Lang, K.; Lu, H.-J.; Hu, Y.; Cui, X.; Wojtas, L.; Zhang, X. P. Angew. Chem., Int. Ed. 2018, 57, 16837. doi: 10.1002/anie.201808923
Fukagawa, S.; Kato, Y.; Tanaka, R.; Kojima, M.; Yoshino, T.; Matsunaga. S. Angew. Chem., Int. Ed. 2019, 58, 1153. doi: 10.1002/anie.201812215

图式 2 钴催化芳烃和氨基甲酸酯C—H胺化反应
Scheme 2 Cobalt-catalyzed aromatic and carbamate C—H amination

图式 3 钴催化芳香烃和二噁唑的胺化反应
Scheme 3 Amination of aromatic hydrocarbons and dioxazole catalyzed by Cobalt

图式 8 钴催化8-氨基喹啉导向C—H胺化
Scheme 8 Cobalt-catalyzed 8-aminoquinoline-directed C—H amination

图式 10 钴催化电氧化苯甲酰胺C—H胺化
Scheme 10 Cobalt-catalyzed electrooxidation of benzamide C—H amination

图式 11 钴催化吲哚和磺酰叠氮化物的胺化反应
Scheme 11 Amination of indole and sulfonyl azide catalyzed by cobalt

图式 12 钴催化吲哚和磷酰基叠氮化物的胺化反应
Scheme 12 Amination of indole and phosphoryl azide catalyzed by cobalt

图式 13 钴催化电氧化烷基胺C—H胺化
Scheme 13 Cobalt-catalyzed electrooxidation of alkylamine C—H amination

图式 21 钴催化氨磺酰叠氮化物分子内C—H胺化反应
Scheme 21 Cobalt-catalyzed C—H amination in the molecule of ammoniasulfonyl azide

图式 22 钴(Ⅱ)金属自由基催化1, 5-C(sp3)—H胺化反应
Scheme 22 Cobalt (Ⅱ) metal free radical catalyzes the 1, 5- C(sp3)—H amination reaction

图式 23 钴催化氨基磺酰叠氮化物分子内1, 6-C(sp3)—H胺化反应
Scheme 23 Cobalt-catalyzed intramolecular 1, 6-C(sp3)—H amination of aminosulfonyl azide

图式 24 非手性钴(Ⅲ)/手性羧酸杂化体系催化胺化反应
Scheme 24 Catalytic amination reaction catalyzed by achiral cobalt(Ⅲ)/chiral carboxylic acid hybrid system

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:













