Scheme 1.
Preparation of aryl ketones
Palladium-Catalyzed Regioselective ortho-Acylation of Azoxybenzenes under Aqueous Conditions
Xiaopei Chen , Zhiwei Ma , Chuanchuan Wang , Juntao Liu , Jinsong Wu

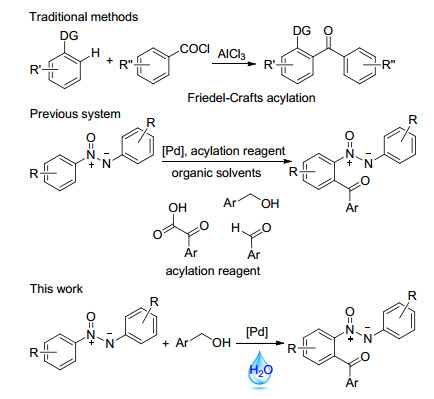
Aryl ketones are important structural motifs in dyes, fragrances, fine chemicals, pharmaceuticals and agrochemicals.[1] Conventionally, building such structure involves Friedel-Crafts acylation and oxidation of secondary alcohols by various oxidants.[2] Although it is effective as documented in almost all organic chemistry textbooks, these processes suffer from some drawbacks, such as the formation of waste materials due to the use of excessive Lewis acids and oxidants (Scheme 1). The coupling of arenes with aliphatic olefins and carbon monoxide using ruthenium as a catalyst has provided an alternative pathway.[3] The initial example developed by Moore and co-workers[4] involves Ru3(CO)12-catalyzed acylation of pyridine with CO and olefins. Recently, palladium-catalyzed acylation of C—H bonds has been one of the most practical, atom- and step-economical methods in synthetic chemistry. In 2009, Cheng et al.[5] first described palladium-catalyzed ortho-acylation reaction of arenes containing a pyridyl as a directing group with aryl aldehydes. Later, palladium-catalyzed C(sp2)—H bond acylation of 2-arylpyridines, [5, 6] oximes, [7] amides, [8] acetanilides, [9] indolizines, [10] azobenzenes[11] and benzothiazoles[12] using aldehydes, alcohols, α-oxocarboxylic acids, carboxylic acids, α-diketones, toluene derivatives and amines as acylation sources has been subsequently developed.
Azoxy compounds are important materials and useful intermediates in electronic devices due to their liquid crystalline properties. They also have wide applications in the fields of pharmaceuticals, analytic chemistry, functional materials, organic synthesis, and the dye industry.[13] In view of their importance, a number of methods have been established to build a structural skeleton.[14] Recently, Wang, Sun and Cui group[15] realized ortho-acylation of azoxybenzenes using aldehydes, α-oxocarboxylic acids and alcohols (Scheme 1). However, most of these processes are performed in organic solvents. From the perspective of green chemistry, water is an ideal chemical solvent, because it has natural, cheap, non-toxic, environmental characteristics.[16] In the past few years, acylation reactions in water have occasionally been reported.[17] Herein, we independently report a facile and efficient protocol to acylated azoxybenzenes via palladium-catalyzed C—H bond activation using alcohols as acylation reagents under aqueous conditions (Scheme 1).
Our initial investigation was focused on the acylation of azoxybenzene (1a) with benzylalcohol (2a). The results were summarized in Table 1. The desired product 3a was obtained in an isolated yield of 15% in water using 10 mol% PdCl2 as catalyst and 70% tert-butyl hydroperoxide (TBHP) in water as oxidant (Entry 1, Table 1). To improve the solubility of the reactants in water, 5 mol% sodium dodecyl sulfate (SDS) was added to the system. Pd(OAc)2 provided a higher yield compared to PdCl2 and Pd(TFA)2 (Entry 4 vs. Entries 2, 3, Table 1). Subsequently, various oxidants were tested in the presence of Pd(OAc)2 under air atmosphere (Entries 5~11). The yields could not be improved by using di-tert-butylperoxide (DTBP) and AgOAc (Entries 5, 6). Only a trace amount of the desired product 3a were observed when K2S2O8 and (NH4)2S2O8 as the oxidants (Entries 7, 8). benzoquinone (BQ), 2, 3-dichloro- 5, 6-dicyano-1, 4-benzoquinone (DDQ) and H2O2 were found to be ineffective for this process (Entries 9~11). Other phase-transfer catalysts (PTC) showed that SDS is superior to other phase-transfer catalysts such as tetrabutylammonium bromide (TBAB), 18-crown-6, or tween 80 (Entries 12~14). Meanwhile, decreasing the temperature resulted in a slightly decrease in the yield (Entry 15). When the reaction was carried out under nitrogen atmosphere, the desired product 3a was also obtained in 75% yield (Entry 16), which indicated that oxygen molecule was tolerated. After surveying a variety of catalysts, oxidants, PTC and reaction temperatures, the optimized reaction conditions were identified as follows: 10 mol% Pd(OAc)2 as catalyst, TBHP as oxidant, SDS as the phase- transfer catalyst, and H2O as solvent, at 100 ℃ under air atmosphere for 24 h.
 下载:
导出CSV
下载:
导出CSV
 | ||||
| Entrya | Catalyst | Oxidant | PTC | Yield b/% |
| 1 | PdCl2 | TBHP | 15 | |
| 2 | PdCl2 | TBHP | SDS | 35 |
| 3 | Pd(TFA)2 | TBHP | SDS | 68 |
| 4 | Pd(OAc)2 | TBHP | SDS | 80 |
| 5 | Pd(OAc)2 | DBHP | SDS | 20 |
| 6 | Pd(OAc)2 | AgOAc | SDS | 15 |
| 7 | Pd(OAc)2 | K2S2O8 | SDS | Trace |
| 8 | Pd(OAc)2 | (NH4)2S2O8 | SDS | Trace |
| 9 | Pd(OAc)2 | BQ | SDS | NDe |
| 10 | Pd(OAc)2 | DDQ | SDS | NDe |
| 11 | Pd(OAc)2 | H2O2 | SDS | NDe |
| 12 | Pd(OAc)2 | TBHP | TBAB | 20 |
| 13 | Pd(OAc)2 | TBHP | 18-Crown-6 | 42 |
| 14 | Pd(OAc)2 | TBHP | Tween 80 | 10 |
| 15c | Pd(OAc)2 | TBHP | SDS | 65 |
| 16d | Pd(OAc)2 | TBHP | SDS | 75 |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), catalyst (10 mol%), oxidant (4.5 equiv.), PTC (5 mol%) and water (2.0 mL), under air, 100 ℃, 24 h. b Isolated yield. c 80 ℃. d Nitrogen protection.e ND is not detected. | ||||
With the optimal reaction conditions in hand, the generality and the scope of the substrates for this transformation were tested(Table 2, 3a~3m). A wide range of primary alcohols were firstly evaluated in reaction with azoxybenzene (1a). In general, the reactions of primary alcohols with electron-donating groups (such as CH3, OCH3) or weak electron-withdrawing groups (such as F, Cl, Br) on the aromatic ring afforded the desired products in 55%~78% yields (3b~3f). The presence of the strong electron-
withdrawing group, such as (4-nitrophenyl)methanol, restrained the reaction, providing the product in only 32% yield (3g). Steric hindrance on the aryl group of primary alcohols had no significant effect, furnishing the corresponding products in moderate yields (3h~3l). Notably, when azoxybenzene (1a) was coupled with (3-nitro-phenyl)methanol (2l) under the optimized reaction conditions, a satisfactory yield of the corresponding product 3l was achieved. Disubstituted benzyl alcohols 3i provided the desired products in 63% yield. Aliphatic alcohol exhibited low efficiency in this transformation. For example, the n-butyl alcohol delivered 34% yield of 3m.
Heteroarylaldehydes, such as 2-pyridinecarboxaldehyde, 2-furanaldehyde and 2-thiophenecarboxaldehyde, were not suitable substrates. To expand the scope of this method, some typical disubstituted azoxybenzenes were examined. The results demonstrated that azoxy derivatives with electron-donating groups gave better yields than those with electron-withdrawing groups. For instance, the reactions of 4, 4'-dimethylazoxybenzene, 4, 4'-diisopropylazoxybenzene 4, 4'-dimethoxyazoxybenzene, 3, 3'-dimethylazoxybenzene, and 2, 2'-dimethylazoxybenzene provided the corresponding products in 68%~73% yields (3n~3r). The electron-withdrawing group in the substrate, such as F and Br, provided 3s and 3t in 32% and 33% yields.
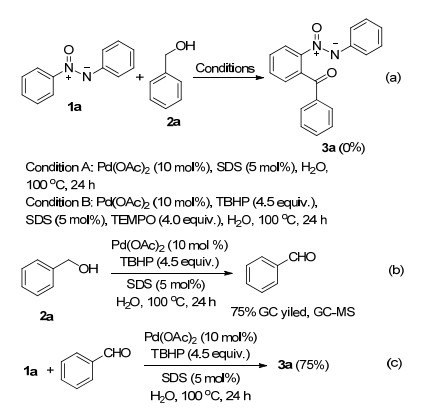
To clarify the reaction mechanism, some control experiments were carried out (Scheme 2). When the palladium-catalyzed acylation of 1a with 2a was carried out in the absence of TBHP (Scheme 2a, Condition A), no acylated product 3a was detected. Moreover, addition of a radical scavenger 2, 2, 6, 6-tetramethylpiperidyl-1-oxyl (TEMPO) to the reaction mixture under the standard reaction conditions made this acylation suppressed, suggesting the possibility of a radical process (Scheme 2a, Condition B). Furthermore, it was found that benzaldehyde was formed under the reaction conditions without the azoxybenzene (Scheme 2b). Then, the reaction between azoxybenzene (1a) and benzaldehyde were examined, and the product 3a was provided in 75% yield (Scheme 2c).
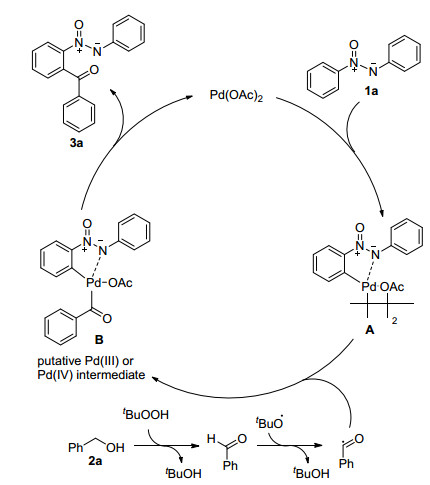
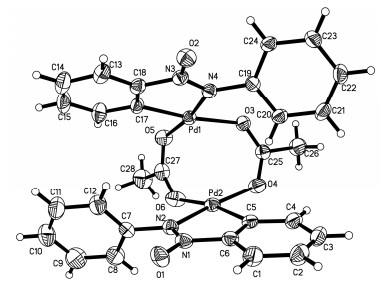
Based on the results obtained and the literature, [15, 17, 18] a tentative reaction mechanism for the palladium-catalyzed ortho-C—H acylation of azoxybenzene (1a) was proposed and depicted in Scheme 3. The palladium catalyst reacted with azoxybenzene by chelation-directed C—H activation to generate complex A (Figure 1). Meanwhile, with the effect of TBHP, the benzaldehyde, which was produced from the oxidation of benzylalcohol (2a), generated an acyl radical. Then, the complex A would react with the acyl radical, affording the oxidative addition product as the dimeric PdIII or reactive PdIV species B.[19] Finally, the reductive elimination of intermediate B afforded coupling product 3a and regenerated Pd(II) for next cycle.
In summary, a simple, efficient and environmentally benign protocol for the synthesis of acylated azoxybenzenes via Pd-catalyzed C—H regioselective functionalization has been developed. The method is characterized by its wide substrate scope, high regioselectivity, mild reaction conditions, and readily available starting materials. Further investigations to expand the substrate scope and application of such chemistry in organic synthesis are underway.
Melting points were measured on a microscopic apparatus and were uncorrected. 1H NMR spectra were recorded on a 400 MHz spectrometer in deuterated chloroform. The chemical shifts (δ) are reported relative to tetramethylsilane. 13C NMR spectra were recorded using a 100 MHz spectrometer. The chemical shifts are reported relative to residual CHCl3 (δC=77.00). High-resolution mass spectrometry (HRMS) was performed on a Q-TOF spectrometer with micromass MS software using electrospray ionization (ESI). X-ray analysis was performed with a single-crystal X-ray diffractometer. Unless otherwise noted, all of the reagents were purchased from commercial suppliers and used without purification.
All of the azoxybenzenes were prepared from arylamines, according to the literature.[20] H2O2 (30%, 0.92 mL, 9.00 mmol) was added to a solution of arylamine (0.27 mL, 3.00 mmol) and SeO2 (33.3 mg, 0.30 mmol) in MeOH (10 mL). The reaction mixture was stirred at room temperature (r.t.) until complete consumption of the starting material, which was followed by thin-layer chromatography (TLC) (20 h). The solvent was evaporated under vacuum. The residue was partitioned between CH2Cl2 (20 mL) and H2O (20 mL). The organic layer was separated, and the aqueous phase was extracted with CH2Cl2 (20 mL×2). The combined organic phase were dried (MgSO4) and concentrated in vacuo. The crude product was purified by column chromatography on silica gel (CH2Cl2-pentane, V:V=1:10) to afford the desired azoxybenzene derivatives.
Azoxybenzene (1a, 39.6 mg, 0.20 mmol), benzyl alcohol (2a, 63 μL, 0.40 mmol), Pd(OAc)2 (4.48 mg, 0.02 mmol), TBHP (126 uL, 70%, 0.9 mmol), SDS (2.88 mg, 0.01 mmol) and H2O (2.0 mL) were mixed in a dry reaction tube. The mixture was stirred at 100 ℃ under air for 24 h. When the reaction was completed, the crude mixture was cooled to room temperature. The mixture was extracted with ethyl acetate three times. The filtrate was concentrated in vacuum and the resulting residue was purified by preparative thin layer chromatography (silica gel, ethyl acetate/petroleum ether, V:V=1:10) to give the desired product 1-(2-benzoylphenyl)-2-phenyldiazene 1-oxide (3a)[15] as a yellow viscous liquid (48.3 mg, 80%). 1H NMR (400 MHz, CDCl3) δ: 8.24~8.22 (m, 1H), 7.76 (d, J=7.4 Hz, 2H), 7.70~7.64 (m, 4H), 7.53~7.51 (m, 1H), 7.46 (t, J=6.2 Hz, 1H), 7.36 (t, J=6.1 Hz, 2H), 7.29 (d, J=3.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 194.0, 147.2, 143.1, 136.9, 134.8, 133.1, 131.4, 130.5, 129.9, 128.9, 128.8, 128.5, 125.0, 123.4; HRMS (ESI) calcd for C19H15N2O2 [M+H]+ 303.1128, found 303.1133.
1-(2-(4-Methylbenzoyl)phenyl)-2-phenyldiazene 1-oxide (3b):[15] yellow liquid (37.9 mg, 60%). 1H NMR (400 MHz, CDCl3) δ: 8.22~8.21 (m, 1H), 7.73~7.71 (m, 2H), 7.65~7.63 (m, 4H), 7.51~7.48 (m, 1H), 7.32~7.27 (m, 3H), 7.16 7.16 (d, J=7.9 Hz, 2H), 2.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 194.1, 144.3, 143.6, 135.4, 134.8, 131.6, 130.7, 130.2, 129.6, 129.4, 128.8, 125.4, 123.7, 22.0; HRMS (ESI) calcd for C20H17N2O2 [M+H]+ 317.1285, found 317.1289.
1-(2-(4-Methoxybenzoyl)phenyl)-2-phenyldiazene 1-oxide (3c):[15] yellow liquid (36.5 mg, 55%). 1H NMR (400 MHz, CDCl3) δ: 8.22~8.20 (m, 1H), 7.75 (d, J=8.4 Hz, 3H), 7.63~7.61 (m, 2H), 7.49~7.47 (m, 1H), 7.31~7.27 (m, 3H), 6.84 (d, J=8.8 Hz, 2H), 3.79 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 193.2, 163.9, 143.6, 135.5, 131.6, 131.6, 130.6, 130.4, 130.2, 129.1, 128.9, 125.4, 123.7, 114.20, 55.8; HRMS (ESI) calcd for C20H17N2O3 [M+H]+ 333.1234, found 333.1238.
1-(2-(4-Fluorobenzoyl)phenyl)-2-phenyldiazene 1-oxide (3d):[15] yellow liquid (46.1 mg, 72%). 1H NMR (400 MHz, CDCl3) δ: 8.24~8.22 (m, 1H), 7.77~7.73 (m, 4H), 7.66 (t, J=9.3 Hz, 2H), 7.50~7.48 (m, 1H), 7.31~7.29 (m, 3H), 7.03 (t, J=10.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 192.9, 167.3, 164.7, 143.5, 134.8, 133.8, 131.8, 131.8, 131.7, 130.5, 129.1, 128.9, 125.4, 123.8, 116.2, 115.9; HRMS (ESI) calcd for C19H14FN2O2 [M+H]+ 321.1034, found 321.1037.
1-(2-(4-Chlorobenzoyl)phenyl)-2-phenyldiazene 1-oxide (3e):[15] yellow liquid (52.4 mg, 78%), m.p. 64.6~65.2 ℃. 1H NMR (400 MHz, CDCl3) δ: 8.26~8.23 (m, 1H), 7.75 (d, J=8.0 Hz, 2H), 7.70 (d, J=8.4 Hz, 2H), 7.65 (t, J=6.6 Hz, 3H), 7.51~7.49 (m, 1H), 7.34~7.31 (m, 5H); 13C NMR (100 MHz, CDCl3) δ: 193.1, 143.4, 139.8, 135.7, 134.6, 131.8, 131.1, 130.5, 130.5, 129.2, 129.1, 129.0, 125.4, 123.8; HRMS (ESI) calcd for C19H14ClN2O2 [M+H]+, 337.0738, found 337.0743.
1-(2-(4-Bromobenzoyl)phenyl)-2-phenyldiazene 1-oxide (3f):[15] yellow liquid (57.0 mg, 75%). m.p. 62.5~63.4 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.25~8.23 (m, 1H), 7.74 (d, J=7.4 Hz, 2H), 7.66 (t, J=4.7 Hz, 2H), 7.61 (d, J=8.3 Hz, 2H), 7.49 (d, J=8.0 Hz, 3H), 7.35~7.30 (m, 3H). 13C NMR (100 MHz, CDCl3) δ: 193.3, 143.4, 136.2, 134.6, 132.2, 131.8, 131.1, 130.6, 130.5, 129.1, 129.0, 128.6, 125.4, 123.8; HRMS (ESI) calcd for C19H14BrN2O2 [M+H]+ 381.0233, found 381.0236.
1-(2-(4-Nitrobenzoyl)phenyl)-2-phenyldiazene 1-oxide (3g):[15] yellow liquid (22.2 mg, 32%). 1H NMR (400 MHz, CDCl3) δ: 8.30~8.28 (m, 4H), 8.18 (d, J=8.6 Hz, 2H), 7.89 (d, J=8.6 Hz, 2H), 7.77~7.70 (m, 4H), 7.56~7.54 (m, 1H), 7.33~7.31 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: 190.4, 150.4, 143.2, 142.0, 133.9, 132.1, 131.6, 130.9, 129.7, 129.2, 125.4, 124.1, 123.8; HRMS (ESI) calcd for C19H14N3O4 [M+H]+ 348.0979, found 348.0981.
1-(2-(2-Methylbenzoyl)phenyl)-2-phenyldiazene 1-oxide (3h):[15] yellow liquid (36.7 mg, 58%). 1H NMR (400 MHz, CDCl3) δ: 8.05~8.03 (m, 1H), 7.64~7.60 (m, 3H), 7.57~7.54 (m, 2H), 7.32~7.25 (m, 5H), 7.14~7.07 (m, 2H), 2.53 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 195.7, 148.3, 143.7, 140.2, 136.6, 136.1, 132.2, 131.1, 131.4, 131.3, 130.7, 130.0, 128.7, 125.7, 123.8, 21.5; HRMS (ESI) calcd for C20H17N2O2 [M+H]+ 317.1285; found 317.1289.
1-(2-(2, 4-Dichlorobenzoyl)phenyl)-2-phenyldiazene 1-oxide (3i): yellow liquid (46.6 mg, 63%). 1H NMR (400 MHz, CDCl3) δ: 8.06 (d, J=7.4 Hz, 1H), 7.79~7.77 (m, 2H), 7.68~7.63 (m, 3H), 7.48 (d, J=8.3 Hz, 1H), 7.37~7.31 (m, 4H), 7.16~7.14 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 191.4, 143.6, 138.6, 134.8, 134.7, 134.6, 132.5, 131.9, 131.3, 130.6, 129.9, 128.9, 127.2, 125.5, 123.9; HRMS (ESI) calcd for C19H13Cl2N2O2 [M+H]+ 371.0349, found 371.0352.
1-(2-(3-Methoxybenzoyl)phenyl)-2-phenyldiazene 1-oxide (3j): yellow liquid (43.1 mg, 65%). 1H NMR (400 MHz, CDCl3) δ: 8.24~8.20 (m, 1H), 7.73~7.71 (m, 2H), 7.66~7.63 (m, 2H), 7.53~7.51 (m, 1H), 7.38 (s, 1H), 7.33~7.31 (m, 3H), 7.25~7.20 (m, 2H), 7.04~6.99 (m, 1H), 3.75 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 194.1, 160.1, 143.5, 138.7, 135.1, 131.7, 130.9, 129.8, 129.2, 128.8, 125.3, 123.7, 122.1, 120.2, 112.9, 55.8; HRMS (ESI) calcd for C20H17N2O3 [M+H]+ 333.1234, found 333.1238.
1-(2-(3-Chlorobenzoyl)phenyl)-2-phenyldiazene 1-oxide (3k):[15] yellow liquid (39.6 mg, 59%). 1H NMR (400 MHz, CDCl3) δ: 8.26~8.24 (m, 1H), 7.77~7.74 (m, 3H), 7.68~7.66 (m, 2H), 7.58 (d, J=7.8 Hz, 1H), 7.51~7.49 (m, 1H), 7.42 (d, J=7.9 Hz, 1H), 7.35~7.25 (m, 4H); 13C NMR (100 MHz, CDCl3) δ: 193.0, 143.4, 139.0, 135.2, 134.5, 133.3, 131.8, 131.2, 130.5, 130.2, 129.1, 129.0, 129.0, 127.2, 125.4, 123.8; HRMS (ESI) calcd for C19H14ClN2O2 [M+H]+ 337.0738, found 337.0743.
1-(2-(3-Nitrobenzoyl)phenyl)-2-phenyldiazene 1-oxide (3l): yellow liquid (38.8 mg, 56%). 1H NMR (400 MHz, CDCl3) δ: 8.57~8.56 (m, 1H), 8.30~8.27 (m, 2H), 8.03 (d, J=7.7 Hz, 1H), 7.78~7.75 (m, 2H), 7.73~7.69 (m, 2H), 7.57~7.52 (m, 2H), 7.34~7.30 (m, 3H); 13C NMR (100 MHz, CDCl3) δ: 192.0, 148.7, 147.5, 143.3, 138.9, 134.3, 133.7, 132.1, 131.6, 130.8, 130.1, 129.1, 129.0, 127.5, 125.5, 124.0, 123.7; HRMS (ESI) calcd for C19H14N3O4 [M+H]+ 348.0979, found 348.0982.
1-(2-Butyrylphenyl)-2-phenyldiazene 1-oxide (3m):[15] yellow liquid (18.1 mg, 34%). 1H NMR (400 MHz, CDCl3) δ: 8.10 (d, J=8.1 Hz, 3H), 7.58~7.56 (m, 2H), 7.49~7.46 (m, 2H), 7.43~7.40 (m, 2H), 2.73 (t, J=7.0 Hz, 2H), 1.71 (q, J=10.4, 3.0 Hz, 2H), 0.92 (t, J=7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ: 203.4, 171.5, 143.9, 137.2, 131.6, 130.6, 130.6, 129.1, 127.8, 125.8, 123.9, 45.3, 18.1, 14.5; HRMS (ESI) calcd for C16H17N2O2 [M+H]+ 269.1285, found 269.1288.
1-(2-Benzoyl-4-methylphenyl)-2-(p-tolyl)diazene 1-oxide (3n):[15] yellow liquid (46.9 mg, 71%). 1H NMR (400 MHz, CDCl3) δ: 8.13 (d, J=8.4 Hz, 1H), 7.76 (d, J=7.4 Hz, 2H), 7.65 (d, J=8.4 Hz, 2H), 7.47~7.41 (m, 2H), 7.34 (t, J=7.7 Hz, 2H), 7.29 (s, 1H), 7.08 (d, J=8.3 Hz, 2H), 2.47 (s, 3H), 2.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 194.7, 145.3, 142.4, 141.3, 137.4, 135.0, 133.3, 131.3, 129.5, 129.4, 129.1, 128.8, 125.5, 123.5, 21.9, 21.6; HRMS (ESI) calcd for C21H19N2O2 [M+H]+ 331.1441, found 331.1445.
1-(2-Benzoyl-4-isopropylphenyl)-2-(4-isopropyl-phenyl)diazene 1-oxide (3o):[15] yellow liquid (52.5 mg, 68%). 1H NMR (400 MHz, CDCl3) δ: 8.16 (d, J=8.5 Hz, 1H), 7.77 (d, J=7.5 Hz, 2H), 7.67 (d, J=8.5 Hz, 2H), 7.50~7.44 (m, 2H), 7.38~7.33 (m, 3H), 7.13 (d, J=8.5 Hz, 2H), 3.08~2.97 (m, 1H), 2.91~2.81 (m, 1H), 1.30 (d, J=6.8 Hz, 6H), 1.19 (d, J=6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ: 194.8, 153.1, 151.5, 145.4, 141.6, 137.5, 135.1, 133.3, 129.2, 128.8, 128.8, 127.1, 126.8, 125.5, 123.6, 34.4, 34.4, 24.0; HRMS (ESI) calcd for C25H27N2O2 [M+H]+ 387.2067, found 387.2072.
1-(2-Benzoyl-4-methoxyphenyl)-2-(4-methoxyphenyl)-diazene 1-oxide (3p):[15] yellow liquid (50.7 mg, 70%). 1H NMR (400 MHz, CDCl3) δ: 8.11 (d, J=9.1 Hz, 1H), 7.75 (d, J=9.0 Hz, 2H), 7.69~7.67 (m, 2H), 7.36 (t, J=8.6 Hz, 1H), 7.27 (d, J=2.7 Hz, 1H), 7.27~7.16 (m, 1H), 7.02~6.99 (m, 1H), 6.84 (d, J=2.6 Hz, 1H), 6.68 (d, J=9.0 Hz, 2H), 3.78 (s, 3H), 3.69 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 194.2, 161.9, 160.8, 137.5, 137.3, 133.3, 129.1, 128.8, 127.9, 127.7, 125.2, 116.0, 113.9, 56.3, 55.8; HRMS (ESI) calcd for C21H19N2O4 [M+H]+ 363.1339, found 363.1344.
1-(2-Benzoyl-5-methylphenyl)-2-(m-tolyl)diazene 1-oxide (3q):[15] yellow liquid (48.2 mg, 73%). 1H NMR (400 MHz, CDCl3) δ: 7.98 (s, 1H), 7.75 (d, J=7.6 Hz, 2H), 7.47~7.40 (m, 5H), 7.34 (t, J=7.6 Hz, 2H), 7.16 (t, J=8.7 Hz, 1H), 7.07 (d, J=7.5 Hz, 1H), 2.52 (s, 3H), 2.27 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 194.5, 147.8, 143.6, 141.7, 138.5, 137.6, 133.2, 132.2, 130.9, 129.3, 129.1, 128.8, 128.6, 125.6, 124.0, 122.3, 21.7, 21.6; HRMS (ESI) calcd for C21H19N2O2 [M+H]+ 331.1441, found 331.1449.
1-(2-Benzoyl-6-methylphenyl)-2-(o-tolyl)diazene 1-oxide (3r):[15] yellow liquid (46.1 mg, 70%). 1H NMR (400 MHz, CDCl3) δ: 7.79 (d, J=7.5 Hz, 2H), 7.64 (d, J=8.0 Hz, 1H), 7.53~7.44 (m, 3H), 7.42~7.33 (m, 3H), 7.20~7.13 (m, 2H), 7.07 (t, J=4.6 Hz, 1H), 2.54 (s, 3H), 2.22 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 194.5, 148.2, 142.4, 136.9, 135.4, 134.4, 134.2, 133.7, 132.3, 130.9, 130.1, 129.5, 129.3, 128.7, 127.6, 126.0, 121.5, 18.7, 18.4; HRMS (ESI) calcd for C21H19N2O2 [M+H]+ 331.1441, found 331.1447.
1-(2-Benzoyl-4-fluorophenyl)-2-(4-fluorophenyl)-diazene 1-oxide (3s):[15] yellow liquid (21.6 mg, 32%). 1H NMR (400 MHz, CDCl3) δ: 8.27 (dd, J=8.1, 3.4 Hz, 1H), 7.81~7.75 (m, 4H), 7.50 (d, J=7.27 Hz, 1H), 7.40~7.31 (m, 3H), 7.20 (dd, J=5.2, 2.5 Hz, 1H), 6.97 (t, J=8.9 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ: 192.7, 164.1 (d, JC-F=254.3 Hz), 163.1 (d, JC-F=251.7 Hz), 143.4, 139.8 (d, JC-F=3.1 Hz), 137.4 (d, JC-F=3.5 Hz), 136.6, 133.8, 129.1 (d, JC-F=11.6 Hz), 127.9 (d, JC-F=8.5 Hz), 126.1 (d, JC-F=8.9 Hz), 117.7 (d, JC-F=23.0 Hz), 116.3 (d, JC-F=24.6 Hz), 115.9 (d, JC-F=22.3 Hz); HRMS (ESI) calcd for C19H13F2N2O2 [M+H]+ 339.0940, found 339.0943.
1-(2-Benzoyl-5-bromophenyl)-2-(3-bromophenyl)-diazene 1-oxide (3t): yellow liquid (30.2 mg, 33%). 1H NMR (400 MHz, CDCl3) δ: 8.37 (d, J=1.69 Hz, 1H), 7.88 (d, J=1.82 Hz, 1H), 7.82~7.80 (m, 1H), 7.75 (d, J=7.8 Hz, 2H), 7.57~7.55 (m, 1H), 7.51 (d, J=7.4 Hz, 1H), 7.43~7.40 (m, 4H), 7.19 (t, J=7.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ: 193.3, 144.2, 136.8, 135.0, 133.9, 133.4, 130.7, 130.6, 130.2, 129.1, 129.1, 127.9, 127.0, 124.6, 124.3, 122.5; HRMS (ESI) calcd for C19H13Br2N2O2 [M+H]+ 458.9338, found 458.9336.
Supporting Information The spectroscopic characterization of the products 3a~3t, X-ray crystallographic data of dimer Pd complex A and GC-MS spectra of Scheme 2c. The Supporting Information is available free of charge via the Internet at http://sioc-journal.cn/.
(a) Surburg, H.; Panten, J. Common Fragrance and Flavour Materials, Wiley Online Library, 2006.
(b) McGrath, N. A.; Brichacek, M.; Njardarson, J. T. J. Chem. Educ. 2010, 87, 1348.
(a) Olah, G. A. Friedel-Crafts Chemistry, Wiley, New York, 1973.
(b) Sartori, G.; Maggi, R. Chem. Rev. 2006, 106, 1077.
(c) Fernandez, M.; Tojo, G. In Oxidation of Alcohols to Aldehydes and Ketones: A Guideto Current Common Practice, Ed.: Tojo, E., Springer, New York, 2006.
(d) Sartori, G.; Maggi, R. Advances in Friedel-Crafts Acylation Reactions, CRC Press, Taylor & Francis Group, 2010.
(a) Moore, E. J.; Pretzer, W. R.; O'Connell, T. J.; Harris, J.; LaBounty, L.; Chou, L.; Grimmer, S. S. J. Am. Chem. Soc. 1992, 114, 5888.
(b) Chatani, N.; Fukuyama, T.; Kakiuchi, F.; Murai, S. J. Am. Chem. Soc. 1996, 118, 493.
(c) Fukuyama, T.; Chatani, N.; Kakiuchi, F.; Murai, S. J. Org. Chem. 1997, 62, 5647.
(d) Chatani, N.; Ie, Y.; Kakiuchi, F.; Murai, S. J. Org. Chem. 1997, 62, 2604.
(e) Ie, Y.; Chatani, N.; Ogo, T.; Marshall, D. R.; Fukuyama, T.; Kakiuchi, F.; Murai, S. J. Org. Chem. 2000, 65, 1475.
Moore, E. J.; Pretzer, W. R.; OConnell, T. J.; Harris, J.; LaBounty, L.; Chou, L.; Grimme, S. S. J. Am. Chem. Soc. 1992, 114, 5888. doi: 10.1021/ja00040a078
Jia, X. F.; Zhang, S. H.; Wang, W. H.; Luo, F.; Cheng, J. Org. Lett. 2009, 11, 3120. doi: 10.1021/ol900934g
(a) Xiao, F. X.; Shuai, Q.; Zhao, F.; Basle, O.; Deng, G. J.; Li, C. J. Org. Lett. 2011, 13, 1614.
(b) Xu, Z. P.; Xiang, B.; Sun, P. P. RSC Adv. 2013, 3, 1679.
(c) Khemnar, A. B.; Bhanage, B. M. Eur. J. Org. Chem. 2014, 6746.
(d) Kishore, R.; Kantam, M. L.; Yadav, J.; Sudhakar, M.; Laha, S.; Venugopal, A. J. Mol. Catal. A: Chem. 2013, 379, 213.
(e) Zhang, Q.; Yang, F.; Wu, Y. J. Chem. Commun. 2013, 49, 6837.
(f) Li, M. Z.; Ge, H. B. Org. Lett. 2010, 12, 3464.
(a) Han, S.; Sharma, S.; Park, J.; Kim, M.; Shin, Y.; Mishra, N. K.; Bae, J. J.; Kwak, J. H.; Jung, Y. H.; Kim, I. S. J. Org. Chem. 2014, 79, 275.
(b) Sharma, S.; Kim, M.; Park, J.; Kim, M.; Kwak, J. H.; Jung, Y. H.; Oh, J. S.; Lee, Y.; Kim, I. S. Eur. J. Org. Chem. 2013, 6656.
Wu, Y. N.; Feng, L. J.; Lu, X.; Kwong, F. Y.; Luo, H. B. Chem. Commun. 2014, 50, 15352. doi: 10.1039/C4CC07440H
(a) Weng, J. Q.; Yu, Z. Q.; Liu, X. H.; Zhang, G. F. Tetrahedron Lett. 2013, 54, 1205.
(b) Fang, P.; Li, M. Z.; Ge, H. B. J. Am. Chem. Soc. 2010, 132, 11898.
(c) Yin, Z. W.; Sun, P. P. J. Org. Chem. 2012, 77, 11339.
(d) Li, C. L.; Wang, L.; Li, P. H.; Zhou, W. Chem.-Eur. J. 2011, 17, 10208.
(e) Wu, Y. N.; Choy, P. Y.; Mao, F.; Kwong, F. Y. Chem. Commun. 2013, 49, 689.
Yang, Y. Z.; Chen, L.; Zhang, Z. G.; Zhang, Y. H. Org. Lett. 2011, 13, 1342. doi: 10.1021/ol200025k
(a) Song, H. Y.; Chen, D.; Pi, C.; Cui, X. L.; Wu, Y. J. J. Org. Chem. 2014, 79, 2955.
(b) Li, H. J.; Li, P. H.; Wang, L. Org. Lett. 2013, 15, 620.
(c) Li, H. J.; Li, P. H.; Tan, H.; Wang, L. Chem.-Eur. J. 2013, 19, 14432.
(d) Li, Z. Y.; Li, D. D.; Wang, G. W. J. Org. Chem. 2013, 78, 10414.
(e) Xiong, F.; Qian, C.; Lin, D. G.; Zeng, W.; Lu, X. X. Org. Lett. 2013, 15, 5444.
Zhao, J. C.; Fang, H.; Xie, C.; Han, J. L.; Li, G. G.; Pan, Y. Asian J. Org. Chem. 2013, 2, 1044. doi: 10.1002/ajoc.201300208
(a) Ikeda, T.; Tsu, O. Science 1995, 268, 1873.
(b) Kimura, K.; Suzuki, T.; Yokoyama, M. J. Phys. Chem. 1990, 94, 6090.
(c) Campbell, D.; Dix, L. R.; Rostron, P. Dyes Pigm. 1995, 29, 77.
(d) Huang, J. M.; Kuo, J. F.; Chen, C. Y. J. Appl. Polym. Sci. 1995, 55, 1217.
(e) Lee, H. K.; Kanazawa, A.; Shiono, T.; Ikeda, T.; Fujisawa, T.; Aizawa, M.; Lee, B. Chem. Mater. 1998, 10, 1402.
(f) Li, H. J.; Li, P. H.; Zhao, Q.; Wang, L. Chem. Commun. 2013, 49, 9170.
For selected examples, see: (a) Hou, Z.; Fujiware, Y.; Taniguchi, H. J. Org. Chem. 1988, 53, 3118.
(b) Sakai, N.; Fuji, K.; Nabeshima, S.; Ikeda, R.; Konakahara, T. Chem. Commun. 2010, 46, 3173.
(c) Wada, S.; Urano, M.; Suzuki, H. J. Org. Chem. 2002, 67, 8254.
(d) Wang, Y.; Cheng, G. L.; Cui, X. L. Chin. J. Org. Chem. 2012, 32, 2018 (in Chinese).
(王勇, 程国林, 崔秀灵, 有机化学, 2012, 32, 2018.)
(a) Sun, M.; Hou, L. K.; Chen, X. X.; Yang, X. J.; Sun, W.; Zang, Y. S. Adv. Synth. Catal. 2014, 356, 3789.
(b) Li, H. J.; Li, P. H.; Zhao, Q.; Wang, L. Chem. Commun. 2013, 49, 9170.
(c) Yi, M. L.; Cui, X. L.; Zhu, C. W.; Pi, C.; Zhu, W. M.; Wu, Y. J. Asian J. Org. Chem. 2015, 4, 38.
(d) Hou, L. K.; Chen, X. X.; Li, S.; Cai, S. X.; Zhao, Y. X.; Sun, M.; Yang, X. J. Org. Biomol. Chem., 2015, 13, 4160.
(a) Yang, J.; Fu, T.; Long, Y.; Zhou, X. G. Chin. J. Org. Chem. 2017, 37, 1111 (in Chinese).
(杨军, 付婷, 龙洋, 周向葛, 有机化学, 2017, 37, 1111.)
(b) Zhou, Z.; Duan, J. F.; Mu, X. J.; Xiao, S. Y. Chin. J. Org. Chem. 2018, 38, 585 (in Chinese).
(周曌, 段建凤, 穆小静, 肖尚友, 有机化学, 2018, 38, 585.)
(c) Qin, H. F.; Li, X. R. Chin. J. Org. Chem. 1992, 12, 309 (in Chinese).
(秦合法, 李萱荣, 有机化学, 1992, 12, 309.)
(a) Szabó, F.; Daru, J.; Simkó, D.; Nagy, T. Z.; Stirling, A.; Novák, Z. Adv. Synth. Catal. 2013, 355, 685.
(b) Szabó, F.; Simkó, D.; Novák, Z. RSC Adv. 2014, 4, 3883.
(c) Xiao, F. H.; Chen, S. Q.; Huang, H. W.; Deng, G. J. Eur. J. Org. Chem. 2015, 7919.
Zhang, D.; Cui, X. L.; Yang, F. F.; Q Zhang, Q. Q.; Zhu, Y.; Wu, Y. J. Org. Chem. Front. 2015, 2, 951. doi: 10.1039/C5QO00120J
(a) Rosewall, C. F.; Sibbald, P. A.; Liskin, D. V.; Michael, F. E. J. Am. Chem. Soc. 2009, 131, 9488.
(b) Xu, L. M.; Li, B. J.; Yang, Z.; Shi, Z. J. Chem. Soc. Rev. 2010, 39, 712.
(c) Sibbald, P. A.; Rosewall, C. F.; Swartz, R. D.; Michael, F. E. J. Am. Chem. Soc. 2009, 131, 15945.
(d) Powers, D. C.; Ritter, T. Nat. Chem. 2009, 1, 302.
(e) Powers, D. C.; Geibel, M. A. L.; Klein, J. E. M. N.; Ritter, T. J. Am. Chem. Soc. 2009, 131, 17050.
(f) Deprez, N. R.; Sanford, M. S. J. Am. Chem. Soc. 2009, 131, 11234.
(g) Racowski, J. M.; Dick, A. R.; Sanford, M. S. J. Am. Chem. Soc. 2009, 131, 10974.
Christin, G.; Beate, P.; Elisabeth, I.; Karola, R. B. Synthesis 2008, 1889.
Table 1. Optimization of the reaction conditionsa
| ||||
| Entrya | Catalyst | Oxidant | PTC | Yield b/% |
| 1 | PdCl2 | TBHP | 15 | |
| 2 | PdCl2 | TBHP | SDS | 35 |
| 3 | Pd(TFA)2 | TBHP | SDS | 68 |
| 4 | Pd(OAc)2 | TBHP | SDS | 80 |
| 5 | Pd(OAc)2 | DBHP | SDS | 20 |
| 6 | Pd(OAc)2 | AgOAc | SDS | 15 |
| 7 | Pd(OAc)2 | K2S2O8 | SDS | Trace |
| 8 | Pd(OAc)2 | (NH4)2S2O8 | SDS | Trace |
| 9 | Pd(OAc)2 | BQ | SDS | NDe |
| 10 | Pd(OAc)2 | DDQ | SDS | NDe |
| 11 | Pd(OAc)2 | H2O2 | SDS | NDe |
| 12 | Pd(OAc)2 | TBHP | TBAB | 20 |
| 13 | Pd(OAc)2 | TBHP | 18-Crown-6 | 42 |
| 14 | Pd(OAc)2 | TBHP | Tween 80 | 10 |
| 15c | Pd(OAc)2 | TBHP | SDS | 65 |
| 16d | Pd(OAc)2 | TBHP | SDS | 75 |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.6 mmol), catalyst (10 mol%), oxidant (4.5 equiv.), PTC (5 mol%) and water (2.0 mL), under air, 100 ℃, 24 h. b Isolated yield. c 80 ℃. d Nitrogen protection.e ND is not detected. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们