交叉脱氢偶联反应(CDC)直接通过两种不同的X—H键结合生成新的化学键, 过程中避免了反应底物的预先官能化, 具有优异的步骤经济性和原子经济性, 已成为化学合成的一种重要的方法[1 5 .其中, 雷爱文课题组在交叉脱氢偶联领域研究广泛, 相继报道了过渡金属催化[6 、自由基介导[7 的C—H官能化反应, 随后又从光化学、热化学和电化学三个角度对代表性反应进行概述[8 .传统交叉脱氢偶联反应通常使用铜[9 、铁[10 11 、钌[12 、钴[13 、铱[14 、锰[15 等过渡金属催化剂, 但是这类催化剂在催化反应的同时也表现出一系列缺点, 例如价格昂贵、反应条件严苛、有一定的毒性以及难以避免的金属残留等, 制约了其在有机合成中的应用[16 18 .因此, 无金属催化的交叉脱氢偶联反应受到人们的关注. 2015年, Antonchick课题组[19 对无金属催化下不同杂化类型C—C键的交叉脱氢偶联反应进行了归纳.随后, 王彬课题组[20 及Parvatkar课题组[21 也分别以溴试剂和分子碘为催化剂对相关交叉脱氢偶联反应进行了总结.
近年来, 可见光因其储量丰富、廉价易得及清洁安全等优点已被广泛应用于有机合成领域[22 30 .其中, 可见光诱导电子转移是实现X—H键直接官能化的有效途径之一.可见光诱导的交叉脱氢偶联反应无疑具有步骤和原子经济性高及反应条件绿色等优点, 因而引起研究者关注. 2018年, 吴骊珠课题组[31 总结了含钴双催化体系的光氧化还原交叉脱氢偶联反应.同年, 雷爱文课题组[32 33 综述了C—H/X—H的光诱导交叉脱氢偶联反应实例.然而, 对于可见光诱导的无过渡金属催化的交叉脱氢偶联反应, 目前尚无文献对其进行总结和讨论.鉴于此, 本文根据成键类型的不同, 结合近几年来文献报道的各种催化体系以及反应底物, 对可见光诱导的无过渡金属催化交叉脱氢偶联反应及其代表性反应机理进行了整理和归纳.
1.
C—C偶联反应
1.1
C(sp3 )—C(sp3 )偶联
2011年, Tan课题组[34 报道了可见光诱导叔胺的α -官能化反应.利用孟加拉玫瑰红(Rose Bengal)染料作光敏剂, 在室温条件下实现了N -芳基四氢异喹啉和硝基烷烃以及酮的交叉脱氢偶联反应(Eqs. 1, 2). Eq. 2中, N -芳基四氢异喹啉在吡咯烷/三氟乙酸(TFA)溶液中直接产生亚胺中间体, 比经典Mannich反应操作更简便.该方法利用价廉易得的有机染料作为催化剂, 具有反应条件温和、环境友好的优点.
同年, König等[35 以Eosin Y染料作光敏剂, 研究了可见光诱导下N -芳基四氢异喹啉和亲核试剂的C(sp3 )—H/C(sp3 )—H偶联反应(Eqs. 3, 4).与前者相比, 该体系具有催化剂用量低, 反应时间短等优点.
2012年, 吴骊珠等[36 扩展了N -芳基四氢异喹啉衍生物范围, 将其和硝基烷烃在波长大于450 nm的光照下反应, 以曙红Y的四丁基铵盐(TBA-Eosin Y)染料作光敏剂, 氧气作氧化剂, 得到了一系列交叉脱氢偶联产物(Eq. 5).该方法进一步减少了光敏剂用量, 同时也提高了光催化反应速率.
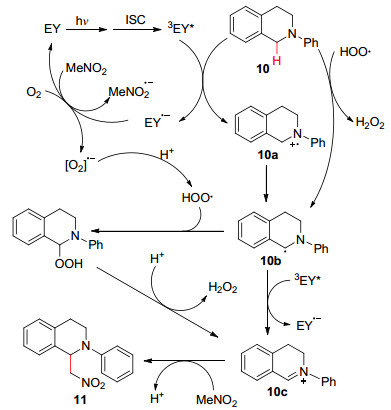
以N -苯基四氢异喹啉(10 )和硝基甲烷为例, 推测其反应机理如下:首先, TBA-Eosin Y在光照条件下被激发生成三线态激发态TBA-Eosin Y*.接着, 光诱导电子从N -苯基四氢异喹啉(10 转移到TBA-Eosin Y*形成N -苯基四氢异喹啉自由基正离子(10a )和TBA-Eosin Y•- .随后, 电子从TBA-Eosin Y•- 转移到氧气和硝基甲烷中, 生成TBA-Eosin Y, 氢过氧自由基和硝基甲烷自由基负离子.同时, 氧气自由基负离子进行质子化, 生成氢过氧自由基.其可以与N -苯基四氢异喹啉(10 )反应, 夺取一个氢原子, 进一步形成N -苯基四氢异喹啉自由基(10b )和H2 O2 .另外, 光诱导电子从N -苯基四氢异喹啉自由基(10b )转移到TBA-Eosin Y*分别产生亚胺离子10c 和TBA-Eosin Y•- .最后, 硝基甲烷与亚胺离子(10c )进行亲核加成, 从而得到产物1-硝基-N -苯基四氢异喹啉(11 ) (Scheme 1
图式 1
2012年, Blechert等[37 采用介孔石墨氮化碳(mpg- C3 N4 )半导体作为非均相光催化剂, 在可见光的照射下, 氧气作氧化剂合成了N -芳基四氢异喹啉衍生物(Eqs. 6, 7).此体系可以进一步结合脯氨酸实现氧化串联光催化.该方法利用非金属固态有机化合物作为催化剂, 具有较大的比表面积和丰富的表面积位点, 提高了光捕获能力和材料的反应吸附能, 又避免了有机染料难以回收的缺点.
2013年, Rueping课题组[38 将连续流反应技术应用到可见光催化氧化叔胺的α -官能化反应中.在绿光的照射下, 以孟加拉玫瑰红(Rose Bengal)染料作光敏剂, 乙腈和水的混合物作溶剂, 在流速为0.03 mL/min的连续反应器中反应5 h即可得到目标产物(Eqs. 8, 9).连续流反应相较于传统的间歇反应, 由于其具有较小的反应通道和较大的比表面积, 使得反应体系在可见光下受到更加均匀有效的照射, 从而大大缩短了反应时间, 并且具有安全、高效的优点.
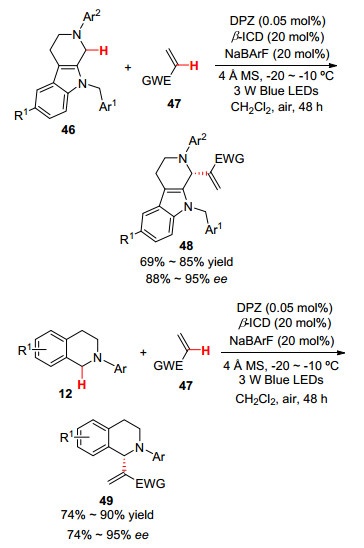
2014年, 江智勇和Bureš等[39 报道了以双氰基吡嗪衍生的发色团(DPZ)作为新型光催化剂的催化氧化反应.其中, 结构中含有给电子基团2-甲氧基噻吩的DPZ衍生物表现出强的可见光吸收, 以其作为光催化剂, 在12 W日光灯照射下, 以N -芳基四氢异喹啉、N -(4-叔丁基苯基)吡咯和二苄胺为底物, 与亲核试剂进行交叉脱氢偶联反应, 获得了良好的产率(Eq. 10).当以亚磷酸二乙酯作为亲核试剂时, 产率也可达81%.该光催化剂在低负载量下表现出高催化效率, 它的电化学特性有助于通过单电子转移产生α -氨基自由基, 更重要的是其平面可极化的π系统有助于自由基阴离子的稳定存在, 降低了能量的消耗, 是一个有望继续发展的绿色催化剂.
2014年, Itoh课题组[40 报道了在光照下, 以2-氯蒽醌(2-Cl-AQN)作光催化剂, 氧气作氧化剂, 在甲醇溶剂中实现了N -芳基四氢异喹啉与亲核试剂硝基烷烃及酮的交叉偶联反应(Eq. 11).此反应体系催化剂用量低及反应条件温和简单, 对吲哚也具有适用性, 但是对于N -烷基四氢异喹啉催化效果不明显, 产物产率低.
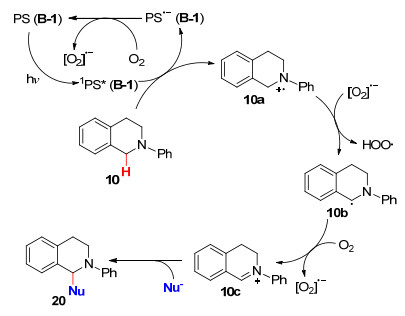
2015年, 吴骊珠课题组[41 报道了氟化硼络合二吡咯甲川类荧光染料(BODIPY)作为光敏剂的交叉脱氢偶联反应.在可见光的照射下, 以1, 3, 5, 7-四甲基-8-苯基- 4, 4-二氟硼烷基胍(B-1 )作光催化剂, 在室温下反应3~6 h即可得到N -芳基四氢异喹啉的衍生物(Eqs. 12, 13), 而且无论富电子还是缺电子亲核试剂, 都获得了高产率的偶联产物.与之前三重激发态3 PS*驱动C—H键活化的研究相反, 该课题组首次提出了BODIPY单重激发态1 PS*促进反应的机理, 为可见光照射下促进有机转化提供了新方法.
以N -苯基四氢异喹啉(10 )和亲核试剂NuH为例, 推测其反应机理如下: PS(B-1)受到光照后转化成单重激发态1 PS*, 其立即被N -苯基四氢异喹啉(10 )还原猝灭, 通过电子转移产生N -苯基四氢异喹啉自由基正离子(10a )和PS•- , 生成的PS•- 将电子转移给氧气以形成氧气自由基负离子, 同时再生基态PS用于催化循环.随后, 氧气从10a 中夺取一个氢原子生成自由基10b 和氢过氧自由基, 10b 进一步被氧气氧化生成亚胺10c , 最后被亲核试剂进攻生成目标产物20 (Scheme 2
图式 2
2015年, Raston等[42 将Rose Bengal染料光催化的N -芳基四氢异喹啉的亲核反应运用到微流体涡流装置(VFD)当中, 开发了一种新型反应设备(Eq. 14).该反应具有时间短和产率高的优点, 且VFD坚固, 易于操作和维护, 在流动过程中不会使化学物质造成通道反应器堵塞, 有利于化学反应的控制.
2015年, Zeitler课题组[43 报道了在CBrCl3 催化下, N -苯基四氢异喹啉转化成溴化亚胺盐, 再与亲核试剂硝基甲烷或丙二酸二甲酯进一步反应得到目标产物(Eq. 15).该催化体系反应条件温和、反应时间短, 对亚磷酸二乙酯和吲哚也有适用性, 为卤代化合物催化有机反应提供了新思路.
2016年, Rios课题组[44 报道了通过交叉脱氢偶联构建含氟结构单元的方法.在绿光的照射下, 以Rose Bengal染料作光敏剂, Bu4 NOAc作碱, N -取代四氢异喹啉和氟代丙二酸酯在CHCl3 溶剂中进行交叉脱氢偶联反应, 以较高产率得到了新型含氟偶联产物(Eq. 16).该方法为合成具有良好的热稳定性、化学稳定性和抗氧化性的含氟化合物提供了新途径, 但是催化效率不高, 反应时间过长.
2017年, 张俊民课题组[45 通过离子交换将Rose Bengal染料固定在聚二甲基硅氧烷(PDMS)海绵上, 获得了负载型催化剂PDMS-RB海绵, 并以其作为光催化剂, 研究了叔胺和硝基烷烃和丙酮的交叉脱氢偶联反应(Eqs. 17, 18).这种新型海绵催化剂在使用过程中表现出优异的光催化活性, 容易回收和再利用, 且在重复使用15次以后仍保持较高催化活性.通过连续流反应器更易按比例放大到工业化规模.该方法操作简便, 绿色环保, 为探索高效率的新型非均相光催化剂开辟了新途径.
同年, 该课题组[46 报道了一种新型双功能的有机海绵光催化剂, 该催化剂通过将Rose Bengal染料光敏剂和烯胺有机催化剂同时固定在聚二甲基硅氧烷(PDMS)海绵上所制得.在该催化剂催化下, 叔胺与酮在水中进行偶联, 获得了不错的产率(Eqs. 19~21).将PDMS sponge 1 的L -脯氨酸甲酯部分用L -脯氨酸钠盐替换, 制得了另一种手性双功能海绵光催化剂PDMS sponge 2 , 并在其催化下实现了叔胺与酮的不对称交叉脱氢偶联反应, 该合成方法具有良好的对映选择性, 且催化剂活性高, 环境友好.
随后, 该课题组[47 又将Rose Bengal染料通过离子键固定到棉纤维上得到新型光催化剂Cot-RB, 将其应用于交叉脱氢偶联反应中, 得到了高产率产物(Eqs. 22, 23).该催化剂利用丰富的天然材料棉纤维, 表现出很好的光催化活性, 且具有可回收重复利用的优点.此课题组的这三项研究为开发更高效环保的负载型光催化剂提供了广阔前景.
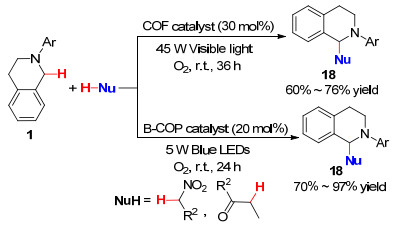
2017年, 苏清课题组[48 在加热条件下将2, 5-二甲氧基对苯二甲酰肼与均苯三甲醛缩合, 合成了基于腙的共价有机骨架(COF), 并以其作为光催化剂, 在45 W的节能灯照射下, 氧气作氧化剂, 研究了N -芳基四氢喹啉和亲核试剂的交叉脱氢偶联反应(Scheme 3
图式 3
次年, 该课题组[49 又通过直接缩聚合成了在聚合物骨架中具有A3 B2 型供体-受体阵列的含二氟硼酸盐的共轭有机聚合物(B-COP), 以其作为光催化剂, 在5 W蓝光的照射下, 氧气氛围中室温下进行反应(Scheme 3
2018年, 温小安等[50 以N -甲基-3(10H )-吖啶酮作为光敏剂, 在甲醇溶剂中, 蓝光的照射下实现了N -芳基四氢异喹啉与各种亲核试剂交叉脱氢偶联反应(Eq. 24).机理研究表明, 该反应是单电子转移诱导的自由基过程.由于该光敏剂稳定且易制备的特点以及其独特的发色团结构和光物理特性, 使得该催化体系具有高原子经济性和良好的水溶性.
2018年, 杨靖亚课题组[51 报道了可见光介导下酮的α -羟甲基化反应.该反应通过自由基途径, 以Rose Bengal染料作光敏剂, K2 CO3 作碱, 甲醇作羟甲基化试剂, 室温下在水中实现交叉脱氢偶联(Eq. 25).此方法合成的α -羟甲基酮具有良好的耐受性, 过程操作简便, 环境友好.
1.2
C(sp3 )—C(sp2 )偶联
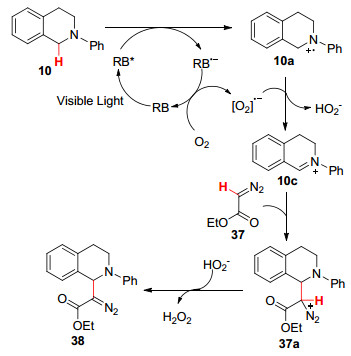
2014年, 周磊课题组[52 首次报道了可见光诱导下叔胺和重氮化合物之间的交叉脱氢偶联反应.在绿光的照射下, 以Rose Bengal染料作光敏剂, 氧气作氧化剂, 室温下在二氯甲烷溶剂中反应12 h得到β -氨基-α -重氮化合物(Eq. 26).所得的产物可以通过过渡金属进一步选择性转化成N -芳基-2, 3-二氢苯并[d ]氮杂䓬.该方法具有广泛的底物范围, 且条件温和, 避免了过氧化物和过渡金属使用下重氮化合物的分解, 为合成杂环化合物提供了一条很好的途径.
以N -苯基四氢异喹啉(10 )和2-重氮基乙酸乙酯(37 )为例, 推测其反应机理如下:首先, RB在可见光的照射下形成激发态RB*, RB*通过单电子转移和叔胺形成N -苯基四氢异喹啉自由基正离子10a 和RB•- .然后, RB•- 被氧气氧化得到氧气自由基负离子, 同时再生RB用于催化循环.随后, 氧气自由基负离子从10a 中夺取一个氢原子生成亚胺离子10c 和氢过氧化物阴离子, 最后10c 和重氮化合物37 结合生成中间体37a , 37a 被氢过氧化物阴离子去质子化生成H2 O2 和α -氨基重氮化合物38 (Scheme 4
图式 4
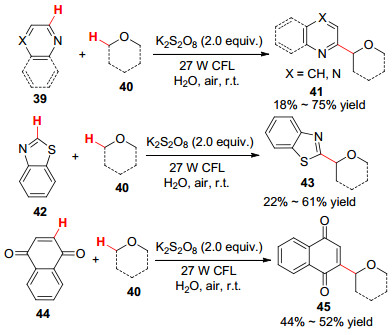
2016年, Shah课题组[53 报道了可见光促进下缺电子芳烃和醚的C—H官能化反应.通过氢原子转移途径, 以K2 S2 O8 作氧化剂, 室温下在水中实现了C(sp2 )—C(sp3 )键的偶联(Scheme 5
图式 5
2016年, 江智勇课题组[54 报道了四氢-β -咔啉和四氢异喹啉的烯烃化反应(Scheme 6 β -ICD和无机盐助催化剂的协同催化作用下表现出优异的区域选择性和对映选择性.
图式 6
2017年, 张俊民课题组[45 研究了负载型催化剂PDMS-RB海绵催化下, N -芳基四氢喹啉和重氮基乙酸乙酯的交叉脱氢偶联反应(Eq. 27).结合前面所述, 可见负载型催化剂PDMS-RB海绵适用性广泛, 不仅能催化实现C(sp3 )—C(sp3 )键的构建, 还能催化得到C(sp2 )—C(sp3 )偶联产物.
2017年, Yamaguchi课题组[55 报道了光催化氧化下由分子碘介导的杂芳烃与羰基化合物的偶联反应.在22 W紧凑型荧光灯的照射下, 以I2 作为催化剂, K3 PO4 作碱, 室温下反应20 h实现了C(sp2 )—C(sp3 )键的偶联(Eq. 28).同时也对这一自由基介导反应机理进行了阐述.该反应体系以廉价的分子碘作为催化剂, 具有稳定性高和环境友好等优点.
2018年, 魏伟课题组[56 报道了在蓝光的照射下, 以Rose Bengal染料作光敏剂, 三乙烯二胺(DABCO)作碱, 叔丁基过氧化氢(TBHP)作氧化剂, 室温下反应24 h, 实现了1-烷基喹喔啉-2(1H )-酮和醚和四氢噻吩的交叉脱氢偶联反应(Eq. 29).该方法产物区域选择性良好, 底物范围广泛, 通常产率较高, 对官能团的耐受性也比较好.
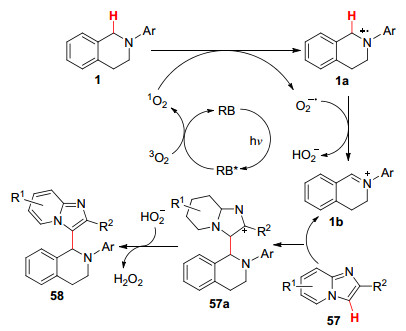
2018年, Hajra课题组[57 报道了以蓝光为光源, 以Rose Bengal染料为光敏剂, 在甲苯溶液中进行了咪唑并[1, 2-a ]吡啶(57 )和N -芳基四氢异喹啉、叔芳胺之间的交叉脱氢偶联反应(Eqs. 30, 31).以咪唑并[1, 2-a ]吡啶(57 )和N -芳基四氢异喹啉(1 )为例, 推测其反应机理如下:基态RB吸收光子后被转换为激发态RB*. RB*将能量转移到基态氧3 O2 , 形成单线态氧1 O2 . N -芳基四氢异喹啉(1 )被单线态氧氧化生成自由基正离子1a 和超氧自由基负离子O2 •- .随后, 进行氢原子转移生成亚胺离子1b 和HO2 - .然后, 1b 加成到咪唑并[1, 2-a ]吡啶57 中得到中间体57a , 最后发生氢原子转移生成化合物58 (Scheme 7
图式 7
随后, 该课题组[58 利用类似催化体系进行了2-烷基-2H -吲唑和醚、四氢噻吩的交叉偶联反应(Eq. 32).该反应可用于克规模合成, 为合成烷杂基化的2H -吲唑开辟了温和简便的路线.
2018年, 金灿等[59 报道了N -芳基甘氨酸酯与N -取代苯胺的光催化交叉脱氢偶联反应.在蓝光的照射下, 以亚甲基蓝作为光催化剂, 无需额外的氧化剂和添加剂, 实现了C(sp2 )—C(sp3 )键的构建(Eq. 33).反应过程包括单电子转移和亲电子加成.该合成方法适用的底物范围广泛, 条件温和且环境友好.
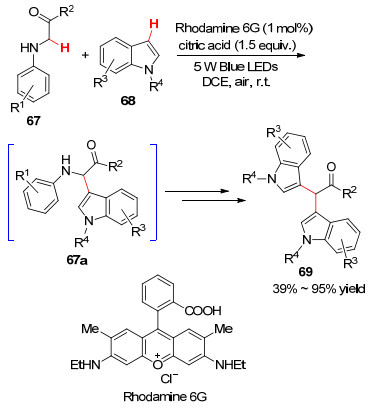
2018年, 张元课题组[60 报道了在光敏剂Rh-6G染料和质子酸柠檬酸的催化作用下甘氨酸衍生物与吲哚衍生物之间的Friedel-Crafts反应(Scheme 8 67a , 随后再进行第二次催化氧化最终得到目标产物.该反应底物范围广泛, 有良好的官能团耐受性, 为构建医药中间体二吲哚甲烷提供了新方法.
图式 8
2019年, 本课题组[61 报道了无过渡金属催化下的苯并噻唑的烷基化反应.以蓝光作为光源, Eosin Y染料为光敏剂, K2 S2 O8 为氧化剂, 室温下反应24 h, 进行自由基介导的苯并噻唑和酰胺衍生物的交叉脱氢偶联反应(Eq. 34).相比Ji课题组[62 苯并噻唑烷基化的方法(Eq. 35), 前者催化体系更简单, 同时仍具有较好产率.
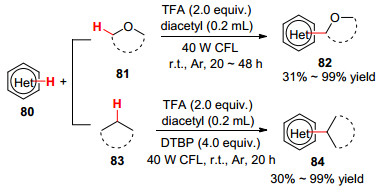
2019年, 雷爱文课题组[63 报道了可见光诱导下醇的α -C—H官能化反应.可见光照射下, 反应以氟试剂作氧化剂, 三氟乙酸(TFA)作添加剂, 在氮气氛中实现了杂芳烃和醇的交叉脱氢偶联反应(Eq. 36).该方法无需光敏剂, 具有官能团耐受性, 且区域选择性良好, 为向杂环引入活性醇羟基提供了新方法.随后, 该课题组[64 也报道了在氟试剂/TFA催化体系下, 室温条件下杂芳烃与环烷烃、醚等的C(sp3 )—H芳基化反应(Eq. 37).
2019年, 李朝军课题组[65 以双乙酰(2, 3-丁二酮)作光敏剂, 在室温条件下实现了交叉脱氢Minisci烷基化反应(Scheme 9 3 )-H底物和双乙酰之间发生了氢原子转移, 其中双乙酰为“无痕”氢原子提取剂.此外, 在二叔丁基过氧化物(DTBP)的辅助下, 该反应可通过双乙酰介导的能量转移扩展到脂肪族的强C—H键.该方法具有很好的稳健性, 能官能化尼古丁、薄荷醇和丙氨酸衍生物等复杂分子.
图式 9
1.3
C(sp2 )—C(sp2 )偶联
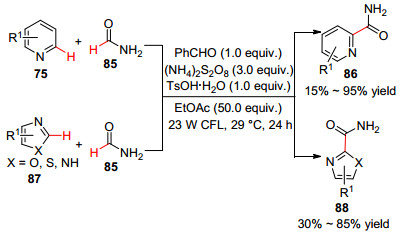
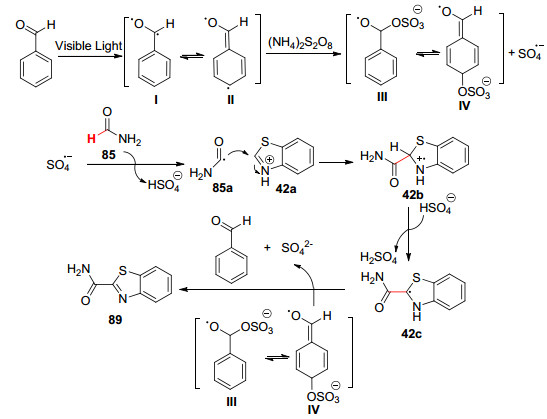
2016年, Ji等[61 提出了光激发下苯甲醛介导(NH4 )2 S2 O8 分解的自由基机理.在家庭23 W紧凑型荧光灯(CFL)照射下, (NH4 )2 S2 O8 作氧化剂, 对甲苯磺酸一水合物(TsOH•H2 O)作添加剂, 实现了苯甲醛催化下唑类杂环化合物与甲酰胺的交叉脱氢偶联反应(Scheme 10 2 )—C(sp2 )键, 有助于促进酰胺衍生物的α -芳基化反应.
图式 10
以苯并噻唑42 与甲酰胺85 为例, 推测其反应机理如下:苯甲醛在可见光的照射下形成I 和II , 其促进(NH4 )2 S2 O8 的分解, 生成SO4 •- 和中间体III 、IV .随后, SO4 •- 从甲酰胺85 中夺取一个氢原子生成自由基85a , 亚胺85a 与C-2位质子化的苯并噻唑42a 发生亲核加成, 然后再进行去质子化和氧化, 得到目标产物89 .其中中间体III 和IV 参与氧化步骤, 从而再生苯甲醛(Scheme 11
图式 11
2018年, 雷爱文课题组[66 报道了光催化下的C(sp2 )—H酰基化反应.反应以叔丁基过氧化氢(TBHP)作氧化剂, TFA作添加剂, 醛作酰基源, 在氮气氛围中室温反应24 h实现了异喹啉的酰基化(Eq. 38).该催化体系无需光催化剂的作用即可以良好的产率得到交叉脱氢偶联产物.
2.
C—N偶联反应
2.1
含N化合物作为亲核试剂
2012年, 肖文精等[67 利用有机染料Na2 -Eosin Y代替Ir(ppy)2 (dtb-bpy)PF6 金属催化剂, t -BuOK作碱, 在光照下实现了分子内交叉脱氢偶联, 得到了产率良好的稠杂环产物(Eq. 38).该方法条件温和, 提供了一种有机染料光敏剂催化下合成杂环化合物的新方法.
2013年, Tan课题组[68 报道了氨基醇和烯烃的交叉脱氢偶联反应.反应在11 W家用荧光灯照射下, 以Rose Bengal染料作光敏剂, 加入一定量的添加剂吡啶以提高反应速率和产率(Eqs. 40, 41).该方法操作简单, 环境友好, 可以避免金属催化剂及其他氧化剂引起的环境污染.但是在进行非末端烯烃的烯丙基胺化反应的时候, 易生成异构体混合物.
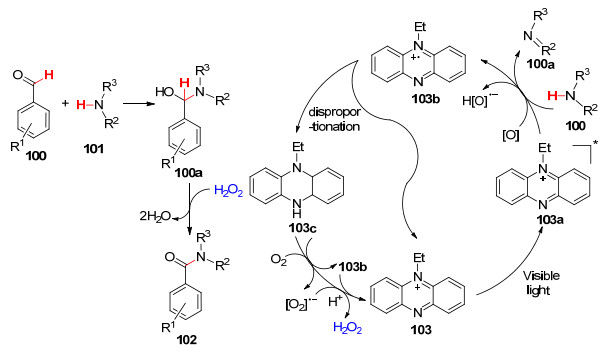
2014年, Leow课题组[69 报道了利用吩嗪盐催化芳香醛衍生物的氧化酰胺化.在可见光的照射下, 以吩嗪乙硫酸盐作催化剂, 室温下进行反应(Eq. 42).该方法原子经济性高, 底物范围广, 为醛的直接酰胺化反应提供了新思路.
吩嗪盐是众所周知的电子受体, 以芳香醛100 和胺101 为例, 推测其反应机理如下Scheme 12 103 在可见光的照射下形成激发态103a , 随即与胺101 发生单电子转移生成自由基正离子化合物103b 和亚胺101a , 然后103b 发生歧化反应生成5-乙基-5, 10-二氢吩嗪(103c )和吩嗪盐103 , 随后103c 被氧气氧化再生光催化剂103 , 同时伴随产生的H2 O2 氧化半缩醛胺100a 成目标产物酰胺102 .
图式 12
2015年, Pandey等[70 报道了可见光催化下苄基C(sp3 )—H的直接胺化.以9, 10-二氰基蒽(DCA)作为光催化剂, 酰胺作氮源, 实现了分子间和分子内的偶联反应(Eqs. 43, 44).该催化体系无需外部氧化剂的参与, 绿色环保, 具有广泛的底物范围和优异的区域选择性.
2015年, Nicewicz等[71 报道了吖啶染料选择性催化的芳基C(sp2 )—H胺化.在455 nm波长的蓝光照射下, 以[(t -Bu)2 -Mes+ -Acr-Ph][BF4 - ]作光催化剂, 2, 2, 6, 6-四甲基哌啶-氮-氧化物(TEMPO)作助催化剂, 在氧气氛围中进行反应(Eq. 45).其中吖啶催化剂促进了关键性中间体芳基阳离子的形成, 且可通过简单的过滤实现催化剂回收再利用.该反应实现了芳烃与各种杂环叔胺的偶联反应, 包括吡唑、三唑等相关衍生物, 底物范围广泛且具有较高的区域选择性和原子经济性.随后, 该课题组[72 以伯胺作为氮源, 在455 nm的光源下, 以吖啶染料[(t -Bu)2 -Mes+ -Acr-Ph][BF4 - ]和[Me2 -Mes+ -Acr-Ph]- [BF4 - ]作为光催化剂, 实现了芳烃的C(sp2 )—H胺化(Eq. 46).该反应条件温和, 且具有较好的官能团耐受性.
2016年, König等[73 报道了吖啶染料催化下磺酰胺和吡咯的偶联反应.通过一锅法在455 nm的可见光照射下, 以[Acr+ -Mes-Me][ClO4 - ]作光催化剂, NaOH作碱, 氧气氛围下在乙腈和水混合溶剂中反应得到偶联产物(Eq. 47).该催化体系条件温和, 具有良好的官能团耐受性和区域选择性, 实现了杂芳烃的磺酰胺化, 可有效地应用于医药化学领域.但是对于苄基或者苯基吡咯反应时产率较低, 可能由于其亲核性低不利于转化.
2017年, Murakami等[74 研究了在蓝光的照射下, 以2, 3-二氯-5, 6-二氰基对苯醌(DDQ)为光敏剂, 萘、噁唑和1, 4-萘醌等芳烃与磺酰亚胺之间的C—H/N—H偶联反应(Eq. 48).该方法操作简便, 提供了一条芳烃磺酰胺化的有效途径.同年, 雷爱文等[75 以DDQ作光敏剂, 亚硝酸叔丁酯(TBN)作电子转移介质, 实现了噻吩和吡唑和吲唑等唑类化合物的C(sp2 )—N偶联(Eq. 49).与单一使用DDQ作催化剂相比, 该方法大大缩短了反应时间.随后, König课题组[76 分别以Boc-胺、氨基甲酸酯、吡唑、磺酰亚胺和脲作为氮源, 在DDQ/TBN催化体系中, 进行了取代苯的胺化反应(Eq. 50).
2.2
含N化合物形成N自由基
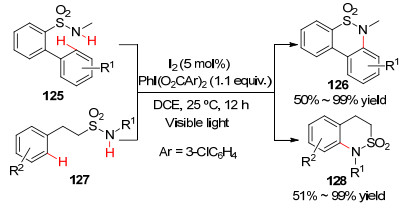
2016年, Muñiz课题组[77 报道了分子内芳基C—H官能化反应.利用分子碘和高价碘PhI(O2 CAr)2 试剂组合的催化体系, 温和的条件下进行芳烃分子内胺化(Scheme 13
图式 13
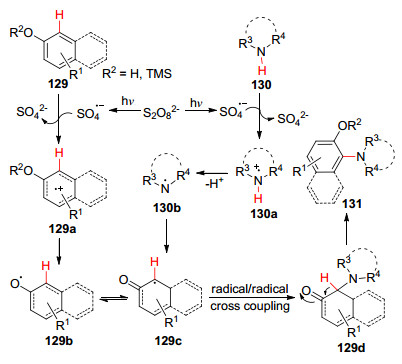
2016年, 夏吾炯课题组[78 报道了在酚和环状胺分子之间的交叉脱氢偶联反应.在蓝光的照射下, 以K2 S2 O8 作为氧化剂, 室温下在乙腈溶剂中进行反应实现了C—N键的构建(Eq. 51).该过程中酚129 和胺130 均被K2 S2 O8 氧化成自由基129c 和130b , 随后发生自由基间的交叉偶联反应得到中间体129d , 最后发生异构化得到最终产物131 (Scheme 14
图式 14
次年, 该课题组[79 又报道了酚和非环状二芳基胺分子间的交叉脱氢偶联反应.在蓝光的照射下, 以(NH4 )2 S2 O8 作为氧化剂, 加入有机光催化剂(OPC), 室温下在邻二氯乙烷溶剂中实现了C(sp2 )—N键的形成(Eq. 52).机理研究表明, 此反应过程是通过自由基间的偶联反应进行的.该反应条件温和, 具有广泛的底物范围和高效的区域选择性, 可用于构建三芳基胺单元.
2017年, Itoh课题组[80 报道了1-甲基-2-烷基吲哚的C—H胺化反应, 反应以邻苯二甲酰亚胺为氮源, 在21 W荧光灯的照射下, 以2-叔丁基蒽醌(2-t -Bu-AQN)作催化剂, K2 CO3 作碱(Eq. 53).该反应过程中在吲哚的C-2位置产生的自由基是其关键中间体, 因此当取代基为给电子基团时产率明显提高.
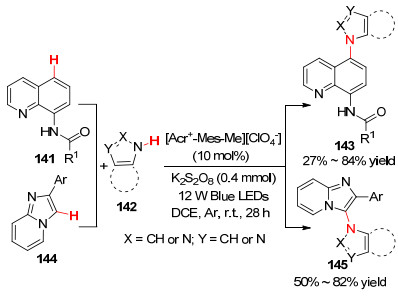
2017年, 雷爱文课题组[81 报道了蓝光照射下, 以[Acr+ -Mes-Me][CIO4 - ]为光催化剂, 四氢呋喃的α - C(sp3 )—H官能化反应(Eq. 54).该方法耐受各种唑类衍生物.同年, Adimurthy课题组[82 报道了相同催化剂催化下, 喹啉酰胺(C-5)和咪唑并吡啶(C-3)的区域选择性C(sp2 )—H胺化(Scheme 15
图式 15
2018年, 魏伟课题组[83 报道了C(sp2 )—H键和N—H键之间的交叉脱氢偶联反应.在蓝光的照射下, 以Eosin Y染料作光敏剂, 在四氢呋喃溶液中, 室温条件下通过自由基介导合成3-氨基喹喔啉-2(1H )-酮(Eq. 55).该方法操作简单, 反应条件温和, 具有较好的原子经济性.
2018年, 何延红课题组[84 报道了唑类和缺电子烯烃的光催化交叉脱氢偶联.在光照条件下, 以Rose Bengal染料作光敏剂, 丙酮溶液中室温反应24 h实现了C(sp2 )—H的胺化反应(Eq. 56).该合成方法操作简便, 反应条件温和, 具有优异的官能团耐受性和区域选择性, 为N -乙烯基吡唑的合成提供了一种新方法.
3.
C—O偶联反应
2013年, Pandey课题组[85 报道了分子内苄基C(sp3 )—H官能化的环醚化反应.反应以1, 4-二氰基萘(DCN)为催化剂, 在光照及室温条件下, 于乙腈溶剂中进行(Eq. 57).通过光诱导电子转移生成的自由基阳离子进行催化反应.该催化体系具有优异的区域选择性, 产物产率良好.
2014年, 吴骊珠课题组[86 报道了在蓝光的照射下, 以TBA-Eosin Y染料作光敏剂, 二氮杂二环(DBU)作碱, 利用分子内交叉脱氢偶联合成2-取代苯并噁唑的反应(Eq. 58).反应过程中, 在TBA-Eosin Y染料的作用下, 底物转化成自由基中间体, 再进一步进行光催化脱氢得到产物, 其中添加碱有助于电子转移, 抑制单线态氧的副反应.该反应为分子内C—O键的构建开辟了有效途径.
2015年, Gonzalez-Gomez等[87 采用吖啶染料作为光催化剂, 在室温下实现了2-芳基苯甲酸的分子内脱氢酯化.该反应在可见光照射下, 首次利用[Acr+ -Mes-Me][ClO4 - ]和(NH4 )2 S2 O8 的催化组合制备了3, 4-苯并香豆素(Eq. 59).反应通过均裂芳香取代机理进行, 具有操作简便和条件温和等优点.随后, 雷爱文课题组[88 同样以[Acr+ -Mes][ClO4 - ]作为光催化剂, 在助催化剂TEMPO的作用下, 得到了烯烃阳离子自由基, 在氮气保护下实现了烯烃与醇的交叉脱氢偶联(Eq. 60).
2016年, Gilmour等[89 以(-)-核黄素作为催化剂, 利用它的两种光化学活化模式, 分别通过能量转移和单电子转移依次进行诱导异构化和环化途径合成香豆素衍生物(Eq. 61), 其中最关键的步骤是E 构型到Z 构型的转化.该催化体系对芳环上的取代基都具有良好的耐受性, 是有机光化学方法的有益拓展.
2017年, Deb等[90 研究了自由基介导下1-氨基烷基-2-萘酚的分子内交叉脱氢偶联反应.该反应以绿光作为光源, Eosin Y染料为光敏剂, 在乙腈溶液中以中等收率得到产物噁嗪(Eq. 62).该方法无需过渡金属催化剂以及其他添加剂, 绿色环保.
2017年, Hajra课题组[91 报道了可见光介导的咪唑并吡啶C-3烷氧基化反应.以醇作氧源, 在Rose Bengal染料的催化下, 室温下进行反应, 以良好的产率得到了C(sp2 )—H烷氧基化产物(Eq. 63).该方法具有官能团耐受性好, 反应条件温和等优点.
4.
C—P偶联反应
2011年, König等[35 在研究了Eosin Y染料催化下N -芳基四氢异喹啉和亲核试剂两者间的C(sp3 )—C(sp3 )键偶联后, 将亲核试剂扩展到二烷基磷酸酯继续进行研究, 得到了高产率的产物(Eq. 64). 2015年, 吴骊珠等[41 以BODIPY染料为光敏剂, 也将亲核试剂扩展到二烷基磷酸酯(Eq. 65).与以往构建C(sp3 )—P键需要金属催化剂和昂贵的氧化剂不同, 这两种催化体系具有条件温和、操作简便及反应时间短等优点, 为C(sp3 )—P键的构建提供了新方法.
2016年, 吴磊课题组[92 首次报道了无金属催化条件下的光催化C(sp2 )—P成键交叉脱氢偶联反应.利用有机小分子染料Eosin B作为光敏剂, 在可见光的照射下反应24 h, 利用苯并噻唑和芳基磷氧化合物之间的交叉脱氢偶联实现了C—H的直接磷酰化(Eq. 66).该过程通过二苯基膦自由基与苯并噻唑亲核加成, 然后去质子化得到产物.该方法在温和的室温条件下即可高效构建C—P键, 氢气为唯一副产物, 无需氧化剂和任何添加剂, 且对各种官能团都具有很好的耐受性.同年, 雷爱文课题组[93 利用Na2 -Eosin Y染料作为光敏剂, 实现了C(sp2 )—P键的形成(Eq. 67).该合成方法既降低了催化剂用量, 又缩短了反应时间.
2017年, 吴养洁等[94 报道了在绿光的照射下, 以Eosin Y染料为光敏剂, 过氧化二苯甲酰(BPO)为氧化剂, 在氩气的保护下, 乙腈溶剂中反应24 h, 实现了香豆素衍生物的C-3位膦酰化(Eq. 68).通过对照实验证明活性中间体以P为中心的自由基在反应过程中起到关键作用.该催化体系对于多取代的香豆素具有广泛的适用性, 且产率优良, 但反应需在氩气保护下进行且反应时间较长.
2018年, Hajra课题组[95 报道了在室温和光照条件下, 以Rose Bengal染料作光敏剂, 二氮杂二环(DBU)作碱, 在乙腈和水溶剂中反应16 h, 实现了2H -吲唑的C(sp2 )—H膦酰化反应(Eq. 69), 产率良好.该过程为合成含磷的芳香族化合物提供了一种有效的方法.
5.
成环反应
2015年, Brasholz课题组[96 报道了在可见光的照射下, 以1-(硝基甲基)-2-芳基四氢异喹啉为底物, 1-氨基蒽醌(1-AAQ)作光催化剂, K3 PO4 作碱, 室温下在乙腈溶剂中通过交叉脱氢-环化合成12-硝基取代的四环吲哚[2, 1-α ]异喹啉衍生物(Eq. 70).目标产物也可直接通过N -芳基四氢异喹啉和硝基甲烷在该催化体系下一锅法进行四重级联转化获得.该研究证明了蒽醌衍生物的高效催化能力, 反应过程中需要碱的参与, 且部分化合物反应时间长达140 h, 尚待进一步改进.
2018年, 江智勇课题组[97 制备了9种二氰基吡嗪类光敏分子(DPZ)和二氰基咪唑衍生物(DCI)作为光催化剂, 在蓝光的照射下, 二氯甲烷溶剂中, 于25 ℃反应6 h实现了N -芳基四氢异喹啉和顺丁烯二酰亚胺的环化反应(Eq. 71).实验证明DPZ比DCI更高效.此催化体系具有条件温和、操作简便和催化效果好等优点, 符合绿色化学的要求.
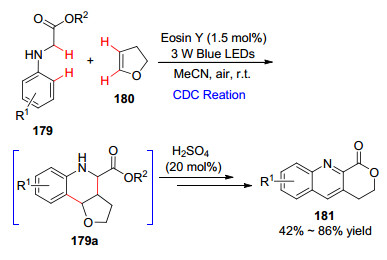
2018年, 张元课题组[98 研究了基于交叉脱氢偶联的环化串联反应.以蓝光为光源, 在光敏剂Eosin Y染料的催化下, 甘氨酸酯与2, 3-二氢呋喃在室温下进行交叉脱氢偶联生成关键中间体179a , 随后该中间体经过酸化、酯交换和芳构化获得一系列喹啉稠合内酯化合物(Scheme 16 α -C—H官能化的一条有效途径.
图式 16
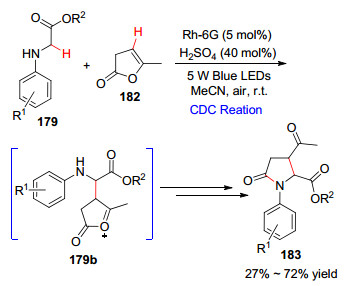
随后, 该课题组[99 在Rh-6G染料和硫酸的催化体系中实现了甘氨酸酯与α -当归内酯的交叉脱氢-环化反应(Scheme 17 α -当归内酯发生C(sp3 )—C(sp2 )偶联反应生成中间体179b , 随后经过分子内亲核取代和去质子化得到目标产物183 .该反应条件温和, 合成的γ -内酰胺可作为重要中间体合成复杂天然产物.
图式 17
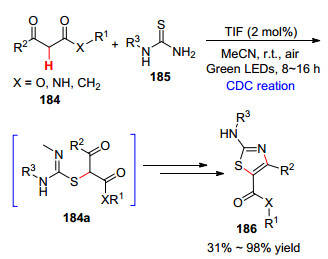
2018年, Chuah课题组[100 报道了1, 3-二羰基化合物和硫脲的偶联反应, 在绿光的照射下, 以四碘荧光素染料作光催化剂, 合成了氨基噻唑(Scheme 18 184a , 随后发生分子内环化反应得到目标产物186 .
图式 18
6.
其他偶联反应(C—Si, N—P, S—S, S—P)
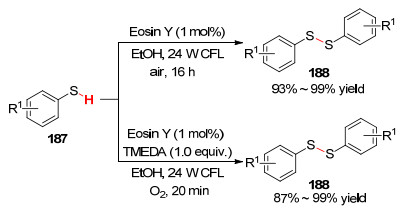
2015年, Noël课题组[101 报道了在光催化条件下硫酚通过分子间脱氢偶联生成二硫化物的反应.在24 W紧凑型荧光灯照射下, 以Eosin Y染料作光敏剂, 乙醇溶剂中反应16 h, 通过一锅法高产率地合成了二硫化物.随后又运用了连续流反应方法, 加入添加剂四甲基乙二胺(TMEDA)进行反应, 仅需20 min便得到高产率二硫化物(Scheme 19
图式 19
2016年, Wacharasindhu课题组[102 报道了在可见光的照射下, 以Rose Bengal染料为光敏剂, 于室温下水溶剂中实现硫醇合成二硫化物的反应(Eq. 72).该催化体系条件温和, 无需金属催化或氧化剂参与, 产物产率较高.
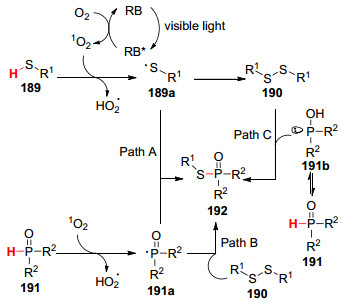
2016年, 李萍课题组[103 报道了在蓝光的照射下, 以Rose Bengal染料作光敏剂, 在室温下实现了硫醇和有机磷化合物的交叉偶联反应(Eq. 73).推测其反应机理如下:首先, RB在光照下被激发成激发态RB*, 其与O2 通过能量转移生成单线态氧(1 O2 ), 同时激发态RB*返回其基态RB.随后, 1 O2 从硫醇189 中夺取氢原子得到硫基自由基189a , 其快速偶联形成二硫化物190 .另一方面, 1 O2 与烷基膦氧化物191 发生氢原子转移生成以P为中心基团的自由基191a .最后, 191a 与硫基自由基189a 偶联(Path A), 或者191a 与二硫化物190 偶联(Path B), 或者可以通过191b 亲核进攻二硫化物190 产生目标产物192 (Path C)(Scheme 20
图式 20
2016年, Rios课题组[104 首次使用有机染料在光催化条件下合成氨基磷酸酯.在绿光的照射下, 以Rose Bengal染料作光敏剂, 亚磷酸二乙酯作磷源, 在70 ℃下反应实现了芳香胺和脂肪族胺的磷酸化(Eqs. 74, 75).该合成反应通过一锅法进行, 是一个体系温和、绿色高效的过程, 为N—P键的构建提供了一条简单易行的途径.
2018年, 吴勇等[105 利用亚甲基蓝作光敏剂, K2 CO3 作碱, 异丙醇作溶剂, 在蓝光、室温和空气条件下合成了硫代磷酸酯(Eq. 76).该合成方法具有底物范围广, 官能团耐受性高的特点, 适用于含氨基、羟基、氰基和三氟甲基等基团的衍生物.
2018年, 姜超课题组[106 报道了二芳基硅烷通过分子内交叉脱氢偶联合成具有特殊有机电致发光特性的二苯并噻咯(硅芴)的反应.该反应以Rose Bengal染料作光敏剂, TBHP作氧化剂, KOH作碱, 在温和的条件下实现了C—Si键的形成(Eq. 77).该方法可广泛用于硅芴衍生物的合成, 以拓展其在光电材料领域中的应用.
7.
结论与展望
列举了可见光诱导的无过渡金属催化的交叉脱氢偶联反应, 其中包括C—C, C—N, C—O, C—P, C—Si, S—S, S—P, N—P键偶联和成环的反应实例.在上述光诱导的交叉脱氢偶联反应中, 通常以氧杂蒽类染料(如Rose Bengal和Eosin Y)、蒽醌类染料(如AAQ和AQN)、吖啶类染料、BODIPY染料、吩嗪类化合物、吡嗪类化合物、固体催化剂(如mpg-C3 N4 )及负载型催化剂(如PDMS-RB和Cot-RB)等作为光催化剂, 避免了过渡金属的参与, 具有绿色环保、温和高效以及原子经济性高的优点.目前, 根据一些研究报道[107 109 , 光催化剂和质子还原助催化剂结合形成的新型双催化体系可增强光吸收效率, 其利用光催化剂引发电子转移, 质子还原催化剂捕获电子, 显著提高了光催化产氢的活性.但是上述质子还原催化剂由于含有过渡金属, 通常能耗大, 污染严重且价格昂贵.因此, 可以进一步研究环境友好的非金属光协同催化剂, 用以开发更绿色、高效和广泛的交叉脱氢偶联反应.
综上所述, 可见光诱导的无过渡金属催化交叉脱氢偶联反应已成为有机合成领域新的研究方向和热点之一, 值得进一步探索与研究.






 下载:
下载:









































































 下载:
下载:





















