图式 1.
基于Julia烯烃化反应合成碘代烯丙基化合物
Scheme 1.
Synthesis of iodo-substituted allylsilanes based on the Julia olefination

烯丙基硅化合物是一种重要的有机合成子, 自1948年Sommer等[1]发现其与亲电试剂的反应以来, 已被广泛应用于C—C键的构建[2]和复杂天然产物的合成研究[3]中.烯丙基硅化合物结构中同时存在硅原子和烯丙基, 弱极性的C—Si键赋予了烯丙基硅化合物相较于其它烯丙基金属试剂更好的稳定性, 便于制备和储存; 同时, 硅原子特殊的电子结构使得烯丙基硅化合物在化学转化过程中能有更优异的表现, 比如硅原子能通过α-效应或β-效应稳定α-碳负离子、α-碳自由基、β-碳正离子和β-碳自由基, 从而控制反应的选择性[2d].正是由于烯丙基硅化合物的这些优势, 越来越多的合成方法被化学家们发展[4]并加以利用, 而光学纯烯丙基硅合成方法的发展[5]进一步推动了烯丙基硅化合物在不对称合成中的应用[6].烯丙基硅化合物的主要合成方法包括Wittig反应、取代硅烷的消除反应、1, 3-二烯的1, 4-硅氢化反应、烯烃与含硅金属试剂的偶联反应等, 但这些合成方法往往不能很好地控制所得烯丙基硅化合物双键的构型而得到混合物, 极大地影响烯丙基硅在有机合成中的应用.因此, 发展高效高立体选择性的烯丙基硅合成方法具有重要意义.
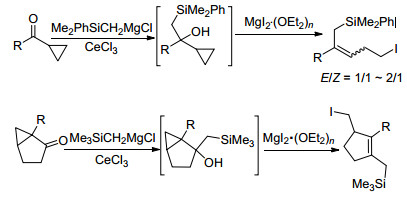
2004年, 我们课题组[7]发展了一种简洁高效的合成碘代烯丙基硅的方法(Scheme 1), 利用带有大位阻基团的硅甲基格氏试剂Me2PhSiCH2MgCl与环丙基酮的亲核反应得到环丙基取代的β-羟基硅烷化合物, 进而在MgI2•(OEt2)2作用下选择性地发生Julia烯烃化反应, 高效合成了带有卤原子的烯丙基硅化合物.该类官能化的烯丙基硅化合物在萜类天然产物全合成中应用成功[8].该方法可应用于环状和链状碘代烯丙基硅化合物的合成, 但合成链状烯丙基硅化合物双键的立体选择性往往并不理想, 得到两种构型的混合产物[7, 8b](Scheme 1).因此, 发展该类型官能化烯丙基硅化合物高效高立体选择性合成方法对其在全合成中应用具有重要意义.
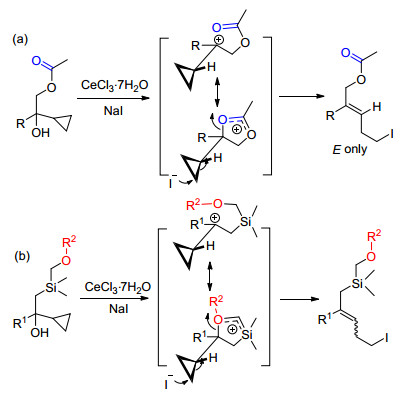
2005年, 我们课题组[9]在利用Julia卤代烯烃化反应合成多取代烯烃过程中, 发现环丙基甲醇化合物结构中羟基β位带有乙酰氧基(OAc)的底物的卤化开环反应能高立体选择性地得到E式构型烯烃.推测可能是反应过程中形成的碳正离子中间体结构中, 酰基与碳正离子形成了亚稳态五元环阳离子结构, 稳定了反应中间体, 同时控制了后续碘离子进攻环丙环的方向, 得到单一构型的产物(Scheme 2a).基于以上反应机理推测, 本研究工作通过在硅原子上引入能稳定碳正离子的取代基(OR2), 探索利用β-环丙基-β-羟基硅化合物的Julia烯烃化反应立体选择性合成官能化烯丙基硅的可行性(Scheme 2b).
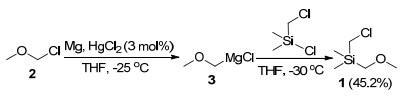
烷氧基取代的β-环丙基-β-羟基硅化合物利用相应的有机硅格氏试剂与环丙基酮的亲核加成反应即可得到.首先探索了合成有机硅格氏试剂需要的烷氧甲基氯甲基二甲基硅1的合成. α-卤代甲基硅化合物的合成可通过卤代卤甲基硅烷的亲核取代反应实现, 常用的亲核试剂是有机锂试剂和格氏试剂[10].设计了利用甲氧基甲基格氏试剂3与氯甲基二甲基氯硅烷的亲核反应合成烷氧甲基取代的氯甲基硅烷化合物.在-30 ℃条件下[11], 四氢呋喃(THF)作为溶剂, 甲氧基甲基格氏试剂与氯甲基二甲基氯硅烷能顺利发生反应, 以45.2%的产率得到氯甲基-甲氧基甲基-二甲基硅烷(1) (Scheme 3).
接下来, 考察带有烷氧基甲基烯丙基硅的合成条件.参考本课题组[7]以往关于烯丙基硅化合物的合成条件, 以CeCl3•7H2O作为催化剂, 由氯甲基-甲氧基甲基-二甲基硅烷1制备的格氏试剂能与环丙基甲基酮(4a)顺利发生反应得到三级醇中间体5a, 无需纯化, 进而以Et2O作为溶剂, MgI2•(OEt)n作为催化剂, 发生Julia开环反应, 以18.6%的产率得到E/Z构型的混合碘代烯丙基硅产物6a, E/Z比例为1:2.1, 同时得到少量Peterson消除产物7(表 1, Entry 1).为了进一步提高目标烯丙基硅产物的产率及其立体选择性, 我们考察了不同溶剂及温度对Julia开环反应的影响(表 1).
 下载:
导出CSV
下载:
导出CSV
 |
|||||
| Entry | Solvent | T/℃ | Yieldb/% | E:Zc | 6a:7ac |
| 1 | Et2O | 50 | 18.6 | 1:2.1 | 3.4:1 |
| 2 | PhH | 50 | 43.3 | 1:1.0 | 2.5:1 |
| 3 | DME | 50 | 24.0 | 1:4.5 | 4.1:1 |
| 4 | 1, 4-Dioxane | 50 | 36.7 | 1:3.4 | 3.3:1 |
| 5 | THF | 50 | 19.5 | 1:2.7 | 3.6:1 |
| 6 | MeCN | 50 | 41.2 | 1:1.5 | 7.1:1 |
| 7 | PhMe | 50 | 39.2 | 1:1.5 | 1.7:1 |
| 8 | Et2O | 30 | 53.0 | 1:1.6 | 1.5:1 |
| 9 | Et2O | 0 | 47.8 | 1:1.5 | 2.9:1 |
| 10 | PhH | 30 | 41.2 | 1:1.1 | 2.3:1 |
| 11 | PhH | 0 | 34.1 | 1:1.8 | 3.5:1 |
| a Reaction conditions: tertiary alcohol 5a prepared from the reaction of 1 (5.0 mmol), 4a (3.4 mmol) under the presence of Mg (5.0 mmol) and CeCl3 (5.0 mmol), MgI2•(OEt)n (1.0 mmol) in dry solvent (20.0 mL). b Isolated yields of 6a based on the loading of 4a. c Determined by 1H NMR. | |||||
从表 1中可以看出, 在50 ℃反应条件下, 在醚类溶剂中(乙二醇二甲醚, 1, 4-二氧六环, THF), 能以更好的选择性得到Z式构型的烯丙基硅化合物(Entries 3~5);以MeCN作为反应介质, 则能显著抑制Peterson消除反应(Entry 6).分别以Et2O和PhH作为反应溶剂, 在不同的温度下反应, 发现降低温度, 该反应的化学选择性(6a/7a)和立体选择性(E/Z)均未有明显改善(Entries 8~11).
以Et2O和PhH作为溶剂[12], 50 ℃反应温度下, 不同环丙基酮衍生的β-羟基硅的Julia开环反应合成带有不同取代基的烯丙基硅化合物的情况如表 2所示.在实验条件下, 原位合成的带有正己基(Entries 1, 2)、高香叶基(Entries 3, 4)和苯基(Entries 5, 6)的环丙基醇5均能选择性地发生Julia开环反应, 得到高碘代烯丙基硅化合物, 且与甲基取代的底物相比, 带有较大取代基的烯丙基硅化合物产率得以显著提高.值得注意的是, 与烷基取代的三级醇底物(5a~5c)相比, 苯基环丙基酮衍生的三级醇5d发生Julia开环反应具有更好的立体选择性, 以7:1的比例得到Z式构型为主的烯丙基硅化合物.但硅原子上烷氧基的引入, 并没有明显改善高香叶基取代的烯丙基硅化合物合成的立体选择性[7a](Entries 3, 4).由此可见, 利用环丙基甲醇化合物的Julia开环反应合成烯丙基硅化合物的立体选择性与底物结构密切相关.
下载:
导出CSV
 |
|||||
| Entry | 6 | Solvent | Yieldb/% | E:Zc | 6:7c |
| 1 |  |
PhH | 72.8 | 1:1.8 | 5.6:1 |
| 2 | Et2O | 54.5 | 1:1.3 | 7.7:1 | |
| 3 |  |
PhH | 59.6 | 1:1.3 | 4.0:1 |
| 4 | Et2O | 66.1 | 1:1.2 | 4.2:1 | |
| 5 |  |
PhH | 69.6 | 1:7.1 | 5.1:1 |
| 6 | Et2O | 61.7 | 1:2.9 | 3.5:1 | |
| a Reaction conditions: tertiary alcohol prepared from the reaction of 1 (5.0 mmol), 4 (3.35 mmol) under the presence of Mg (5.0 mmol) and CeCl3 (5.0 mmol), MgI2•(OEt)n (1.0 mmol) in dry solvent (20.0 mL) at 50 ℃. b Isolated yields of 6 based on the loading of 4. c Determined by 1H NMR. | |||||
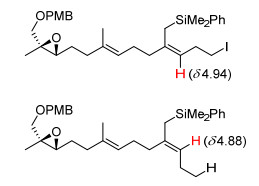
由于E、Z构型的烯丙基硅产物极性相似, 通过柱层析无法实现其分离.构型的确定是通过对比两者1H NMR谱图中形成的双键氢化学位移的相对大小, 并与已知类似化合物的谱图对照, 结合1D NOE谱图确定的.参照已知类似化合物的谱图数据, 对于高碘代烯丙基硅化合物, 碘代乙基与硅甲基位于双键同侧(Z式构型)的异构体中双键氢处于更低场[8b](图 1), 通过对比双键氢积分比例, 从而确定该反应产物的E/Z比例.
利用带有烷氧甲基硅基的环丙基甲醇化合物的Julia烯烃化反应, 成功制备了一系列新型烯丙基硅化合物.研究表明, 以MgI2•(OEt)n作为催化剂, 烷基和芳基取代的环丙基醇底物均能得到Z式构型为主的产物.反应的立体选择性与底物结构有关, 芳基取代的底物能以更好的选择性得到Z式构型烯丙基硅.所得到的该类烯丙基硅化合物同时存在卤原子、C—C双键和硅原子反应位点, 是一种潜在的重要有机合成中间体.
核磁共振氢谱和碳谱采用美国安捷伦Aglilent DD2核磁共振仪测定, CDCl3为溶剂, TMS为内标; 高分辨质谱采用Acquity SQD液相色谱-串联质谱联用仪测定.所用化学试剂和溶剂均为商业可得, 未经特殊说明, 溶剂均按照标准操作干燥处理后使用, 试剂均是购买后直接使用.使用薄层色谱(TLC, GF254硅胶板)技术监测反应进程, 显色剂采用磷钼酸、香草醛、单质碘和高锰酸钾溶液等.原料4a和4d可商业购买得到, 4b[12]、4c[9]可参照已有文献方法得到.
干燥的150 mL三口瓶中加入新鲜打磨并剪碎的Mg条(200.0 mmol, 4.8 g, 1.0 equiv.)和HgCl2 (6.0 mmol, 1.6 g, 0.03 equiv.), 置换气体后加入THF溶液(20 mL).反应混合液置于-20 ℃低温浴槽中冷却后, 滴加几滴氯甲基甲基醚(MOMCl, 2)的干燥THF溶液(40 mL)引发格氏反应, 随后将反应液冷却至-25 ℃, 滴加剩下的MOMCl溶液(15.2 mL, 16.1 g, 200 mmol, 1.0 equiv.), 滴加完毕后, 反应混合液在该温度下继续搅拌反应2 h得到格氏试剂3, 直接用于下一步反应.
将上述制备得到的格氏试剂3溶液降温至-30 ℃, N2保护下滴加入氯甲基二甲基氯硅烷(15.8 mL, 17.2 g, 120 mmol, 0.6 equiv.)的干燥THF溶液(40 mL), 滴加完毕后, 继续搅拌反应过夜.薄层色谱(TLC)监测反应完毕后, 加入甲醇(20 mL)淬灭反应, 乙酸乙酯萃取, 合并有机相后依次用水和饱和食盐水洗涤, 无水Na2SO4干燥, 旋蒸除去溶剂后, 减压蒸馏(0.09 MPa), 收集65~67 ℃馏分, 得到8.4 g无色液体1, 产率为46.0%. 1H NMR (CDCl3, 400 MHz) δ: 3.34 (s, 3H), 3.20 (s, 2H), 2.84 (s, 2H), 0.15 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ: 64.3, 63.4, 28.7, -6.1; IR (KBr) ν: 3850, 3744, 1518, 1244, 456, 420 cm-1.
N2保护下25 mL干燥三口瓶中加入新鲜打磨的镁屑(5.0 mmol, 120.2 mg)、一粒碘和干燥Et2O (2.0 mL), 室温下滴加几滴氯甲基甲氧基甲基二甲基硅烷(1, 溶解于3.0 mL干燥Et2O中), 加热引发格氏反应(碘颜色褪去, 呈白色浑浊), 然后缓慢滴加剩下的1 (5.0 mmol, 763.5 mg)的Et2O溶液, 继续搅拌反应直至固体镁屑完全消失, 直接用于下一步反应.
干燥的50 mL三口瓶中加入无水CeCl3 (5.0 mmol, 1233.0 mg), 真空条件下加热至170 ℃剧烈搅拌2 h (CeCl3呈类似液体流动的白色固体粉末), 体系中充入N2, 并冷却至室温后, 加入干燥THF (8.0 mL), 剧烈搅拌2 h形成CeCl3的均匀悬浊液(保证无结块), 而后加入环丙基甲基酮(4, 3.4 mmol)的干燥THF溶液(6.0 mL), 室温下搅拌反应2 h后, 反应混合液冷却至0 ℃, 加入格氏试剂3.滴加完毕后, 反应体系缓慢升至室温, TLC监测反应直至4完全消失(约30 min), 加入饱和NaHCO3溶液(3.0 mL)淬灭反应, 乙酸乙酯萃取, 合并有机相, 饱和食盐水洗涤, 无水MgSO4干燥, 浓缩, 所得粗产物5直接用于下一步反应.
N2保护下, 将上述制备得到的三级醇5粗产物溶解于干燥溶剂(20.0 mL)中, 50 ℃下加入MgI2•(OEt)n [0.0625 mol/L in Et2O/Benzene (V/V=1/1), 16.0 mL]溶液, 滴加完毕后, 继续搅拌反应至TLC监测原料完全消失.饱和NaHCO3溶液(6.0 mL)淬灭反应, 乙酸乙酯萃取, 合并有机相, 依次用饱和Na2S2O3溶液、H2O和饱和食盐水洗涤, 无水MgSO4干燥, 浓缩, 柱层析[V(石油醚):V(乙酸乙酯)=100:1]得到产物6.
2-环丙基-1-[(甲氧基甲基)二甲基硅基]辛-2-醇(5b):无色油状物. 1H NMR (CDCl3, 400 MHz) δ: 3.37 (s, 3H), 3.16 (s, 2H), 2.17 (s, 1H), 1.49~1.54 (m, 2H), 1.29~1.43 (m, 8H), 1.08 (s, 2H), 0.82~0.89 (m, 4H), 0.39~0.41 (m, 2H), 0.29~0.32 (m, 2H), 0.10 (s, 3H), 0.07 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 71.7, 67.4, 63.3, 44.6, 32.0, 30.0, 28.7, 24.4, 22.7, 21.7, 14.1, 1.2, 1.0, -2.5, -2.6; IR (KBr) ν: 3360, 2851, 2379, 1657, 1633 cm-1; HRMS calcd for C15H32NaO2Si [M+Na]+ 295.2064, found 259.2059.
(E, E)-2-环丙基-1-[(甲氧基甲基)二甲基硅基]-6, 10-二甲基十一-5, 9-二烯-2-醇(5c):无色油状物. 1H NMR (CDCl3, 400 MHz) δ: 5.14 (t, J=6.8 Hz, 1H), 5.09 (t, J=6.8 Hz, 1H), 3.37 (s, 3H), 3.18 (br, 1H), 3.16 (s, 2H), 2.04~2.18 (m, 4H), 1.96~2.00 (m, 2H), 1.68 (s, 3H), 1.52~1.62 (m, 8H), 1.11 (s, 2H), 0.81~0.88 (m, 1H), 0.38~0.42 (m, 2H), 0.28~0.36 (m, 2H), 0.10 (s, 3H), 0.08 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 134.7, 131.2, 124.8, 124.3, 71.6, 67.3, 63.2, 44.4, 39.5, 28.8, 26.7, 25.6, 23.0, 21.6, 17.6, 15.9, 1.2, 1.1, -2.5, -2.6; IR (KBr) ν: 3447, 3084, 2964, 2926, 1445, 1250, 1097 cm-1; HRMS calcd for C20H38NaO2Si [M+Na]+ 361.2539, found 361.2530.
1-环丙基-2-[(甲氧基甲基)二甲基硅基]苯乙醇(5d):无色油状物. 1H NMR (CDCl3, 400 MHz) δ: 7.45~7.48 (m, 2H), 7.29~7.32 (m, 2H), 7.18~7.22 (m, 1H), 4.20 (s, 1H), 3.33 (s, 3H), 3.02 (d, J=12.4 Hz, 1H), 2.88 (d, J=12.8 Hz, 1H), 1.58 (d. J=14.8 Hz, 1H), 1.47 (d, J=14.8 Hz, 1H), 1.20~1.27 (m, 1H), 0.32~0.53 (m, 3H), 0.23~0.29 (m, 1H), 0.01 (s, 3H), -0.39 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 149.2, 127.7, 126.1, 125.1, 73.6, 66.9, 63.2, 30.8, 25.6, 2.1, 1.4, -2.7, -3.6; IR (KBr) ν: 3405, 2815, 1446, 1250, 1224 cm-1. HRMS calcd for C15H24NaO2Si [M+Na]+ 287.1443, found 287.1453.
(E/Z)-(5-碘-2-甲基-戊-2-烯-1-基)(甲氧基甲基)二甲基硅烷[(E/Z)-6a]:无色油状物; 1H NMR (CDCl3, 400 MHz) δ: 4.98 [t, J=4.8 Hz, 0.5H, =C—H(Z)], 4.94 [t,J=4.8 Hz, 0.5H, =C—H(E)], 3.35 (s, 1.5H), 3.34 (s, 1.5H), 3.09~3.11 (m, 4H), 2.58 (q, J=4.8 Hz, 1H), 2.52 (q, J=4.8 Hz, 1H), 1.68 (s, 1.5H), 1.61 (s, 1.5H), 1.58 (s, 1H), 1.56 (s, 1H), 0.07 (s, 3H), 0.06 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ: 135.6, 135.5, 121.3, 121.1, 66.5, 66.3, 63.4, 63.4, 32.8, 32.7, 26.9, 26.1, 20.6, 18.9, 6.6, 6.0, -4.0, -4.4; IR (KBr) ν: 3842, 3459, 2923, 1638, 1259, 1044, 800, 548, 476 cm-1; HRMS calcd for C10H21INaOSi [M+Na]+ 335.0304, found 335.0298.
(E/Z)-(2-(3-碘代亚丙基)辛基)(甲氧基甲基)二甲基硅烷[(E/Z)-6b]:无色油状物; 1H NMR (CDCl3, 400 MHz) δ: 4.96 [t, J=4.8 Hz, 0.5H, =C—H(Z)], 4.91 (t, J=4.8 Hz, 0.5 H, =C—H(E)), 3.35 (s, 1.5H), 3.34 (s, 1.5H), 3.07~3.11 (m, 4H), 2.51~2.59 (m, 2H), 1.90~1.95 (m, 2H), 1.57 (s, 1H), 1.54 (s, 1H), 1.26~1.30 (m, 8H), 0.87~0.90 (m, 3H), 0.05 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ: 136.1, 126.7, 69.5, 62.1, 36.8, 33.5, 31.3, 29.2, 27.5, 23.0, 14.1, 5.6, 4.0, -3.7; IR (KBr) ν: 3728, 3703, 2921, 1654, 1521, 1167 cm-1. HRMS calcd for C15H32IOSi [M+H]+ 383.1267, found 383.1274.
(Z)-[2-(3-碘代亚丙基)-6, 10-二甲基-5, 9-十一二烯-1-基](甲氧基甲基)二甲基硅烷[(Z)-6c]:无色油状物. 1H NMR (CDCl3, 400 MHz) δ: 5.08~5.13 (m, 2H), 4.99 (t, J=6.8 Hz, 1H), 3.34 (s, 3H), 3.08~3.11 (m, 4H), 2.50~2.56 (m, 2H), 2.04~2.14 (m, 4H), 1.92~1.99 (m, 4H), 1.68 (s, 3H), 1.59~1.60 (m, 8H), 0.06 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ: 139.2, 135.1, 131.2, 124.3, 124.0, 120.6, 66.5, 63.4, 39.7, 39.0, 32.7, 26.8, 26.6, 25.7, 18.7, 17.7, 16.1, 6.0, -4.0; IR (KBr) ν: 3363, 3205, 2853, 2379, 1636, 837, 736 cm-1. HRMS calcd for C20H37INaO-Si [M+Na]+ 471.1556, found 471.1555.
(Z)-(5-碘-2-苯基-戊-2-烯-1-基)(甲氧基甲基)二甲基硅烷[(Z)-6d]:无色油状物; 1H NMR (CDCl3, 400 MHz) δ: 7.23~7.36 (m, 5H), 5.46 (t, J=7.0 Hz, 1H), 3.20~3.23 (m, 5H), 2.88 (s, 2H), 2.72~2.77 (m, 2H), 2.06 (s, 2H), -0.11 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ: 144.0, 139.9, 128.1, 127.0, 126.5, 124.4, 66.1, 63.2, 33.2, 18.4, 5.3, -4.3; IR (KBr) ν: 3455, 2965, 2928, 1639, 1072, 977, 804 cm-1. HRMS calcd for C15H24INaOSi [M+Na]+ 397.0461, found 397.0468.
辅助材料(Supporting Information) 化合物1, 5b~5d和6a~6d的1H NMR和13C NMR谱图.这些材料可以免费从本刊网站(http://sioc-journal.cn/)上下载.
Sommer, L. H.; Tyler, L. J.; Whitmore, F. C. J. Am. Chem. Soc. 1948, 70, 2872. doi: 10.1021/ja01189a010
For recent reviews, see: (a) Ramachandran, P. V.; Nicponski, D. R.; Gagare, P. D. In Comprehensive Organic Synthesis II, Vol. 2, Eds.: Knochel, P.; Molander, G. A., Elsevier, Amsterdam, 2014, p. 72.
(b) Díez-Poza, C.; Barbero, A. Eur. J. Org. Chem. 2017, 2017, 4651.
(c) Schmidt, A. W.; Knölker, H.-J. Synlett 2010, 2207.
(d) Chabaud, L.; James, P.; Landais, Y. Eur. J. Org. Chem. 2004, 3173.
(e) Barbero, A.; Pulido, F. J. Acc. Chem. Res. 2004, 37, 817.
(f) Méndez, M.; Echavarren, A. M. Eur. J. Org. Chem. 2002, 15.
(g) Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063.
Langkopf, E.; Schinzer, D. Chem. Rev. 1995, 95, 1375. doi: 10.1021/cr00037a011
For reviews on the synthesis of allylsilanes see 2a, and also see: (a) Sarkar, T. K. Synthesis 1990, 969.
(b) Sarkar, T. K. Synthesis 1990, 1101.
(c) Suginome, M.; Ito, Y. J. Organomet. Chem. 2003, 680, 43.
(d) Barbero, A.; Pulido, F. J. Synthesis 2004, 779.
Han, J. W.; Hayashi, T. Tetrahedron: Asymmetry 2010, 21, 2193 and references therein. doi: 10.1016/j.tetasy.2010.07.034
(a) Chan, T. H.; Wang, D. Chem. Rev. 1992, 92, 995.
(b) Masse, C. E.; Panek, J. S. Chem. Rev. 1995, 95, 1293. For selected examples on transformations of chiral allylsilanes, see:
(c) Hayashi, T.; Konishi, M.; Kumada, M. J. Am. Chem. Soc. 1982, 104, 4963.
(d) Wang, D. Chin. J. Org. Chem. 2001, 21, 1090(in Chinese). (王东, 有机化学, 2001, 21, 1090.)
(e) Wu, J.; Pu, Y.; Panek, J. S. J. Am. Chem. Soc. 2012, 134, 18440.
(a) Li, W.-D. Z.; Yang, J.-H. Org. Lett. 2004, 6, 1849.
(b) Yang, J. H.; Li, W. D. Z. Chin. Chem. Lett. 2005, 16, 433.
(a) Shen, S.-J.; Li, W.-D. Z. J. Org. Chem. 2013, 78, 7112.
(b) Gao, H.-T.; Wang, B.-L.; Li, W.-D. Z. Tetrahedron 2014, 70, 9436.
Li, W.-D. Z.; Peng, Y. Org. Lett. 2005, 7, 3069. doi: 10.1021/ol051051+
Lawrence, N. J. Science of Synthesis (Houben-Weyl), Georg Thieme Verlag, Stuttgart, Germany, 2002, pp. 579~594.
Methoxymethyl magnesium chloride is known to be obtained at low temperature but decomposes above -20℃ to release ethylene gas, see: (a) Normant, H.; Castro, B. C. R. Acad. Sci. 1963, 257, 2115.
(b) Castro, B. Bull. Soc. Chim. Fr. 1967, 1533.
Scheiper, B.; Bonnekessel, M.; Krause, H.; Fürstner, A. J. Org. Chem. 2004, 69, 3943. doi: 10.1021/jo0498866

图式 1 基于Julia烯烃化反应合成碘代烯丙基化合物
Scheme 1 Synthesis of iodo-substituted allylsilanes based on the Julia olefination

图式 2 官能化烯丙基硅化合物的立体选择性合成
Scheme 2 Stereoselective synthesis of functionalized allylsilane compounds
表 1 溶剂和温度对反应的影响a
Table 1. Effect of solvent and temperature on the reaction
|
|||||
| Entry | Solvent | T/℃ | Yieldb/% | E:Zc | 6a:7ac |
| 1 | Et2O | 50 | 18.6 | 1:2.1 | 3.4:1 |
| 2 | PhH | 50 | 43.3 | 1:1.0 | 2.5:1 |
| 3 | DME | 50 | 24.0 | 1:4.5 | 4.1:1 |
| 4 | 1, 4-Dioxane | 50 | 36.7 | 1:3.4 | 3.3:1 |
| 5 | THF | 50 | 19.5 | 1:2.7 | 3.6:1 |
| 6 | MeCN | 50 | 41.2 | 1:1.5 | 7.1:1 |
| 7 | PhMe | 50 | 39.2 | 1:1.5 | 1.7:1 |
| 8 | Et2O | 30 | 53.0 | 1:1.6 | 1.5:1 |
| 9 | Et2O | 0 | 47.8 | 1:1.5 | 2.9:1 |
| 10 | PhH | 30 | 41.2 | 1:1.1 | 2.3:1 |
| 11 | PhH | 0 | 34.1 | 1:1.8 | 3.5:1 |
| a Reaction conditions: tertiary alcohol 5a prepared from the reaction of 1 (5.0 mmol), 4a (3.4 mmol) under the presence of Mg (5.0 mmol) and CeCl3 (5.0 mmol), MgI2•(OEt)n (1.0 mmol) in dry solvent (20.0 mL). b Isolated yields of 6a based on the loading of 4a. c Determined by 1H NMR. | |||||
 下载: 导出CSV
下载: 导出CSV
表 2 带有不同取代基的烯丙基硅的合成a
Table 2. Synthesis of allylsilanes with different substituents
|
|||||
| Entry | 6 | Solvent | Yieldb/% | E:Zc | 6:7c |
| 1 | |
PhH | 72.8 | 1:1.8 | 5.6:1 |
| 2 | Et2O | 54.5 | 1:1.3 | 7.7:1 | |
| 3 | |
PhH | 59.6 | 1:1.3 | 4.0:1 |
| 4 | Et2O | 66.1 | 1:1.2 | 4.2:1 | |
| 5 | |
PhH | 69.6 | 1:7.1 | 5.1:1 |
| 6 | Et2O | 61.7 | 1:2.9 | 3.5:1 | |
| a Reaction conditions: tertiary alcohol prepared from the reaction of 1 (5.0 mmol), 4 (3.35 mmol) under the presence of Mg (5.0 mmol) and CeCl3 (5.0 mmol), MgI2•(OEt)n (1.0 mmol) in dry solvent (20.0 mL) at 50 ℃. b Isolated yields of 6 based on the loading of 4. c Determined by 1H NMR. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们