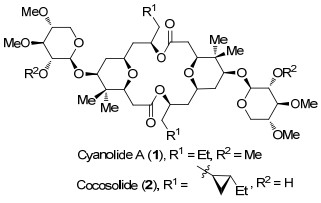
图 1.
Cyanolide A和cocosolide的结构
Figure 1.
Structures of cyanolide A and cocosolide

天然产物在现代药物领域中占据了非常重要的地位.在人类的衍生进化过程中, 自然界创造了成千上万种生物活性化合物, 它们是传统药物的主要来源.同时, 在人类社会的发展进程中, 在药物使用和推广领域逐渐建立了一套从自然界发现到商业化生产的完整体系.据估计, 过去四十年批准的所有小分子新药中, 65%是天然产物及其衍生物[1], 交叉学科的发展已经研究了这些重要分子的生态意义、生物学作用和治疗潜力.大环内酯是一类非常重要的天然产物, 具有抗菌、抗癌等良好的生理活性, 目前, 注册的大环类药物约68种, 处于临床试验的有35种[2], 如用于治疗感染的红霉素A[3]和用于抗癌治疗的埃普西隆B[4]等.由于拥有非常突出的生理活性, 大环内酯化合物引起了合成化学家和生物学家的极大兴趣.
抗生素的耐药性已经众所周知, 并且在全球范围内引起了重大的关注. 2016年, Myers等[5]提出了一种全新的合成大环内酯的方法.
Cyanolide A (1)和cocosolide (2)是两个具有对称结构的木喃糖苷类大环内酯(图 1), 前者由Gerwick等[6]于2010年从巴布亚新几内亚的Lyngbya bouillonii提取物中分离得到, 后者是2016年从关岛的一种金黄色的蓝藻提取物中分离得到[7], 它们均与2002年分离得到的天然产物Clavosolide A[8]具有相似的对称双内酯结构.
血吸虫病仍然是世界范围内最流行的寄生虫感染疾病之一, 可引起人肠道感染和泌尿系统病变, 估计受感染人数为2.07亿, 另有7.79亿人口具有被感染的风险[9~11].血吸虫病的传染主要是由宿主螺类动物引起, 通过杀死螺类软体动物是有效阻止血吸虫病传染的方法.生物活性研究表明, cyanolide A具有很好的灭螺效果, 是一种潜在的良好灭螺剂, 尤其对光滑双脐螺有很好的效果(LC50=1.2 μmol·L-1), 对血吸虫病的防治有重要的意义.
Cocosolide则可以很好抑制IL-2的产生, 研究表明, 分子结构中的糖苷基团和对称双内酯构架对其生物活性的表现有重要的意义.此外, Cocosolide对抗CD-3刺激的T-细胞也具有抗增殖作用[7].
Cyanolide A和cocosolide新颖的结构和良好的生物活性引起了合成化学家的极大兴趣[12, 13].鉴于Cyanolide A和cocosolide具有高度的结构相似性, 在四氢吡喃环上都有偕二甲基结构, 对它们的合成工作进行梳理和总结, 有利于cyanolide A和cocosolide研究工作的进一步开展.本综述根据四氢吡喃环构筑方法的不同, 如氧杂迈克尔加成反应、氧鎓离子环化反应和过渡金属催化的环化反应等, 对cyanolide A和cocosolide的合成进展情况进行总结, 在此基础上, 提出了cocosolide的研究计划.
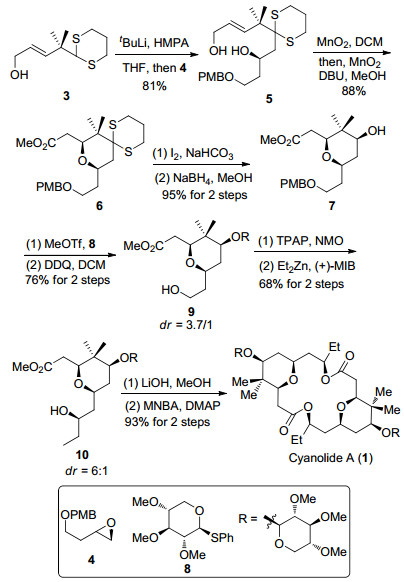
2010年, Hong等[12a]利用烯丙醇氧化-串联迈克尔加成反应率先完成了cyanolide A的全合成, 并确定其绝对构型(Scheme 1).噻烷化合物3[14]和环氧化合物4[15]反应得到二醇化合物5, 后者利用二氧化锰将一级羟基氧化为醛, 随后被氧化为相应的甲酯继而发生迈克尔加成反应, 得到四氢吡喃环化合物6.脱除四氢吡喃环上的1, 3-二噻烷结构, 接着发生羰基的立体选择性还原得到羟基化合物7.化合物7和糖苷基8[16]在路易斯酸催化下发生糖苷化反应, 以3.7/1的非对映选择性得到β构型为主的糖苷化合物, 再经过PMB基团的脱除得到伯醇9.后者在Parikh-Doering氧化条件下转化为醛, 在(+)-MIB手性环境中利用二乙基锌构建手性羟基(dr=6:1)[17]得到单体化合物10.最后经过酯基水解反应和Shiina酯化反应[18]完成天然产物cyanolide A的合成.
2011年, Reddy小组[12b]以不对称aldol反应、氧杂迈克尔加成反应和Yamaguchi酯化反应等为关键步骤, 完成了天然产物cyanolide A双内酯大环骨架的合成(Scheme 2).五元内酯11[19]经过苄基保护、酯基还原、Wittig反应、TBS保护、硼氢化氧化反应和Dess-Martin氧化反应等6个步骤, 转化为醛12, 随后发生路易斯酸控制的不对称aldol缩合反应, 以95%的非对映选择性合成了β-羟基酮14.在儿茶酚硼烷作用下, β-羟基酮发生1, 3-cis还原反应, 并进一步转化为缩丙酮15.再依次经过TBS脱除、Swern氧化反应和Horner-Wittig反应, 得到α, β-不饱和酯16.在酸性条件下进行缩丙酮水解的同时发生氧杂迈克尔加成反应[20], 顺利构建四氢吡喃环, 再经过酯基水解得到化合物17.最后经过Yamaguchi大环内酯化反应[21]和催化加氢反应即可完成cyanolide A的双内酯大环骨架18的合成.
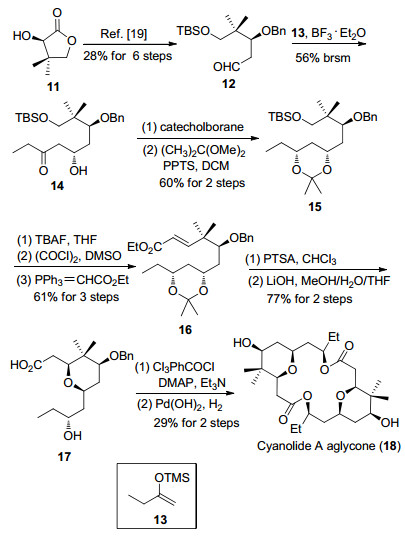
2011年, Pabbaraja小组[12d]报道了cyanolide A的双内酯大环骨架的合成(Scheme 3).首先从五元内酯11开始, 经过苄基保护、酯基还原和Wittig反应生成伯醇19.后者经过氧化反应后与手性试剂20发生Nagao-aldol反应[22], 以9:1的非对映选择性得到手性醇21, 接着将手性辅基转化为Weinreb酰胺, 在锂萘体系中脱除苄基, 再在碱性条件下将Weinreb酰胺转化为内酯化合物22.内酯22经过还原反应生成的半缩醛和磷叶立德23发生Wittig反应串联分子内氧杂迈克尔加成, 以7:2的非对映选择性制备了四氢吡喃环24.利用叔丁基二甲基硅烷保护羟基, 羰基经过科里-巴克什-柴田(Corey-Bakshi-Shibata, CBS)还原[23]生成手性醇26, 最后将双键转化为羧酸化合物27, 再利用Shiina酯化完成cyanolide A的双内酯大环骨架的合成.
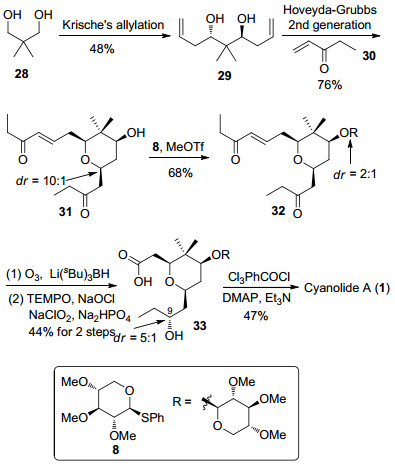
Krische课题组[24]开发了一种基于铱催化的不对称烯丙基化反应, 与传统烯丙基化方法相比, 该反应可以直接对一级醇进行不对称烯丙基化反应. 2013年, Krische等[12h]利用该反应完成了cyanolide A的全合成(Scheme 4).以新戊二醇28为原料, 经过不对称烯丙基化反应制备1, 3-anti二醇29, 随后和乙基乙烯基酮30发生烯烃复分解[25]串联氧杂迈克尔加成反应[26], 由于分子的对称性, 可以得到唯一的四氢吡喃环化合物31.四氢吡喃环中的羟基与化合物8进行糖苷化反应生成32. 32经过臭氧化反应生成的醛酮双羰基化合物发生三仲丁基硼氢化锂还原反应得到二醇化合物, 其中C(9)位置的酮羰基以5:1立体选择性得到天然产物中所需的手性仲醇结构[27].再将上述还原反应中制备的一级伯醇经过2, 2, 6, 6-四甲基哌啶-氧化物(TEMPO)氧化成为羧酸化合物33, 最后经过Yamaguchi大环内酯化反应顺利得到天然产物cyanolide A.
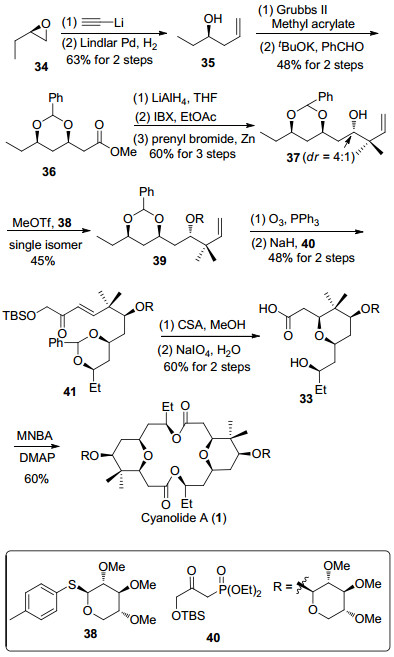
2014年, Bates课题组[12i]利用氧杂迈克尔加成反应构筑了四氢吡喃环, 并完成了cyanolide A的全合成(Scheme 5).在碳链构筑方式和1, 3-cis二羟基结构单元的制备中, 与Jennings小组的合成路线非常相似.从R-环氧丁烷开始, 经过开环反应和炔键还原生成烯丙醇35, 随后发生烯烃复分解反应和分子内氧杂迈克尔加成反应, 构筑1, 3-cis二羟基结构单元.酯36经过四氢铝锂还原和2-碘酰基苯甲酸(IBX)氧化转化为醛, 接着与3, 3-二甲基烯丙基溴发生Barbier-type反应, 以中等的立体选择性(4:1)得到化合物37.后者与38在路易斯酸下进行糖苷化反应, 得到单一的糖苷化合物39. 39经过臭氧化反应生成的醛与磷酸酯40发生Horner-Wittig反应, 生成不饱和酮41. 41在酸性条件下发生缩醛的水解以及氧杂迈克尔加成反应构筑四氢吡喃环, 同时脱除叔丁基二甲基硅烷生成α-羟基酮.随后利用高碘酸钠切断α-羟基酮得到单体33, 最后经由Shiina酯化完成cyanolide A的全合成.
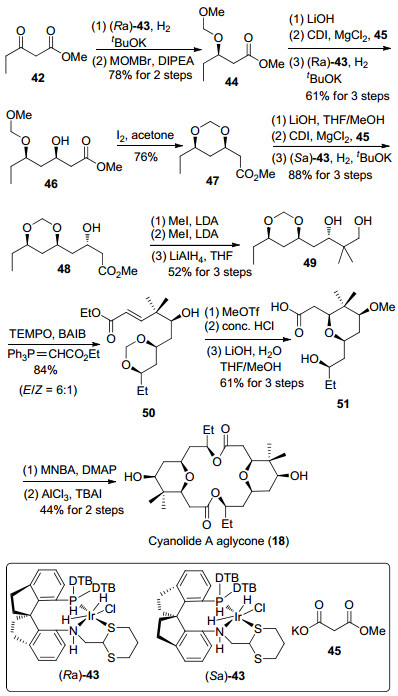
2019年, 周其林等[28]利用不对称催化加氢反应构筑了C(5), C(7)和C(9)三个手性羟基, 并利用氧杂迈克尔加成反应, 完成了cyanolide A双内酯大环骨架的合成(Scheme 6)[12j].首先利用催化剂(Ra)-43对β-羰基酯进行催化加氢, 得到的羟基进行甲氧基甲基(MOM)保护得到化合物44, 接着依次发生酯基水解、酯缩合反应和不对称催化加氢反应, 顺利制备了化合物46.在碘的丙酮溶液中, 先将MOM转化为缩甲醛, 再经过酯基水解、酯缩合反应和不对称催化加氢反应得到β-羟基酯48. 48经过两次甲基化反应和四氢铝锂还原生成1, 3-二醇49.通过选择性氧化伯醇和Wittig反应, 可以将二醇49转化为α, β-不饱和酯50.羟基发生甲醚化后在浓盐酸条件下发生缩甲醛的水解以及分子内氧杂迈克尔加成反应, 再经过酯基水解反应可以制备羧酸化合物51. 51发生Shiina酯化反应生成的双内酯化合物在三氯化铝条件下发生甲基醚的分解[30], 完成了cyanolide A双内酯大环骨架的合成.
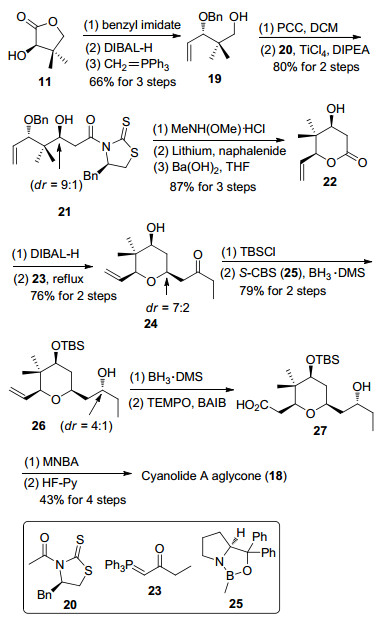
2011年, Rychnovsky小组[12e]利用无保护基合成策略完成了cyanolide A的大环内酯骨架合成, 以Prins环化反应构筑四氢吡喃环结构(Scheme 7).从原料R-环氧丁烷出发, 经缩硫醛开环氧反应得到醇53[30], 随后, 缩硫醛结构在碘作用下发生氧化脱除, 转化为二甲氧基缩醛化合物54.在羧酸片段的构筑中, 起始原料3, 3-二乙氧基-2, 2-二甲基丙酸乙酯(55)和三甲基硅基甲基锂试剂反应得到相应的甲基酮, 随后将甲基酮转化为三氟甲磺酸酯57[31].后者与三甲基硅基甲基格氏试剂发生Kumada偶联反应, 接着在酸性条件下将缩醛水解得到醛58. (-)-Sparteine诱导的不对称aldol反应和皂化反应顺利将醛58转变成β-羟基羧酸化合物60[32].通过Yamaguchi酯化反应以中等的收率顺利将醇54和羧酸60连接起来.酸催化的Prins环化反应在顺利构筑四氢吡喃环的同时生成双内酯大环骨架62.最后, 经过环外双键的氧化断裂[33]以及底物控制的羰基还原反应, 完成cyanolide A双内酯大环骨架的合成.
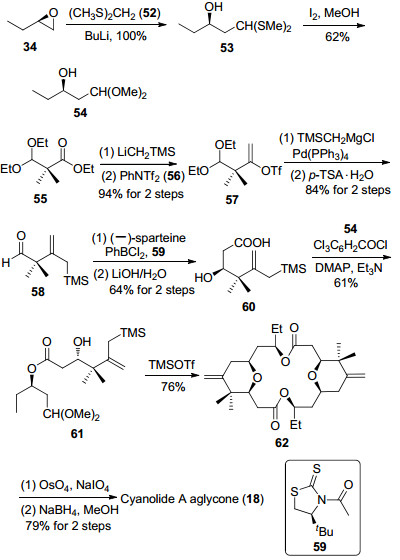
2011年, Jennings等[12f]利用氧鎓离子还原方法构筑四氢吡喃环结构, 并完成了cyanolide A大环骨架的合成(Scheme 8).从手性烯丙基型化合物35出发, 经过烯烃复分解反应和分子内氧杂迈克尔加成反应34得到醇63, 随后将其中的酯基转化为醛基, 接着与烯醇硅醚64发生Mukaiyama-aldol反应, 以4.2:1的非对映选择性完成了C(5)手性羟基的构筑[35].甲氧基甲基保护和催化加氢反应可以将碳链转化为内酯66.内酯66与烯丙基溴化镁反应生成的半缩酮在三氟乙酸和三乙基硅氢条件下发生氧鎓离子还原反应, 得到四氢吡喃67.末端双键经过臭氧化切断反应和Pinnick氧化反应转化为羧酸68.最后经过Yamaguchi大环内酯化反应及甲氧基甲基的脱除, 完成了cyanolide A双内酯大环骨架的合成.
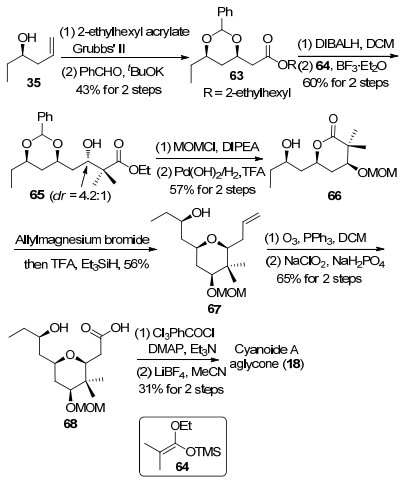
2013年, Rychnovsky等[12g]基于本课题组开发的一种构筑四氢吡喃酮的方法, 对cyanolide A进行了第二次全合成研究(Scheme 9).烯醇硅醚69与2-甲基-2溴丙酰溴(70)反应生成β, γ-不饱和酯后转化为Weinreb酰胺71.苯基硫醚72在丁基锂作用下转变成锂试剂后与Weinreb酰胺(71)反应生成β-羟基酮, 随后发生羰基不对称还原反应[36]得到1, 3-cis二醇73. 73与醛74在路易斯酸条件下发生Prins类型的环化反应, 得到四氢吡喃酮75.对羟基进行PMB保护后将酮羰基还原, 生成的化合物76与糖苷8发生糖苷化反应, 脱除PMB保护后得到的伯醇经过TEMPO氧化转化为羧酸, 再利用Shiina酯化构建双内酯大环骨架, 完成cyanolide A的全合成.
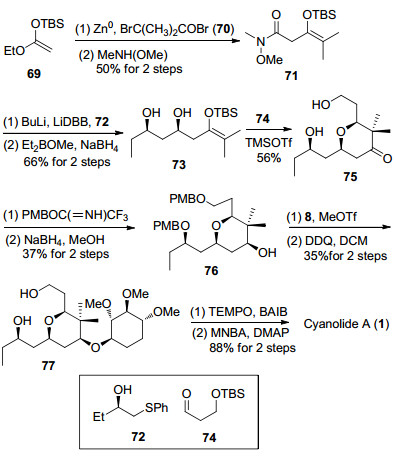
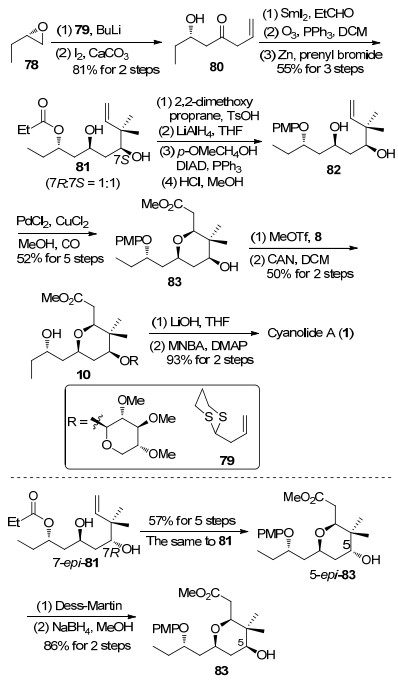
2011年, 厍学功课题组[12c]利用钯催化的烷氧羰基化反应构筑四氢吡喃环, 完成了天然产物cyanolide A的全合成工作(Scheme 10). S-环氧丁烷和噻烷79发生开环氧反应, 脱除噻烷保护后得到β-羟基酮80. β-羟基酮80经过Evans-Tishchenko还原反应[37]生成1, 3-anti二醇, 接着发生双键臭氧化切断反应, 生成的醛与3, 3-二甲基烯丙基溴发生Barbier-type反应[38]得到化合物81.将化合物81中的1, 3-二醇转化成缩丙酮, 随后将丙酰基脱除后进行C(9)位羟基的手性翻转, 构建天然产物结构中所需的羟基手性, 再利用盐酸水解缩丙酮后得到化合物82.二醇化合物82发生钯催化的双键烷氧羰基化反应[39], 构筑四氢吡喃环结构, 得到的83与糖苷8进行糖苷化反应后在硝酸铈铵条件下脱除PMP[40], 生成化合物10.最后经过酯基的水解和Shiina酯化反应, 完成cyanolide A的全合成.在上述合成工作中, 非对映异构体7-epi-81通过羟基的手性翻转可以转化化合物83.
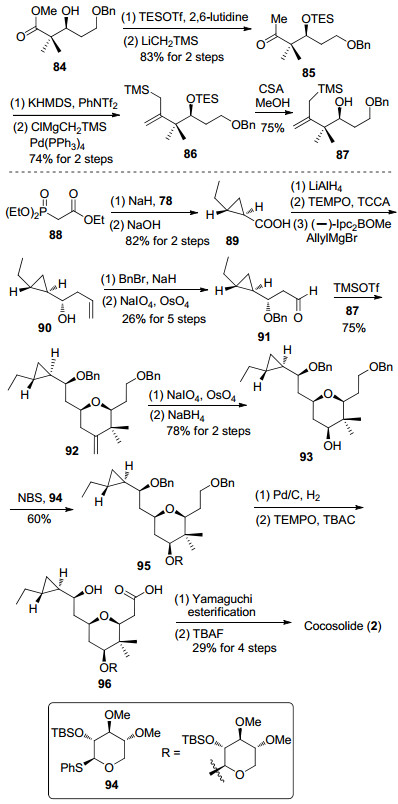
2016年, Ye等[13a]报道了cocosolide的分离, 合成以及生物活性研究等工作, 利用Prins环化反应构筑四氢吡喃环, 以Simmon-Smith反应生成手性环丙烷结构, 最后通过Yamaguchi大环内酯化反应完成合成工作(Scheme 11).在烯丙基硅烷87的制备中, 与Rychnovsky小组在cyanolide A的合成工作中相似[12e], 主要利用Kumada偶联反应来完成.
β-羟基酯84[41]经过三乙基硅烷保护后与三甲基硅基甲基锂反应制备甲基酮85, 接着在双(三甲基硅基)氨基钾(KHMDS)中将甲基酮转化为三氟甲磺酸烯醇酯, 然后与三甲基硅基甲基氯化镁发生Kumada偶联反应生成86, 最后通过樟脑磺酸(CSA)酸性条件解除三乙基硅烷保护, 完成烯丙基硅烷87的制备.商业化的原料88和S-环氧丁烷78发生Simmon-Smith反应的同时发生皂化反应, 得到环丙基甲酸化合物89[42].后者经过四氢铝锂还原、TEMPO氧化和不对称烯丙基化反应生成烯丙醇化合物90.接着发生羟基的苄基保护和末端双键的氧化断裂得到醛91.醛91和烯丙基硅烷87在路易斯酸条件下发生Prins环化反应生成四氢吡喃化合物92.环外双键氧化断裂成醛后, 在低温下被硼氢化钠还原生成四氢吡喃醇93. 93与糖苷94发生糖苷化反应, 然后利用催化加氢脱除苄基, 在TEMPO中进行选择性氧化, 将一级醇转化为羧酸得到96.最终经过Yamaguchi酯化反应和叔丁基二甲基硅烷保护基的脱除, 完成cocosolide的全合成.
大环内酯作为一种重要的天然产物类型, 广泛存在于海洋生物中.由于其结构复杂, 通常具有多手性中心, 尤其是多个手性羟基, 在合成上有较大的难度, 比如leiodelide A和neopeltolide等.因此尽管有些天然产物具有非常优秀的生理活性, 但是研究进度依然非常缓慢[43], 成为临床药物还有很远的距离.对已知大环内酯化合物的结构进行修饰是制备新的大环内酯化合物的有效方法[44], 比如克拉霉素和阿奇霉素均是红霉素衍生物.与此同时, 大环内酯类化合物通常很难通过生物发酵的方法制备, 因此, 从简单而廉价的原料开始, 通过人工合成的方法是获取大环内酯的重要手段之一.自2016年Meyers等发表了利用模块合成大环内酯的方法后, 该领域对大环内酯类天然产物及其衍生物的合成有了新的突破.
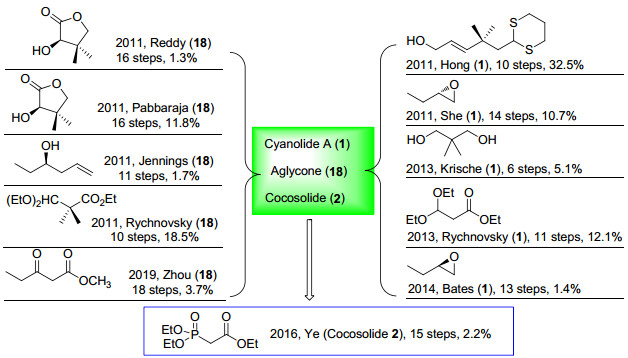
本文主要综述了具有高度结构相似性的对称双内酯天然产物cyanolide A和cocosolide的合成工作, 并根据四氢吡喃环合成方法的不同(氧杂迈克尔加成, 氧鎓离子环化反应和过渡金属催化的环化反应等)进行分类描述.到目前为止, 关于cyanolide A的合成有10篇文献报道, 而关于cocosolide则只有1篇(Scheme 12).尽管有许多的合成研究报道, 但依然存在各种各样的问题.如Hong小组的合成有步骤简短和产率较高等优点, 但起始原料需要经过一些复杂的步骤来制备. Krische的合成策略是目前最为简短的, 但是合成产率较低, 并且在合成过程中只有中等偏下的立体选择性. Zhou利用甲氧基甲醚进行1, 3-二羟基保护的方法, 尽管新颖, 但是合成步骤冗长, 合成效率并不好.而在cocosolide的合成中, Ye小组利用Prins环化反应和Yamaguchi大环内酯化反应完成它的首次合成, 但是产率较低.因此, 关于cyanolide A和cocosolide及其类似结构化合物的合成有必要进行更深入的研究.
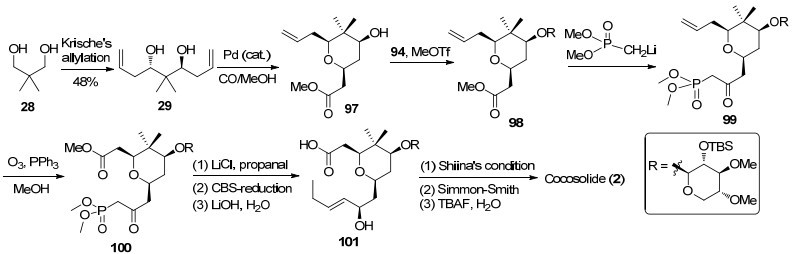
基于对cyanolide A合成工作的深入研究和cocosolide的结构分析, 我们提出了针对cocosolide的合成方案.如Scheme 13所示, 利用Krische不对称烯丙基化将新戊二醇转化为手性二醇化合物29, 随后发生钯催化的双键烷氧羰基化反应制备四氢吡喃环97, 由于二醇结构的对称性可以得到单一产物.四氢吡喃环中的羟基与糖苷8发生糖苷化反应生成化合物98.甲基膦酸二甲酯锂盐可以将化合物98中的酯基转化为羰基得到β-羰基膦酸酯99, 接着末端双键在臭氧化条件下转化为酯基化合物100.化合物100依次经过Horner-Wadsworth-Emmons反应、CBS还原反应以及酯基的水解反应得到大环内酯化前驱体羧酸化合物101.在Shiina酯化条件下发生大环化反应生成双内酯大环骨架, 再将其中的双键进行Simmon-Smith反应构筑手性环丙烷结构, 最后脱除硅烷保护完成天然产物cocosolide的不对称合成, 目前, 已经取得了一定的进展.在后续研究中, 我们将围绕对称的双内酯大环骨架及相关化合物的合成展开进一步研究工作.
Newman, D. J.; Cragg, G. M. J. Nat. Prod. 2016, 79, 629. doi: 10.1021/acs.jnatprod.5b01055
Giordanetto, F.; Kihlberg, J. J. Med. Chem. 2014, 57, 278. doi: 10.1021/jm400887j
McGuire, J. M.; Bunch, R. L.; Anderson, R. C.; Boaz, H. E.; Flynn, E. H.; Powell, H. M.; Smith, J. W. Antibiot. Chemother. 1952, 2, 281.
Altmann, K.-H.; Gaugaz, F. Z.; Schiess, R. Mol. Diversity 2011, 15, 383. doi: 10.1007/s11030-010-9291-0
Seiple, I. B.; Zhang, Z. Y.; Jakubec, P.; Langlois-Mercier, A.; Wright, P. M.; Hog, D. T.; Yabu, K. Z.; Allu, S. R.; Fukuzaki, T.; Carlsen, P. N.; Kitamura, Y.; Zhou, X.; Condakes, M. L.; Szczypiński, F. T.; Green, W. D.; Myers, A. G. Nature 2016, 533, 338. doi: 10.1038/nature17967
Pereira, A. R.; McCue, C. F.; Gerwick, W. H. J. Nat. Prod. 2010, 73, 217. doi: 10.1021/np9008128
Gunaseker, S. P.; Li, Y.; Ratnayake, R.; Luo, D. M.; Lo, J.; Reibenspies, J. H.; Xu, Z. S.; Clare-Salzler, M. J.; Ye, T.; Paul, V. J.; Luesch, H. Chem.-Eur. J. 2016, 22, 8158. doi: 10.1002/chem.201600674
Rao, R. M.; Faulkner, D. J. J. Nat. Prod. 2002, 65, 386. doi: 10.1021/np010495l
Steinmann, P.; Keiser, J.; Bos, R.; Tanner, M.; Utzinger, J. Lancet. Infect. Dis. 2006, 6, 411. doi: 10.1016/S1473-3099(06)70521-7
Chitsulo, L.; Engels, D.; Montresor, A.; Savioli, L. Acta Trop. 2000, 77, 41. doi: 10.1016/S0001-706X(00)00122-4
World Health Organization Report of the Scientific Working Group Meeting on Schistosomiasis, Geneva, Switzerland, November 14~16, 2005.
(a) Hong, J.; Kim, H. Org. Lett. 2010, 12, 2880.
(b) Hajare, A. K.; Ravikumar, V.; Khaleel, S.; Bhuniya, D.; Reddy, D. S. J. Org. Chem. 2011, 76, 963.
(c) Yang, Z.; Xie, X.; Jing, P.; Zhao, G.; Zheng, J.; Zhao, C.; She, X. Org. Biomol. Chem. 2011, 9, 984.
(d) Pabbaraja, S.; Satyanarayana, K.; Ganganna, B.; Yadav, J. S. J. Org. Chem. 2011, 76, 1922.
(e) Gesinski, M. R.; Rychnovsky, S. D. J. Am. Chem. Soc. 2011, 133, 9727.
(f) Sharpe, R. J.; Jennings, M. P. J. Org. Chem. 2011, 76, 8027.
(g) Tay, G. C.; Gesinski, M. R.; Rychnovsky, S. D. Org. Lett. 2013, 15, 4536.
(h) Waldeck, A. R.; Krische, M. J. Angew. Chem., Int. Ed. 2013, 52, 4470.
(i) Bates, R. W.; Lek, T. G. Synthesis 2014, 46, 1731.
(j) Che, W.; Li, Y. Z.; Liu, J. C.; Zhu, S. F.; Xie, J. H.; Zhou, Q. L. Org. Lett. 2019, 21, 2369.
(j) Lee, K.; Lanier, M. L.; Kwak, J. H.; Kim, H.; Hong, J. Y. Nat. Prod. Rep. 2016, 33, 1393.
(a) Ye, T.; Xu, Z.; Li, Y.; Luesch, H.; Paul, V. J.; Gunasekera, S. P. CN 105884843, 2016.
(b) Luesch, H.; Paul, V. J.; Gunasekera, S. WO 2017152099, 2017.
Armesto, D.; Horspool, W. M.; Gallego, M. G.; Agarrabeitia, A. R. J. Chem. Soc., Perkin Trans. 1 1992, 163.
Mohapatra, D. K.; Das, P. P.; Reddy, D. S.; Yadav, J. S. Tetrahedron Lett. 2009, 50, 5941. doi: 10.1016/j.tetlet.2009.08.028
Barry, C. S.; Bushby, N.; Charmant, J. P. H.; Elsworth, J. D.; Harding, J. R.; Willis, C. L. Chem. Commun. 2005, 5097
(a) Nugent, W. A. Chem. Commun. 1999, 1369.
(b) Chen, Y. K.; Lurain, A. E.; Walsh, P. J. J. Am. Chem. Soc. 2002, 124, 12225.
Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. J. Org. Chem. 2004, 69, 1822. doi: 10.1021/jo030367x
Ball, M.; Baron, A.; Bradshaw, B.; Omori, H.; MacCormick, S.; Thomas, E. J. Tetrahedron Lett. 2004, 45, 8737. doi: 10.1016/j.tetlet.2004.09.124
Liu, J.; Yang, J. H.; Ko, C.; Hsung, R. P. Tetrahedron Lett. 2006, 47, 6121. doi: 10.1016/j.tetlet.2006.06.067
Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn. 1979, 52, 1989. doi: 10.1246/bcsj.52.1989
Crimmins, M. T.; Emmitte, K. A. Org. Lett. 1999, 1, 2029. doi: 10.1021/ol991201e
Corey, E. J.; Bakshi, R. K.; Shibata, S. J. Am. Chem. Soc. 1987, 109, 5551. doi: 10.1021/ja00252a056
(a) Bower, J. F.; Kim, I. S.; Patman, R. L.; Krische, M. J. Angew. Chem., Int. Ed. 2008, 48, 34.
(b) Patman, R. L.; Bower, J. F.; Kim, I. S.; Krische, M. J. Aldrichim. Acta 2008, 41, 95.
(c) Han, S. B.; Kim, I. S.; Krische, M. J. Chem. Commun. 2009, 7278.
Fuwa, H.; Noto, K.; Sasaki, M. Org. Lett. 2011, 13, 1820. doi: 10.1021/ol200333p
Fuwa, H. Heterocycles 2012, 85, 1255. doi: 10.3987/REV-12-730
Pirrung, M. C.; Kenney, P. M. J. Org. Chem. 1987, 52, 2335. doi: 10.1021/jo00387a053
(a) Che, W.; Wen, D. C.; Zhu, S. F.; Zhou, Q. L. Org. Lett. 2018, 20, 3305.
(b) Bao, D. H.; Wu, H. L.; Liu, C.-L.; Xie, J. H.; Zhou, Q. L. Angew. Chem., Int. Ed. 2015, 54, 8791.
Sun, J.; Dong, Y.; Cao, L.; Wang, X.; Wang, S.; Hu, Y., J. Org. Chem. 2004, 69, 8932. doi: 10.1021/jo0486239
Schaus, S. E.; Brandes, B. D.; Larrow, J. F.; Tokunaga, M.; Hansen, K. B.; Gould, A. E.; Furrow, M. E.; Jacobsen, E. N. J. Am. Chem. Soc. 2002, 124, 1307. doi: 10.1021/ja016737l
Nicolaou, K. C.; Li, A.; Edmonds, D. J.; Tria, S.; Ellery, S. P. J. Am. Chem. Soc. 2009, 131, 16905. doi: 10.1021/ja9068003
Zhang, Y.; Sammakia, T. J. Org. Chem. 2006, 71, 6262. doi: 10.1021/jo0605694
VanRheenen, V.; Kelly, R. C.; Cha, D. Y. Tetrahedron Lett. 1976, 17, 1973 doi: 10.1016/S0040-4039(00)78093-2
(a) Evans, D. A.; Gauchet-Prunet, J. A. J. Org. Chem., 1993, 58, 2446.
(b) Rotulo-Sims, D.; Prunet, J. Org. Lett. 2007, 9, 4147
Evans, D. A.; Dart, M. J.; Duffy, J. L.; Yang, M. G. J. Am. Chem. Soc. 1996, 118, 4322. doi: 10.1021/ja953901u
Narasaka, K.; Pai, F. C. Tetrahedron 1984, 40, 2233. doi: 10.1016/0040-4020(84)80006-X
Evans, D. A.; Hoveyda, A. H. J. Am. Chem. Soc. 1990, 112, 6447 doi: 10.1021/ja00173a071
Petrier, C.; Luche, J. L. J. Org. Chem. 1985, 50, 910. doi: 10.1021/jo00206a047
White, J. D.; Hong, J.; Robarge, L. A. Tetrahedron Lett. 1999, 40, 1463. doi: 10.1016/S0040-4039(98)02693-8
Gao, D.; O'Doherty, G. A. J. Org. Chem. 2005, 70, 9932. doi: 10.1021/jo051681p
Reiff, E. A.; Nair, S. K.; Henri, J. T.; Greiner, J. F.; Reddy, B. S.; Chakrasali, R.; David, S. A.; Chiu, T.-L.; Amin, E. A.; Himes, R. H.; Vander Velde, D. G.; Georg, G. I. J. Org. Chem. 2010, 75, 86. doi: 10.1021/jo901752v
(a) Armstrong, A.; Scutt, J. N. Org. Lett. 2003, 5, 2331.
(b) Armstrong, A.; Scutt, J. N. Chem. Commun. 2004, 510.
(c) Delhaye, L.; Merschaert, A.; Delbeke, P.; Briúne, W. Org. Process Res. Dev. 2007, 11, 689.
(d) Bray, C. D.; Minicone, F. Chem. Commun. 2010, 46, 5867.
(e) Kumar, P.; Dubey, A.; Harbindu, A. Org. Biomol. Chem. 2012, 10, 6987.
(a) Ren, R. G.; Mao, Z. Y.; Wei, B. G.; Lin, G. G. Chin. J. Org. Chem. 2015, 35, 2313 (in Chinese).
(任荣国, 毛卓亚, 魏邦国, 林国强, 有机化学, 2015, 35, 2313.)
(b) Yu, J. F.; Feng, R. K.; Yang, Z. Chin. J. Org. Chem. 2017, 37, 2526 (in Chinese).
(于江帆, 冯若昆, 杨震, 有机化学, 2017, 37, 2526.
史大昕, 冯雪, 庄晓磊, 柴洪新, 刘霆, 张奇, 李加荣, 有机化学, 2014, 34, 2543. http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344571.shtmlShi, D. X.; Feng, X.; Zhuang, X. L.; Chai, H. X.; Liu, T.; Zhang, Q.; Li, J. R. Chin. J. Org. Chem. 2014, 34, 2543 (in Chinese). http://sioc-journal.cn/Jwk_yjhx/CN/abstract/abstract344571.shtml

图式 3 Pabbaraja小组cyanolide A双内酯大环骨架的合成
Scheme 3 Pabbaraja's synthesis of cyanolide A aglycone

图式 7 Rychnovsky第一代cyanolide A双内酯大环骨架的合成
Scheme 7 Rychnovsky's first generation synthesis of cyanolide A aglycone

图式 8 Jennings小组cyanolide A双内酯大环骨架的合成
Scheme 8 Jennings's synthesis of cyanolide A aglycone

图式 9 Rychnovsky’s第二代cyanolide A全合成
Scheme 9 Rychnovsky's second generation synthesis of cyanolide A

图式 12 Cyanolide A (1)和cocosolide (2)的合成
Scheme 12 Synthesis of cyanolide A (1) and cocosolide (2)

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:







