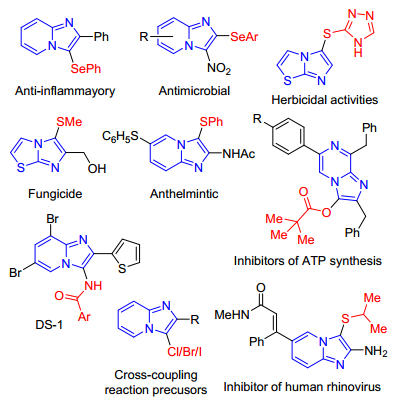
Figure 1.
Representative examples of bioactive imidazole-fused heterocycles

Nitrogen-containing heterocycles are often recognized as ubiquitous structural motif present in numerous natural products, bioactive molecules and functional materials.[1] In particular, imidazole-fused heterocycles, such as imidazopyridines and imidazothiazoles are the most important classes compounds which show a broad spectrum of biological activities and has been widely used in the organic synthesis and materials science.[2] Recently, seeking efficient and novel methods for the synthesis of functionalized imidazole-fused heterocycles has attracted much attention in organic and pharmaceutical chemistry since it can lead to a new class of active compounds with salient bioactivity like antiviral, antibacterial, antiulcer, anticancer, etc. (Figure 1).[3]
In order to access functionalized imidazole-fused heterocycles, further modification through C—H functionalization is a practical and intriguing strategy.[4] In generally, transition-metal-catalyzed cross-couplings between C—X/C—H and different heteroatomic reagents are well known.[5] However, either functionalized substrates or stoichiometric amount of chemical oxidants such as hypervalent metal salts, organic halides, peroxides and oxygen are required in these transformations. Therefore, from the viewpoint of green chemistry, direct transition metal-free C—H bond functionalization provides an ideal way to functionalize imidazole-fused heterocycles and has attracted much attention in recent years, and many excellent and significant works have been presented. The recent progress in the incorporation of heteroatom into imidazole-fused heterocycles through transition metal-free C—H functionalization is introduced and their mechanisms from a new perspective in order to provide reference for related research are also elaborated.
Among the various functionalized imidazole-fused heterocycles, the synthesis of C-3 aminated imidazole-fused heterocycles has gained much attention because of their various biological properties in medicinal chemistry.[6] However, few reports are available for the expeditious synthesis of these molecules via direct incorporation of amine or amine derivatives to a carbon atom through the formation of C—N bond under transition metal-free conditions.[7]
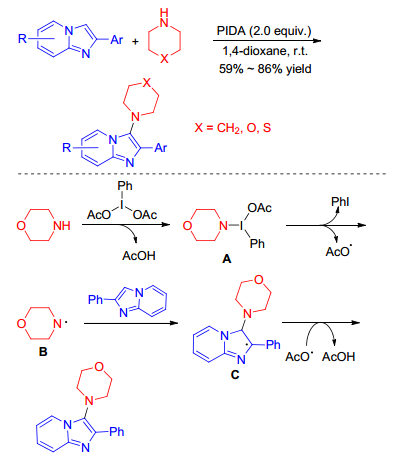
In 2017, Hajra group[8] achieved the PIDA-mediated direct oxidative C—H amination of imidazopyridines under transition metal-free conditions at room temperature. This methodology has broad substrate scopes and provides the corresponding 3-amino substituted imidazopyridines in moderate to high yields in short reaction times. Experimental results suggest that the reaction likely proceeds through a radical pathway (Scheme 1). Initially, N-iodo-amido species A is formed from the morpholine which subsequently give morpholine radical B. The resulting morpholine radical B reacts with imidazo[1, 2-a]pyridine moiety to produce the radical intermediate C which consequently affords the desired product through the elimination of AcOH. The present methodology is the first report for the direct intermolecular amination of imidazopyridines and will gain much importance in organic chemistry.
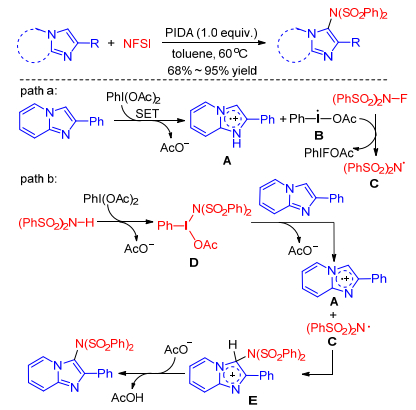
Subsequently, the same group[9] developed the PIDA-mediated regioselective imidation of imidazole-fused heterocycles using commercially available N-fluorobenze-nesulfonimide as an imidating reagent as well in 2018. Similar to the above mentioned method, this protocol can afford various imidated imidazopyridines with good to excellent yields under mild conditions in short times and also proceeds through a radical pathway.
Probably, the first step is the PIDA-mediated oxidation of imidazopyridine to radical cation species A through a single electron-transfer process along with the formation of radical B which is oxidized by NFSI affording the bis-sulfonylamidyl radical C. On the other hand, diben-zenesulfonimide could also produce N-iodoamido species D in the presence of PIDA which subsequently reacts with imidazopyridine to give bis-sulfonylamidyl radical C and imidazopyridine radical cation A. Finally, the resulting imidazopyridine radical cation A regioselectively coupled with the bis-sulfonylamidyl radical C to produce the imidazolenium ion E which consequently affords the product through the elimination of AcOH (Scheme 2).
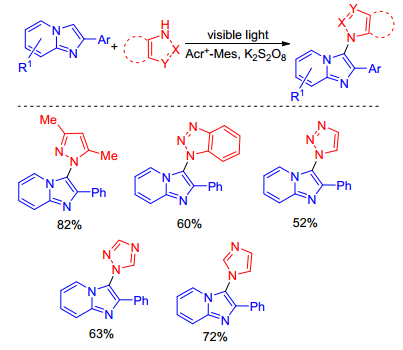
In 2017, Adimurthy and co-workers[10] reported an efficient visible light induced C—H amination of imidazopyridines under transition-metal-free conditions. This method was discovered when they investigated C—H amination of quinoline amides and also has been extended to other heteroamines with good functional group tolerance (Sheme 3). Experimental results indicate that the reaction also proceeds via a radical pathway.
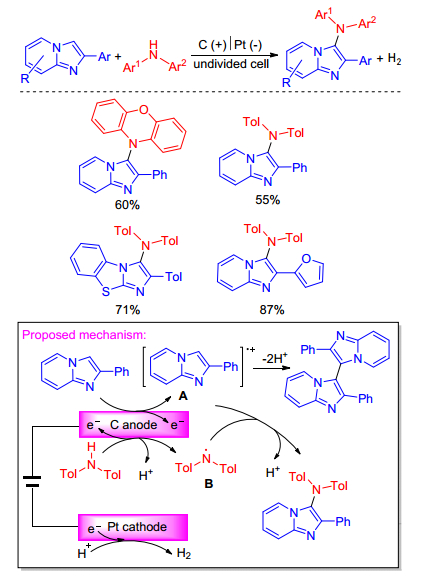
Recently, Lei group[11] presented an effective electrochemical system for the direct C—H amination of imidazopyridines with diarylamines in a simple undivided cell without using metal catalyst or stoichiometric amount of external chemical oxidants. This protocol provided a simple and efficient way to access a variety of heterocycles containing triarylamines under transition metal-free conditions. Mechanism studies indicate that firstly, imidazopyridines could be oxidized at anode to generate radical cation intermediate A. At the same time, diarylamines might be oxidized by carbon anode to generate nitrogen radical B. C—N bond was likely to be formed through the radical/radical cross coupling. Following deprotonation would afford the desired product. Concomitant cathodic reduction could release hydrogen gas during the reaction process (Scheme 4).
Imidazole-fused heterocycles containing alkoxyl group are widely used in biological and medicinal fields, in which C-3 alkoxyl groups containing imidazo[1, 2-a]pyridines have been used as the potent inhibitors of mycobacterial adenosine triphosphate and to measure the luciferase activity in living cells.[12] Even so, there are only few approaches to synthesize imidazopyridine ethers, especially no method for direct C—H alkoxylation on imidazopyridine moieties.[13]
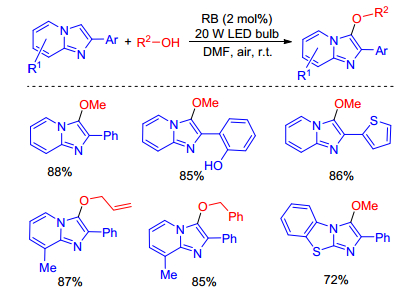
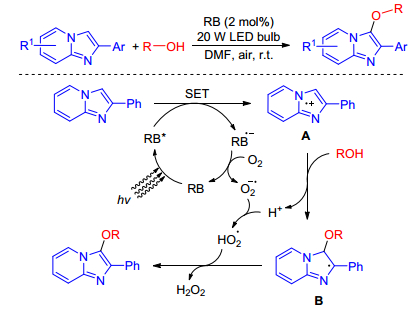
In 2017, Hajra and co-workers[14] reported a direct and environmentally benign method for the alkoxylation of imidazopyridines at room temperature using rose bengal (RB) as an organic photoredox catalyst and ambient air as the terminal oxidant. This methodology tolerates a wide range of functional groups under standard reaction conditions tooffer alkoxylated products in high yields, but not applicable for the C-2 alkyl-substituted and unsubstituted imidazo[1, 2-a]pyridine, sterically hindered tertiary alcohols, and aliphatic thiols (Scheme 5). To our surprise, this is the first and only report for the direct C—H alkoxylation of imidazopyridines so far.
They proposed a plausible mechanistic pathway in Scheme 6 on the basis of experimental result and reported literatures. A single electron transfer (SET) from imidazopyridine to the excited state of rose bengal (RB*) afforded the imidazopyridine radical cation A and photocatalyst radical anion (RB•−). The resulting imidazopyridine radical cation reacts with alcohol to yield the alkoxide adduct radical B. The photoredox cycle is completed by the reduction of O2 to O2•−, which can abstract a proton to produce HO2•. Hydrogen abstraction by HO2• from intermediate B affords the alkoxylation product with H2O2.
Among the functionalization of imidazole-fused heterocycles, introducing organosulfur group into imidazole-fused heterocycles has been considered to be important since it can lead to a new class of biologically active compounds.[15] In generally, transition-metal-catalyzed cross-couplings between C—X/C—H and different sulfenylating reagents is well known.[16] However, from the viewpoint of green chemistry, direct transition metal-free C—H bond sulfenylation provides an ideal way to sulfenylated imidazole-fused heterocycles.
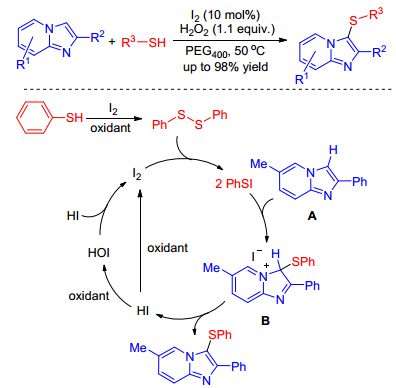
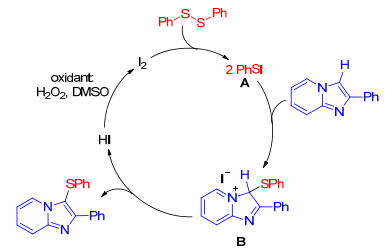
As the easily available and high atom economy sulfur sources, thiols or thiones are often employed for the sulfenylation of imidazole-fused heterocycles. In 2015, Hiebel and co-workers[17] developed a benign and efficient iodine-catalyzed regioselective sulfenylation of imidazole-fused heterocycles using hydrogen peroxide as oxidizing agent. This method tolerated various functional groups and enabled the formation of 3-sulfenylimidazole-fused heterocycles in moderate to excellent yields in PEG400. A plausible mechanism proposed for this transformation is outlined in Scheme 7. Initially, phenyl disulfide which is generated from thiophenol in the presence of I2 or H2O2 reacted with I2 affording the electrophilic PhSI. An electrophilic attack of PhSI on the C-3 position of A can occur leading to an imidazolenium intermediate B. Releasing hydroiodic acid forms target molecules. Then, part of HI can be oxidized by H2O2 into hypoiodous acid, which reacts another portion of HI to regenerate I2 with the formation of water.
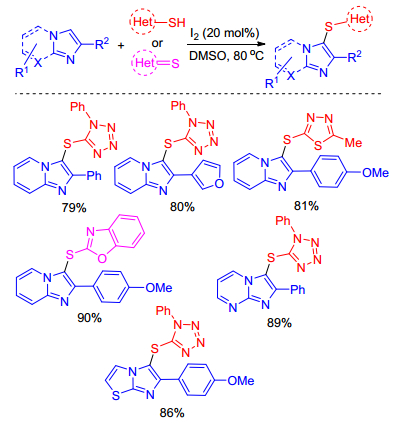
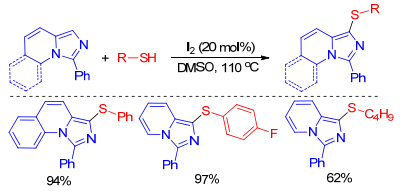
On the basis of analogous mechanism, iodine-catalyzed sulfenylation of imidazole-fused heterocycles could undergo well just using DMSO or air instead of hydrogen peroxide as oxidizing agents. For instance, Prabhu group[18] described a simple synthetic approach for regioselective sulfenylation of imidazole-fused heterocycles using iodine as catalyst and DMSO as oxidant in 2016. This is the first report of sulfenylation of imidazole-fused heterocycles with heterocyclic thiols or thiones under transition metal-free conditions and it presents an efficient, mild, and inexpensive method for sulfenylation of imidazole-fused heterocycles with diverse heterocyclic thiols and heterocyclic thiones (Scheme 8).
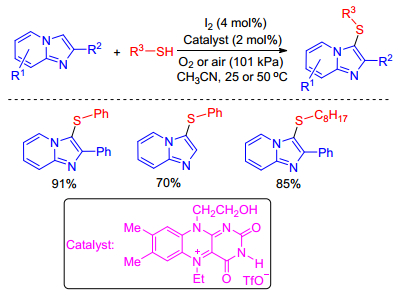
Recently, Iida group[19] performed a green, aerobic sulfenylation of imidazo[1, 2-a]pyridines with thiols using a flavin-and-iodine dual catalytic system, and environmentally benign molecular oxygen as the only sacrificial reagent. The dual transition metal-free catalysts smoothly promote a unique stepwise tandem process, beginning with the aerobic oxidation of a thiol to afford a disulfide that is utilized in Hiebel's work. This methodology enables O2-driven sulfenylation in the absence of stoichiometric amounts of expensive sacrificial reagents and toxic metals, and provides attractive green sulfenylation chemistry to access biologically important 3-sulfenylimidazo[1, 2-a]-pyridines in good yields (Scheme 9).
In 2017, Feng and co-workers[20] completed a new iodine-catalyzed convenient and regioselective protocol to synthesize 3-sulfenylimidazo[1, 5-a]quinoline derivatives by employing thiophenols as sulfenylating agents. Under transition metal and oxidant-free reaction conditions, 3-sulfenylimidazo[1, 5-a]quinoline derivatives were obtained in good to excellent yields with broad functional group tolerance (Scheme 10).
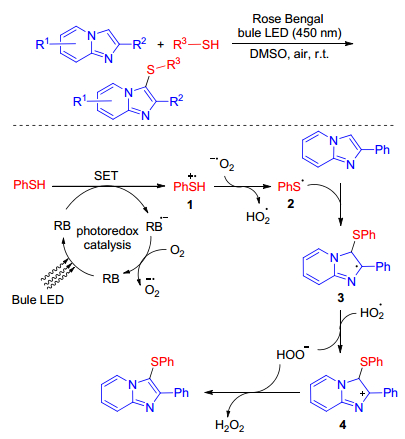
Due to environmental compatibility, excellent functional group tolerance, high reactivity and versatility, visible-light mediated photoredox catalysis in organic synthetic chemistry has extensively been exploited. Barman andco-workers[21] firstly reported visible light induced sulfenylation of imidazole-fused heterocycles with thiols. A series of biologically important C(3) sulfenylated imidazole-fused heterocycles was obtained conveniently in good to excellent yields using readily available materials. The presented method is endowed with several important features including operational simplicity, high atom efficiency and green solvent under ambient conditions.
Authors proposed a plausible reaction pathway for the photocatalytic sulfenylation of imidazole-fused heterocycles shown in Scheme 11. Initially, Rose Bengal was excited under the presence of blue LED light to produce RB*. Then a single electron transfer (SET) from thiol to RB* afforded the radical cation 1 and RB•−, which was oxidized by air generating the ground state Rose Bengal and O2•−. The deprotonation of radical cation 1 by O2•− leads to the formation of stabilized thiyl radical 2 that reacts with imidazo derivatives to produce the radical intermediate 3. Finally, along with the generation of HO2−, 3 is oxidized to intermediate 4, after deprotonation of which affording the desired product along with the release of H2O2. To complete the photoredox cycle, aerobic oxygen probably plays a crucial role to oxidize RB radical anions to the ground state.
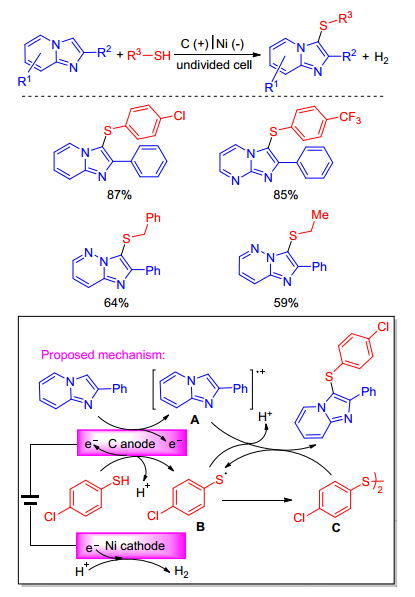
Electrochemistry has also been applied for C—H sulfenylation of imidazole-fused heterocycles. Recently, Lei group[22] achieved selective oxidative C—H sulfenylation of imidazole-fused heterocycles using an undivided electrolytic cell. The reaction avoids the use of stoichiometric amount of external chemical oxidant and produces hydrogen gas as the only byproduct. A plausible mechanism for the reaction is shown in Scheme 12. Oxidation of imidazoheterocycle at anode could generate intermediate A. Oxidation of thiols at anode could form sulfur radical B. The generated thiyl radical could undergo dimerization to give disulfide C. The radical cross-coupling between A and B or A and C all could form C-S bond. Addition of B to imidazo derivatives could not be fully ruled out. Final deprotonation would lead to the target product. At the cathode, the co-solvent methanol could be reduced to give hydrogen gas during the reaction.
In most proposed mechanisms, disulfides can react with iodine to afford electrophilic species RSI which is nucleophilic attacked by various substrates producing sulfenylated products with the formation of HI. Thus disulfides is a kind of frequently employed sulfenylating reagents.
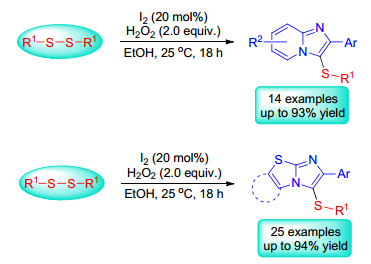
In recent years, Tang, Braga, Wang, et al. achieved the direct sulfenylation of imidazole-fused heterocycles with disulfides in iodine-oxidants systems. For example, Tang and co-workers[23] reported an iodine/hydrogen peroxide-promoted regioselective sulfenylation of imidazole-fused heterocycles with disulfides in ethanol at room temperature in 2015. This green strategy provides a convenient and efficient route for the preparation of diverse sulfenylated imidazole-fused heterocycles under transition metal-free conditions (Scheme 13).
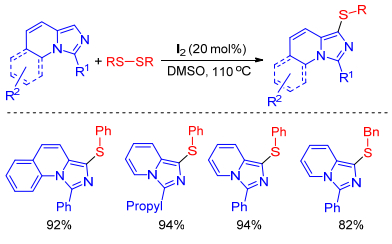
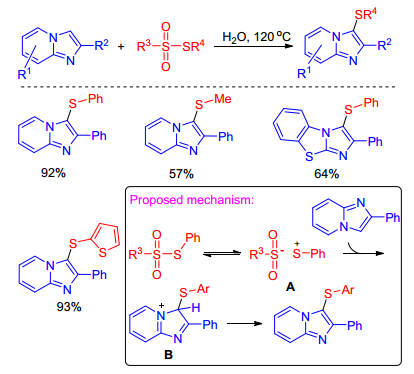
Braga group successively achieved I2-catalyzed[24] and NH4I-catalyzed[25] sulfenylation of imidazole-fused heterocycles with disulfides using DMSO as oxidant in the absence of transition metal catalyst. This mild, green approach allowed the preparation of different types of sulfenylated imidazole-fused heterocycles with structural diversity in good to excellent yields. Furthermore, the current protocol was also extended to other N-heterocyclic cores (Scheme 14).
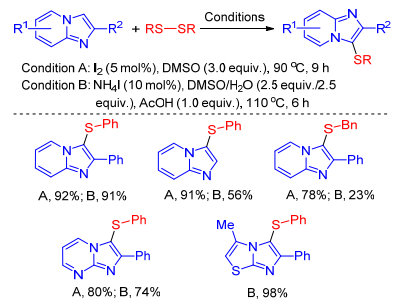
Feng's work[20] demonstrated that the iodine-catalyzed regioselective sulfenylation of imidazo[1, 5-a]quinolones can undergo well with not only thiophenols but also disulfides. Commercially available disulfides could be employed as efficient sulfenylating agents to synthesize diverse 3-sulfe-nylated imidazo[1, 5-a]quinolones (Scheme 15).
The mechanism of the aforementioned sulfenylation is alike and depicted in Scheme 16. Diphenyl disulfide firstly reacts with iodine to generate PhSI in situ, followed by Friedel-Crafts reaction with substrate to produce intermediate B. Intermediate B then undergoes a dehydrogenation process to afford the product and hydrogen iodide. Finally, hydrogen iodide can be readily oxidized by oxidants to regenerate iodine.
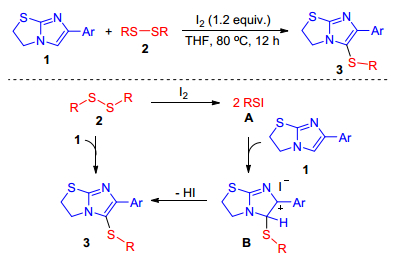
Notably, this reaction also could be promoted using stoichiometric amount iodine in the absence of oxidants. In 2016, Wang group[26] discovered a novel and facile method for the direct sulfenylation of 2, 3-dihydroimidazo-[2, 1-b]thiazoles with disulfides. This transformation is promoted by iodine under transition metal-free conditions, providing the sulfenylated products in moderate to good yields. A plausible mechanism is illustrated in Scheme 17. Initially, disulfides 2 reacts with I2 to generate an elecro philic species RSI A. Subsequently, an electrophilic attack of RS+on imidazo ring forms intermediate B. Finally, deprotonation of B provides the desired product 3.
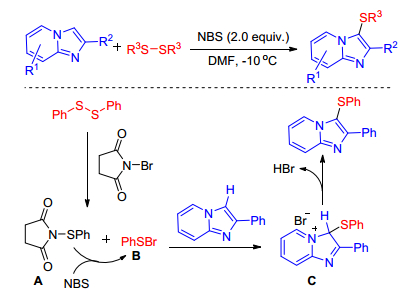
In 2017, Meshram group[27] developed the N-bromo-succinimide promoted direct sulfenylation of imidazole-fused heterocycles with disulfides. A wide range of heterocycles and disulfides were tested, and the desired sulfenylated imidazo compounds were obtained in good to high yields in the presence of inexpensive NBS. A probable mechanism for the formation of sulfenylated imidazo compounds is proposed in Scheme 18. Initially, intermediates A and B were formed by the reaction of disulfides with NBS. Subsequently, the C(3) position of imidazole was attacked by intermediate B to furnish the imidazolenium intermediate C, which on dehydrobromination gave the final product. Furthermore, N-phenyl thiosuccinimide A might also be converted into the highly reactive phenylsulfenyl bromide B in presence of NBS. Yang and co-workers[28] sequentially found NIS also can facilitate sulfenylation of imidazole-fused heterocycles.
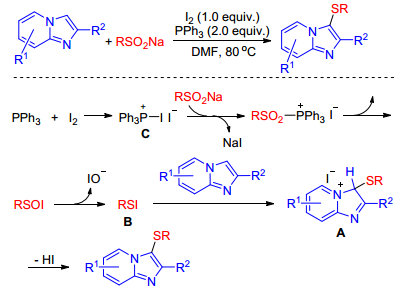
As we known, sodium sulfinates have widely been used as sulfenylating agents because of their stability, easy to handle and convenient preparation.[29] An efficient I2-PPh3 mediated sulfenylation of imidazole-fused heterocycles using sodium sulfinates as sulfenylating reagent was developed by Li and co-workers.[30] Authors speculated that iodine firstly reacts with PPh3 to form intermediate C, which then reacts with sodium sulfinate generating sulfenyl iodide B. Subsequently, the imidazolenium intermediate A is formed by regioselective electrophilic attack of sulfenyl iodide on imidazole-fused heterocycles. Finally, the elimination of HI from the intermediate A affords the desired product (Scheme 19). In 2018, Zhu group[31] also achieved the sulfenylation of imidazole-fused heterocycles with sodium sulfinates using the same catalytic system and just different solvent. These reactions proceed smoothly under transition-metal-free conditions with a broad range of substrate scope, giving the desired products in moderate to excellent yields.
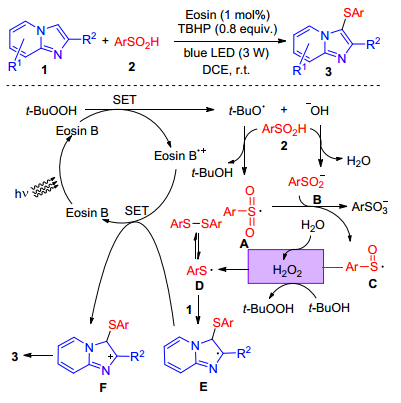
Visible light as an ideal promoter for the sulfenylation of imidazole-fused heterocycles with sulfinic acids was reported by Wang group.[32] This is the first example for the sulfenylation of C(sp2)—H bond using arylsulfinic acids as the sulfur reagents under visible-light-induced conditions. This protocol afforded diverse sulfenylated imidazole derivatives with high yields and exhibited a new model of C—S bond formation. A plausible pathway was proposed in Scheme 20. Firstly, Eosin B was excited by visible light irradiation leading to the excited Eosin B*, which underwent single electron transfer (SET) process to TBHP generating tert-butoxyl radical and a hydroxyl anion. The tert-butoxyl radical abstracted a hydrogen from arylsulfinic acid to afford the sulfonyl radical A, which reacted with arylsulfinate B to furnish the corresponding sulfinyl radical C. Then that was reducted by H2O or t-BuOH to form thiyl radical D, which reacted with imidazole-fused heterocycles to form the carbon centered radical E that could be further transformed into the carbocation intermediate F through SET with eosin B•+. Finally, the sulfonic acid anion attacked the β-H of intermediate E resulting in the desired product.
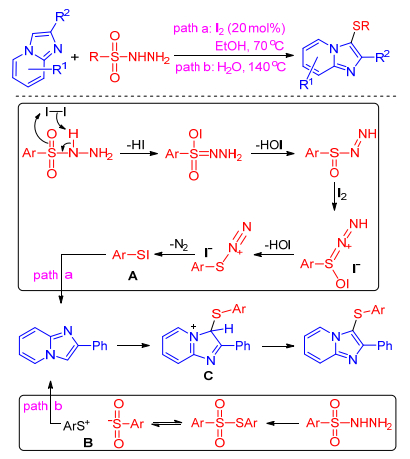
In 2015, Hajra and co-workers[33] presented iodine-cata-lyzed regioselective sulfenylation of imidazole-fused heterocycles using sulfonyl hydrazides as a thiol surrogate. This methodology is applicable to both alkyl and aryl sulfonyl hydrazides, and a library of imidazole-fused heterocycles with broad functionalities under transition metal and oxidant-free, and environmentally benign reaction conditions.
Based on experiment result, they proposed a plausible mechanism in which sulfenyl iodide A is the crucial electrophile species formed by the reaction between sulfonyl hydrazide and I2 along with the elimination of HOI, H2O, and N2. Subsequently the intermediate C is formed by the electrophilic attack by sulfenyl iodide A on imidazole-fused heterocycles. Finally, the intermediate C afforded the desired product by the elimination of HI. The HI reacts with HOI to regenerate the catalyst I2 (Scheme 21, path a). Three years later, Maity group[34] found that the sulfenylation of imidazole-fused heterocycles with sulfonyl hydrazides can undergo well without catalyst-iodine in water. They presented sulfonothioate B is the active electrophile species formed by the split of sulfonyl hydrazides. Then an electrophilic attack of B on the imidazo compounds can lead to C, which is converted to the product upon interaction with sulfonate (Scheme 21, path b).
Based on the comprehension of Maity's work, sulfonothioates as the active electrophile intermediate should also reserve as a sort of sulfenylating reagent. In 2017, Adimurthy group described analogous work that N-heteroarenes were sulfenylated under catalyst-free conditions using sulfonothioates as sulfur source in water.[35] This protocol was used for a variety of N-heteroarenes and arene-/alkane-sulfonothioates. The sulfonothioates were activated exclusively in aqueous medium rather than in an organic solvent. A plausible reaction mechanism is proposed in Scheme 22. In the presence of water, sulfonothioate disassociate into a sulfur anion and PhS+A. Subsequent attack of imidazos on electrophile species A generates imidazolium intermediates B. Finally, elimination of sulfinic acid from intermediate B provides the desired product.
Organoselenium compounds have frequently been found in several new synthetic and biological active molecules, selenium-containing imidazole-fused heterocycles present interesting biological activities among these compounds.[36] Due to the chemical and biological importance, intensive efforts have been made to develop novel and practical synthetic methods to synthesize the selenylated imidazole-fused heterocycles.[37] From the viewpoint of green chemistry, the direct C—H selenylation of imidazole-fused heterocycles is an ideal and prospective strategy among them.
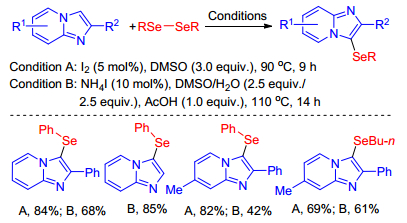
As early as 2015, Tang et al.[23] have discovered the iodine/ hydrogen peroxide catalytic system that promoted sulfenylation of imidazole-fused heterocycles was also applicable for the C—H selenylation of imidazole-fused heterocycles. However, Braga group achieved I2-cata-lyzed[24] and NH4I-catalyzed[25] selenylation of imidazopyridines with diselenides using DMSO instead of H2O2 as oxidant in the absence of transition metal catalyst in 2016 (Scheme 23). This reaction mechanism is the same as above C—H sulfenylation depicted in Scheme 16, using selenium instead of sulfur. This mild and green approach allowed the preparation of 3-selenyl-imidazo[1, 2-a]pyridine derivatives with structural diversity in good to excellent yields.
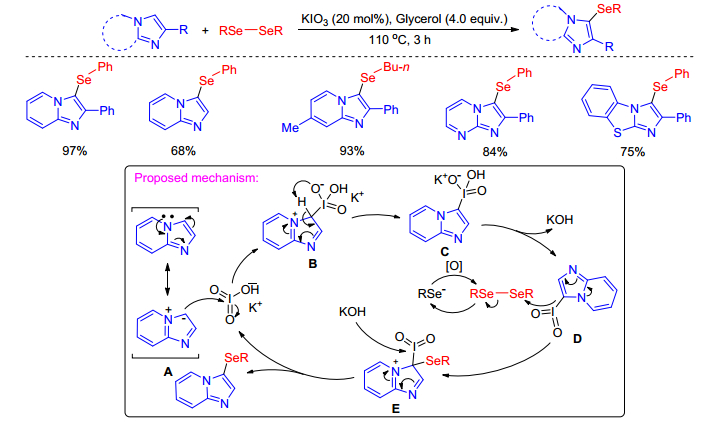
Similar to the Liu's work about direct C—H sulfenylation of indoles[38], Braga group[39] performed the selenylation of imidazole-fused heterocycles with diselenides using simple inorganic iodine salt KIO3 as the sole catalyst and stoichiometric amount of glycerol. A plausible mechanism for this transformation was proposed (Scheme 24). Initially, a nucleophilic attack of species A on hypervalent iodate affords the species B, which by intramolecular proton transfer leads to species C that releasing KOH forms the intermediate D. A nucleophilic attack of D on diselenides results in selenylated species E and selenolate anion which suffer oxidation by air resulting in the diselenide.
Finally, the species E in the presence of KOH forms the desired product with the subsequent regeneration of the catalyst in the reaction medium. The reaction features are high yields, atom economy, metal- and solvent-free approach as well as applicable to sulfenylation reaction and different types of N-heteroarenes.
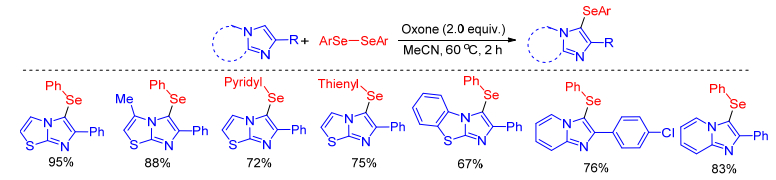
In 2018, Schumacher group[40] presented an efficient oxone-mediated selenylation of imidazo derivatives with diselenides under mild transition metal-free conditions. The methodology affords the selenylated imidazoheterocycle compounds in good to excellent yields with a broad scope of substrates (Scheme 25). Alternatively, they explored that ultrasound irradiation as a tool for fast and efficient energy transfer can significantly reduce the reaction time.
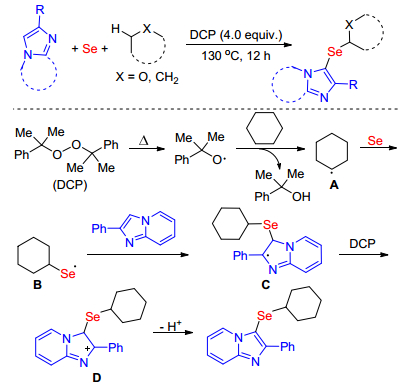
Recently, Guo and co-workers[41] developed a novel DCP-mediated approach for the synthesis of selenylated imidazole-fused heterocycle derivatives via oxidative radical coupling reaction of imidazole-fused heterocycles with ethers/alkanes and Se powder. This procedure uses odorless, stable, and commercially available Se powder as selenylating reagents and affords a wide range of structurally diverse 3-selenyl-imidazoheterocycles with moderate to good yields under mild conditions. Meanwhile, a possible reaction mechanism was illustrated in Scheme 26. Initially, DCP decomposes to cumyl radical under heating, which further reacts with cyclohexane to produce cyclo-hexane radical intermediate A. Next, A reacts with selenium powder to form key intermediate B that reacts with imidazopyridine producing the radical C. The radical C is then captured by DCP to afford the cation intermediate D, which further lose a proton to provide selenated product.The aromatic ring at the 2-position of imidazopyridines is a key factor in stabilization of radical C and cation intermediate D.
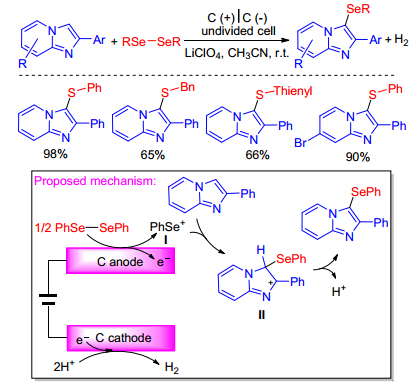
In 2019, Kim group[42] demonstrated a new electrochemical oxidative selenylation of imidazo[1, 2-a]pyridines with diselenides. The selenylation reaction proceeds in an undivided cell equipped with glassy carbon electrodes, employing LiClO4 as supporting electrolyte, and uses shelf-stable diselenides as selenium source and electrons as oxidizing reagents. The proposed reaction mechanism was shown in Scheme 27. Diphenyl diselenide is oxidized to generate phenylselenium cation I. This intermediate cation I then reacts with imidazo[1, 2-a]pyridine yielding intermediate II which is deprotonated to afford desired product.
Halogenated (hetero)aromatic compounds play pivotal role in drug and natural-product syntheses.[43] Particularly, halogenated imidazole-fused heterocycles represent an important class of synthetic intermediates and building blocks in organic chemistry for the construction of diverse and highly functionalized imidazole-fused heterocycles and show unique bioactivities.[44] In the past, the halogenation of imidazole-fused heterocycles generally used toxic and hazardous solvents or reagents, vigorous reaction conditions and expensive catalytic systems. However, with the advancement of chemical science, the direct halogenation of C—H bond provides a straightforward strategy to obtain halogenated imidazole-fused heterocycle compounds in recent years.
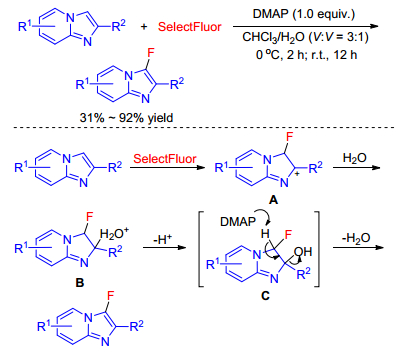
In 2015, the synthesis of 3-fluorinated imidazo[1, 2-a]-pyridines using SelectFluor as the fluorinating reagent in aqueous condition was described by Sun group.[45] In this reaction, DMAP was a benefit to the monofluorinated transformation and caused obvious suppression to the difluorinated product. They proposed that the reaction proceeded through an electrophilic fluorinated mechanism (Scheme 28). Initially, reaction of imidazo[1, 2-a]pyridine with Selectfluor yielded the unstable cation A, followed by addition to water to form B. Next, deprotonation took place to generate intermediate C, and then the proton was extracted by the base DMAP quickly to furnish the monofluorinated product.
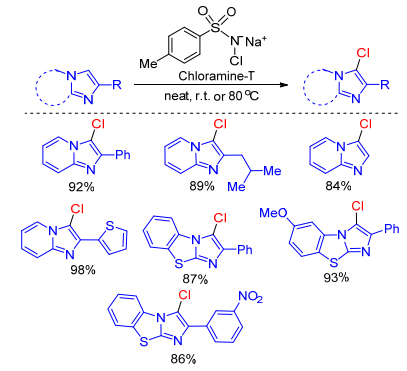
Hajra group[46] successfully developed an environment benign and efficient method for the chlorination of imidazole-fused heterocycles using chloramine-T, a novel chlorinating reagent. A bunch of 3-chloro-substituted imidazo-[1, 2-a]pyridines and imidazo[2, 1-b]thiazoles were synthesized in good yields under neat condition within short time (Scheme 29). General applicability, operational simplicity, open-air, metal-free, environment-friendly reaction conditions and excellent yields are the notable advantages of the present protocol. They speculated that this reaction also proceeds a rapid nucleophilic attack by imidazopyridine to Cl+ cation generated from chloramine-T.
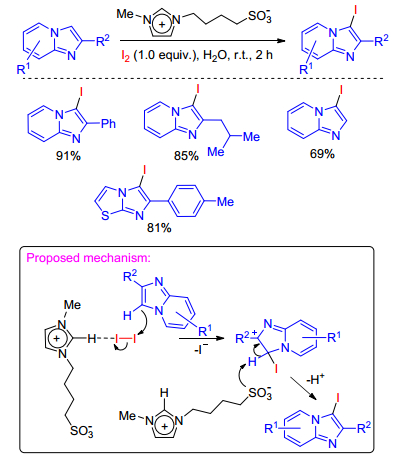
The same group has also achieved the iodination of imidazole-fused heterocycles using a zwitterion-type molten salt as the catalyst and molecular iodine as the iodinating reagent in 2016.[47] The present method provides a straightforward and environmentally benign strategy for the iodination of imidazole-fused heterocycles, shows broad substrate scope applicability and tolerates a wide range of functional groups. A probable mechanistic path is proposed in Scheme 30. Initially, C(2)—H of the imidazolium salt activates the molecular iodine facilitating an electrophilic attack at the C(3) position of the imidazopyridine. Consequently, the sulfonate part of the catalyst promotes elimination of the C(3) hydrogen to furnish the desired iodoimidazopyridine.
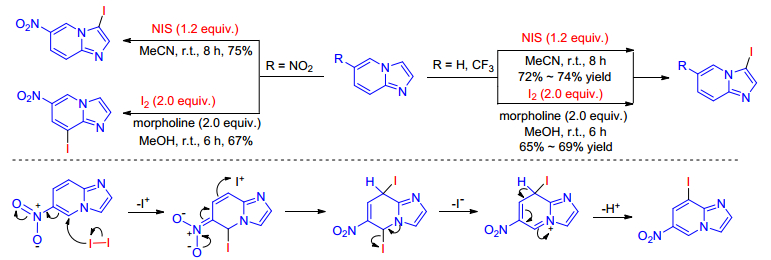
In 2018, Zhao and co-workers[48] investigated the chemose- lectivity of the iodination of 6-substituted imidazo[1, 2-a]- pyridine through experiment and theoretical calculations. The iodination reaction occurred at C-3 position both under action of NIS and I2-morpholine when 6-unsubstituted and trifluoromethyl-imidazo[1, 2-a]pyri- dines were used as starting materials, while using 6-nitro- imidazo[1, 2-a]pyridine as substrates, reaction with NIS afforded the 3-iodo-6-nitroimidazo[1, 2-a]pyridine and 8-iodo-6-nitroimidazo[1, 2-a]pyridine was obtained in the presence of I2-morpholine. Theoretical calculations indicates that this is due to the inductive effect of the nitro group and a plausible reaction mechanism for synthesis of 8-iodo-6- nitroimidazo[1, 2-a]pyridine was put forward based on the calculated MEP results (Scheme 31).
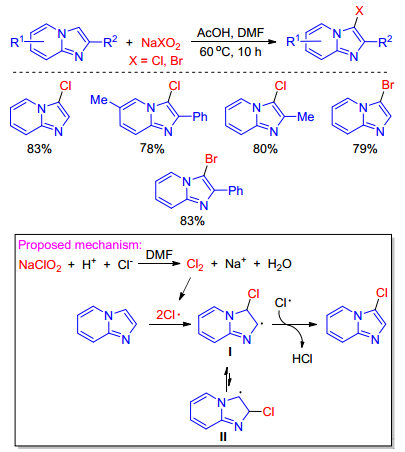
A facile transition metal-free regioselective halogenation of imidazo[1, 2-a]pyridines using cheap sodium chlorite/bromite as the halogen source and oxidant is described by Yu and co-workers.[49] This methodology provides a new selective route to synthesize 3-Cl- or 3-Br- imidazo[1, 2-a]pyridines under mild conditions with good yields. Base on the experimental results and previous works, a possible mechanism was proposed in Scheme 32. Firstly, oxidation-reduction reaction of sodium chlorite happened in the presence of AcOH to produce chlorine. Subsequently, the chlorine radical was easily formed via homolysis of chlorine, which then attack imidazo-[1, 2-a]pyridine resulting in free radical intermediate I. Finally, the free radical intermediate I underwent an aromatization with chlorine free radical to give the target product.
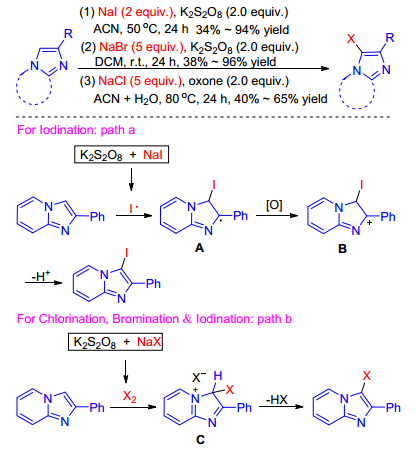
In 2019, Adimurthy et al.[50] reported an novel method for the halogenation of imidazofused heterocycles using readily available sodium salts (NaCl/NaBr/NaI) as halogen source and K2S2O8 (or) oxone as promoter. A variety of halogenated imidazole-fused heterocycles were obtained under mild reaction conditions. It is the first report on the use of simple sodium halide salts (NaI/NaBr/NaCl) for the halogenation. Based on the experiment results, two proposed plausible reaction paths that the iodination may proceed via both ionic and radical pathways, but bromination and chlorination may follow ionic pathways were illustrated in Scheme 33. In path a: initially, K2S2O8 thermally decomposes into sulfate radical which in turn reacts with NaI to generate iodine radical (I•). Addition of iodine radical (I•) to the C-3 position of imidazopyridines generates radical intermediate A. Subsequently, single electron transfer (SET) and oxidative aromatization may deliver the desired product through the intermediate B. In path b: the molecular halogen generated in situ by the oxidant may electrophilically attack on the C-3 position of imidazopyridines to generate another imidazolonium intermediate C, and then elimination of HX from C gives the desired halogenated products.
In summary, the incorporation of heteroatom into imidazole-fused heterocycles through direct C—H functionalization under transition metal-free conditions is very vibrant and has rapidly developed in recent years. Although enormous efforts have been devoted and tremendous progresses have also been made, lots of challenges need to be addressed. Based on the reported literatures, most of transition metal-free C—H functionalization of imidazole-fused heterocycles is focused on the sulfenylation, while the formation of C—O, C—N, C—Se bond is reported less and even there is no report on the construction of C—P bond without transition metal catalyst which will be a hot research area. Furthermore, in terms of eco-friend and atom economy, electricity and visible light are the ideal promoter for the functionalization of imidazole-fused heterocycles, which provides a new approach to access such compounds in spite of few related literature now. Electricity and visible light catalyzed C—H functionalization of imidazole-fused heterocycles are still in the early stage of research and worth to be further studied. Moreover, the mechanisms of these reactions need to be further explored. More specific reaction mechanisms might help us to design and achieve more efficient and eco-friendly functionalization of imidazole-fused heterocycles. All of these are significant for enriching the library of nitrogen hetero-fused rings and further expanding its application in medicine, materials and dyes. Direct functionalization of C—H bond on imidazole-fused heterocycles without transition metal is still a vigorous research field with challenges and prospects. We believe that more and more efforts will be devoted to this, and more and more innovative achievements will emerge in the future.
(a) Majumdar, P.; Pati, A.; Patra, M.; Behera, R. K.; Behera, A. K. Chem. Rev. 2014, 114, 2942.
(b) Frizon, T. E. A.; Rafique, J.; Saba, S.; Bechtold, I. V.; Gallardo, H.; Braga, A. L. Eur. J. Org. Chem. 2015, 16, 3470.
(c) Li, G.-H.; Dong, D.-Q.; Yu, X.-Y.; Wang, Z.-L. New J. Chem. 2019, 43, 1667.
(d) Xie, L.-Y.; Li, Y.-J.; Qu, J.; Duan, Y.; Hu, J.; Liu, K.-J.; Cao, Z.; He, W.-M. Green Chem. 2017, 19, 5642.
(e) Li, G.-H.; Dong, D.-Q.; Deng, Q.; Yan, S.-Q.; Wang, Z.-L. Synthesis 2019, 51, 3313.
(f) Xie, L.-Y.; Peng, S.; Liu, F.; Liu, Y.-F.; Sun, M.; Tang, Z.; Jiang, S.; Cao, Z.; He, W.-M. ACS Sustainable Chem. Eng. 2019, 7, 7193.
(a) Xu, X.-M.; Chen, D.-M.; Wang, Z.-L. Chin. Chem. Lett. 2019, DOI: 10.1016/j.cclet.2019.05.048.
(b)Lhassani,M.;Chavignon,O.;Chezal,J.-M.;Teulade,J.-C.;Chapat,J.-P.;Snoeck,R.;Andrei,G.;Balzarini,J.;Clercq,E.D.;Gueiffier,A.Eur.J.Med.Chem.1999,34,271.
(a) Chitrakar, R.; Subbarayappa, A. Chem. Rec. 2017, 17, 1.
(b) Dong, D.-Q.; Hao, S.-H.; Yang, D.-S.; Li, L.-X.; Wang, Z.-L. Eur. J. Org. Chem. 2017, 2017, 6576.
(c) Sun, F.; Liu, X.; Chen, X.; Qian, C.; Ge, X. Chin. J. Org. Chem. 2017, 37, 2211.
(d) Jin, C.-A.; Xu, Q.; Feng, G.-F.; Jin, Y.; Zhang, L.-Y. Chin. J. Org. Chem. 2018, 38, 775.
(a) Liu, Y.-Y.; Xiong, J.; Wei, L. Chin. J. Org. Chem. 2017, 37, 1667.
(b) Fu, L; Wan, J.-P. Asian J. Org. Chem. 2019, 8, 767.
(c) Jiang, X.; Wang, S.; Guo, G.; Lu, B. Chin. J. Org. Chem. 2017, 37, 841.
(d) Wang, Y.; Liu, Y.-Y. Acta Chim. Sinica 2019, 77, 418.
(e) Guo, Y; Xiang, Y.; Wei, L.; Wan, J.-P. Org. Lett. 2018, 20, 3971.
(f) Gao, Y; Liu, Y.-Y.; Wan, J.-P. J. Org. Chem. 2019, 84, 2243.
(a) Saidi, O.; Marafie, J.; Ledger, A. E.; Liu, P. M.; Mahon, M. F.; Kociok-Kohn, G.; Whittlesey, M. K.; Frost, C. G. J. Am. Chem. Soc. 2011, 133, 19298.
(b) Umierski, N.; Manolikakes, G. Org. Lett. 2013, 15, 4972.
(c) Wu, Z.; Song, H.; Cui, X.; Pi, C.; Du, W.; Wu, Y. Org. Lett. 2013, 15, 1270.
(d) Yi, H.; Chen, H.; Bian, C.; Tang, Z.; Singh, A. K.; Qi, X.; Yue, X.; Yu, L.; Lee, J.-F.; Lei, A. Chem. Commun. 2017, 53, 6736.
(e) Yin, Y.; Xie, J.; Huang, F.-Q.; Qi, L.-W.; Zhang, B. Adv. Synth. Catal. 2017, 359, 1037.
(a) Nordqvist, A.; Nilsson, M. T.; Lagerlund, O.; Muthas, D.; Gising, J.; Yahiaoui, S.; Odell, L. R.; Srinivasa, B. R.; Larhed, M.; Mowbray, S. L.; Karlen, A. Med. Chem. Commun. 2012, 3, 620.
(b) Elleder, D.; Baiga, T. J.; Russell, R. L.; Naughton, J. A.; Hughes, S. H.; Noel, J. P.; Young, J. A. Virol. J. 2012, 9, 305.
(c) Odell, L. R.; Nilsson, M. T.; Gising,, J.; Lagerlund O.; Muthas, D.; Nordqvist, A.; Karlen, A.; Larhed, M. Bioorg. Med. Chem. Lett. 2009, 19, 4790.
Wang, Y.; Frett, B.; McConnell, N.; Li, H. Org. Biomol. Chem. 2015, 13, 2958. doi: 10.1039/C4OB02284J
Mondal, S.; Samanta, S.; Jana, S.; Hajra, A. J. Org. Chem. 2017, 82, 4504. doi: 10.1021/acs.joc.7b00564
Singsardar, M.; Mondal, S.; Sarkar, R.; Hajra, A. ACS Omega 2018, 3, 12505. doi: 10.1021/acsomega.8b02088
Samanta, S.; Ravi, C.; Rao, S. N.; Joshi, A.; Adimurthy, S. Org. Biomol. Chem. 2017, 15, 9590. doi: 10.1039/C7OB02504A
Liu, K.; Wu, J.; Deng, Y.; Song, C.; Song, W.; Lei, A. ChemElectroChem 2019, 6, 4173. doi: 10.1002/celc.201900138
Coutant, E. P.; Janin, Y. L. Chem. Eur. J. 2015, 21, 17158. doi: 10.1002/chem.201501531
Zhang, J.; Lu, X.; Li, T.; Wang, S.; Zhong, G. J. Org. Chem. 2017, 82, 5222. doi: 10.1021/acs.joc.7b00480
Kibriya, G.; Samanta, S.; Jana, S.; Mondal, S.; Hajra, A. J. Org. Chem. 2017, 82, 13722. doi: 10.1021/acs.joc.7b02582
(a) Hamdouchi, C.; Blas, J.; Prado, M.; Gruber, J.; Heinz, B. A.; Vance, L. J. Med. Chem. 1999, 42, 50.
(b) Bochis, R. J.; Olen, L. E.; Fisher, M. H.; Reamer, R. A.; Wilks, G.; Taylor, J. E.; Olson, G. J. Med. Chem. 1981, 24, 1483.
(a) Chen, X.; Hao, X.-S.; Goodhue, C. E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 6790.
(b) Martinek, M.; Korf, M.; Srogl, J. Chem. Commun. 2010, 46, 4387.
(c) Sahoo, S. K.; Banerjee, A.; Chakraborty, S.; Patel, B. K. ACS Catal. 2012, 2, 544.
(d) Saidi, O.; Marafie, J.; Ledger, A. E.; Liu, P. M.; Mahon, M. F.; Kociok-Kohn, G.; Whittlesey, M. K.; Frost, C. G. J. Am. Chem. Soc. 2011, 133, 19298.
(e) Umierski, N.; Manolikakes, G. Org. Lett. 2013, 15, 4972.
(f) Wu, Z.; Song, H.; Cui, X.; Pi, C.; Du, W.; Wu, Y. Org. Lett. 2013, 15, 1270.
(g) Zhao, X.; Dimitrijevic, E.; Dong, V. M. J. Am. Chem. Soc. 2009, 131, 3466.
(h) Niu, B.; Xu, L.; Xie, P.; Wang, M.; Zhao, W.; Pittman, C. U.; Zhou, A. ACS Comb. Sci. 2014, 16, 454.
Hiebel, M.; Berteina-Raboin, S. Green Chem. 2015, 17, 937. doi: 10.1039/C4GC01462F
Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2016, 81, 7838. doi: 10.1021/acs.joc.6b01487
Iida, H.; Demizu, R.; Ohkado, R. J. Org. Chem. 2018, 83, 12291. doi: 10.1021/acs.joc.8b01878
Wu, S.; Feng, C.; Hu, D.; Huang, Y.; Li, Z.; Luo, Z.; Ma, S. Org. Biomol. Chem. 2017, 15, 1680. doi: 10.1039/C6OB02736A
Rahaman, R.; Das, S.; Barman, P. Green Chem. 2018, 20, 141. doi: 10.1039/C7GC02906C
Yuan, Y.; Cao, Y.; Qiao, J.; Lin, Y.; Jiang, X.; Weng, Y.; Tang, S.; Lei, A. Chin. J. Chem. 2019, 37, 49. doi: 10.1002/cjoc.201800405
Ji, X.-M.; Zhou, S.-J.; Chen, F.; Zhang, X.-G.; Tang, R.-Y. Synthesis 2015, 47, 659. doi: 10.1055/s-0034-1379941
Rafique, J.; Saba, S.; Rosrio, A.; R. Braga, A. L. Chem. Eur. J. 2016, 22, 1. doi: 10.1002/chem.201504553
Bettanin, L.; Saba, S.; Doerner, C. V.; Franco, M. S.; Godoi, M.; Rafique, J.; Braga, A. L. Tetrahedron 2018, 74, 3971. doi: 10.1016/j.tet.2018.05.084
Liu, W.-J.; Wang, S.; Chen, Y.-Z.; Huang, Y.; Li, Z.; Wang, A.-D. Phosphorus, Sulfur Silicon Relat. Elem. 2016, 191, 689. doi: 10.1080/10426507.2015.1073278
Maddi, R. R.; Shirsat, P. K.; Kumar, S.; Meshram, H. M. ChemistrySelect 2017, 2, 1544. doi: 10.1002/slct.201601460
安艳妮, 李建晓, 李蒙, 李春生, 杨少容, 有机化学, 2017, 37, 720. http://www.cqvip.com/QK/93463X/201703/671747168.htmlAn, Y.; Li, J.-X.; Li, M.; Li, C.-S.; Yang, S. Chin. J. Org. Chem. 2017, 37, 720(in Chinese). http://www.cqvip.com/QK/93463X/201703/671747168.html
(a) Xiao, F. H.; Xie, H.; Liu, S. W.; Deng, G. J. Adv. Synth. Catal. 2014, 356, 364.
(b) Katrun, P.; Hongthong, S.; Hlekhlai, S.; Pohmakotr, M.; Reutrakul, V.; Jaipetch, T.; Kuhakarn, C. RSC Adv. 2014, 4, 18933.
(c) Rao, H. H.; Wang, P.; Wang, J. C.; Li, Z. F.; Sun, X. Z.; Cao, S. L. RSC Adv. 2014, 4, 49165.
Huang, X.; Wang, S.; Li, B.; Wang, X.; Ge, Z.; Li, R. RSC Adv. 2015, 5, 22654. doi: 10.1039/C4RA17237J
Guo, Y.-J.; Lu, S.; Tian, L.-L.; Huang, E.-L.; Hao, X.-Q.; Zhu, X.; Shao, T.; Song, M.-P. J. Org. Chem. 2018, 83, 338. doi: 10.1021/acs.joc.7b02734
Sun, P.; Yang, D.; Wei, W.; Jiang, M.; Wang, Z.; Zhang, L.; Zhang, H.; Zhang, Z.; Wang, Y.; Wang, H. Green Chem. 2017, 19, 4785. doi: 10.1039/C7GC01891F
Hajra, A.; Bagdi, A. K.; Mitra, S.; Ghosh, M. Org. Biomol. Chem. 2015, 13, 3314. doi: 10.1039/C5OB00033E
Chowdhury, S. R.; Fadikar, P.; Hoque, I. U.; Maity, S. Asian J. Org. Chem. 2018, 7, 332. doi: 10.1002/ajoc.201700670
Ravi, C.; Joshi, A.; Adimurthy, S. Eur. J. Org. Chem. 2017, 2017, 3646. doi: 10.1002/ejoc.201700487
(a) Pang, Y.; An, B.; Lou, L.; Zhang, J.; Yan, J.; Huang, L.; Li, X.; Yin, S. J. Med. Chem. 2017, 60, 7300.
(b) Kumar, S.; Johansson, H.; Kanda, T.; Engman, L.; Muller, T.; Bergenudd, H.; Jonsson, M. Eur. J. Org. Chem. 2017, 2017, 3055.
(c) Kumar, S.; Sharma, N.; Maurya, I.; Bhasin, A. K. K.; Wangoo, N.; Brandao, P.; Fleix, V.; Kumar, R. K. Eur. J. Med. Chem. 2016, 123, 916.
(a) Perin, G.; Lenardao, E. J.; Jacob, R. G.; Panatieri, R. B. Chem. Rev. 2009, 109, 1277.
(b) Freudendahl, D. M.; Santoro, S.; Shahzad, S. A.; Santi, C.; Wirth, T. Angew. Chem. Int. Ed. 2009, 48, 8409.
(c) Abdo, M.; Zhang, Y.; Schramm, V. L.; Knapp, S. Org. Lett. 2010, 12, 2982.
(d) Rafique, J.; Saba, S.; Rosario, A. R.; Braga, A. L. Chem. Eur. J. 2016, 22, 11854.
(e) Shu, S.; Fan, Z.; Yao, Q.; Zhang, A. J. Org. Chem. 2016, 81, 5263.
(f) Sun, K.; Wang, X.; Lv, Y.; Li, G.; Jiao, H.; Dai, C.; Li, Y.; Zhang, C.; Liu, L. Chem. Commun. 2016, 52, 8471.
(a) Bai, F.-C.; Zhang, S.; Wei, L.; Liu, Y.-Y. Asian J. Org. Chem. 2018, 7, 371.
(b); Zhong, S.; Xie, L.; Cao, X.; Liu, Y.-Y.; Wei, L. Org. Lett. 2016, 18, 584.
(c) Zhong, S.; Liu, Y.-Y.; Cao, X.; Wei, L.; Wan, J.-P. ChemCatChem 2017, 9, 465.
Rafique, J.; Saba, S.; Franco, M. S.; Bettanin, L.; Schneider, A. R.; Silva, L. T.; Braga, A. L. Chem. Eur. J. 2018, 24, 4173. doi: 10.1002/chem.201705404
Rodrigues, I.; Barcellos, A. M.; Belladona, A. L.; Roehrs, J. A.; Cargnelutti, R.; Alves, D.; Perin, G.; Schumacher, R. F. Tetrahedron 2018, 74, 4242. doi: 10.1016/j.tet.2018.06.046
Guo, T.; Wei, X.; Liu, Y.; Zhang, P.; Zhao, Y. Org. Chem. Front. 2019, 6, 1414. doi: 10.1039/C9QO00198K
Kim, Y. J.; Kim, D. Y. Tetrahedron Lett. 2019, 60, 739. doi: 10.1016/j.tetlet.2019.02.001
(a) Winterton, N. Green Chem. 2000, 2, 173.
(b) Gribble, G. W. J. Chem. Educ. 2004, 81, 1441.
(c) Liu, C.; Zhang, B. Chem. Record. 2016, 16, 667.
(d) Fujimori, D. G.; Walsh, C. T. Curr. Opin. Chem. Biol. 2007, 11, 553.
(e) Butler, A.; Walker, J. V. Chem. Rev. 1993, 93, 1937.
(a) Nicolaou, K. C.; Bulger, P. G.; Sarlah, D. Angew. Chem., Int. Ed. 2005, 44, 4442.
(b) Rout, L.; Sen, T. K.; Punniyamurthy, T. Angew. Chem., Int. Ed. 2007, 46, 5583.
Liu, P.; Gao, Y.; Gu, W.; Shen, Z.; Sun, P. J. Org. Chem. 2015, 80, 11559. doi: 10.1021/acs.joc.5b01961
Dey, A.; Singsardar, M.; Sarkar, R.; Hajra, A. ACS Omega 2018, 3, 3513. doi: 10.1021/acsomega.7b01844
Mondal, S.; Samanta, S.; Singsardar, M.; Mishra, S.; Mitra, S.; Hajra, A. Synthesis 2016, 48, 4009. doi: 10.1055/s-0035-1562492
Zhao, C.; Li, F.; Yang, S.; Liu, L.; Huang, Z.; Chai, H. Chem. Heterocycl. Compd. 2018, 54, 568. doi: 10.1007/s10593-018-2307-x
Li, J.; Tang, J.; Wu, Y.; He, Q.; Yu, Y. RSC Adv. 2018, 8, 5058. doi: 10.1039/C7RA12100H
Semwal, R.; Ravi, C.; Kumar, R.; Meena, R.; Adimurthy, S. J. Org. Chem. 2019, 84, 792. doi: 10.1021/acs.joc.8b02637

Scheme 14 Sulfenylation of imidazole-fused heterocycles with disulfides using DMSO as oxidant

Scheme 32 Halogenation of imidazole-fused heterocycles with cheap sodium chlorite/bromite

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:






























