Scheme 1.
Previous related works and present work
Palladium-Catalyzed Thiazole-Directed mono-Selective C(sp2)-H Bond Iodination Reaction
Lihao Xing , Lingyan Shao , Xiaopan Fu , Kezuan Deng , Jinyue Yang , Yafei Ji

Aryl iodides represent widespread structural motifs distributed in a range of pharmaceuticals, agrochemicals and natural products.[1] Over the past few decades, aryl iodides have been extensively used as aromatic synthons for constructing complex organic structures via nucleophilic substitution[2] or a variety of cross-coupling reactions (such as Suzuki, [3] Heck[4] and Ullmann reaction, [5] etc.). The traditional strategies to access aryl iodides are direct electrophilic substitution, Sandmeyer reaction and ortho-lithation/ halogenation, which were limited in application by poor regioselectivities, harsh reaction conditions and tedious long procedures.[6]
To overcome these above mentioned disadvantages, diverse C—I bond formation reactions promoted by rear- metal catalysts or more recently investigated base-metal catalysts[7] have been developed. Ever since the first Pd-catalyzed C(sp2)—H bond iodination reaction disclosed by Sanford, [8] a series of protocols using Suárez reagent, [9] N-iodosuccinimide (NIS), [10] sole I2[11] or other iodine sour- ces[12] have emerged to introduce iodine atom to different aryl substrates. A variety of directing groups have also been employed for ortho-C—H bond activation/iodine- tion.[13] However, the thiazole based auxiliary-directed io- dination has rarely been reported. Recently, Petel et al.[14] disclosed Pd-catalyzed benzothiazole-directed C(sp2)—H bond halogenation, in which only four disubstituted examples of iodination were included and the mono-selectivity was not obtained (Scheme 1A). Considering that highly functionalized thiazoles serve as important pharmacophores in drug discovery[15], it is highly desirable to exploit the non-fused thiazole-directed C(sp2)—H bond mono- selective iodination reaction (Scheme 1B).
Given our recent success in thiazole-directed C(sp2)—H bond activation/acyloxylation, [16] our research was initiated by briefly evaluating the substituted pattern of various thoizole directing groups. As indicated in Table 1, direct electrophilic iodination preferred to occurring on the thiazole bearing active site[17] (DG1), which restrained the aimed ortho-iodination. Introducing methyl group to C-5 site of thiazole drastically promoted the reaction both on reaction rate and yield (DG2). Replacing methyl group with ethyl group (DG3) performed similar efficiency. However the larger steric hinderance may potentially limit substrate scope, which means DG2 is still the suitable directing group. To our surprise, changing carboxylate group on the C-2 site of thiazole to methyl (DG4) notably inhibited the reaction, indicating that carboxylate is vital in this catalytic system. Furthermore, DG5 resulted in inferior efficiencies and DG6 was inefficient, both demonstrating that the appropriate electron density of DG2 could promote the C—H bond activation. Thus, the fully substituted DG2 was selected as the optimal directing group for further iodination investigations.
Upon the preferable directing group confirmed, the optimization of reaction conditions utilizing 1b as the model substrate was followed (Table 2). Firstly, a control experiment revealed that the C(sp2)—H bond activation/ iodination process does not occur in the absence of the catalyst (Entry 1). Compared to Pd(OAc)2, other Pd salts led to lower efficiency (Entres 2 vs 3~6). Lowering the loading of Pd(OAc)2 to 5 mol% distinctly depressed the reaction (Entry 7), howbeit further increasing the dosage of Pd(OAc)2 did not result in significently better performance (Entry 8). Subsequently, screening of solvents, including DCE/HFIP (V:V=4:1), DCE, HFIP, DMF, DCM and TCM uncovered that DCE exhibited the best result (Entries 9 vs 2, 10~13). Afterwards, adjusting the amount of NIS showed 2.5 equiv. is more favorable and economic loading (Entres 15 vs 14, 16). Finally, different scales of solvent were applied to this reaction, and 0.1 mol•L-1 was the approprite concentration (Entries 15 vs 17~18). Hence, Entry 15 was selected as the standard reaction conditions for the subsequent functionalization.
 下载:
导出CSV
下载:
导出CSV
 | ||||
| Entry | Catalyst | NIS (equiv.) | Solvent | Yieldb/% |
| 1 | None | 2.0 | DCE/HFIP (V:V=4:1) |
0 |
| 2 | Pd(OAc)2 | 2.0 | DCE/HFIP (V:V=4:1) |
76 |
| 3 | Pd(CH3CN)2Cl2 | 2.0 | DCE/HFIP (V:V=4:1) |
64 |
| 4 | Pd(TFA)2 | 2.0 | DCE/HFIP (V:V=4:1) |
57 |
| 5 | PdCl2 | 2.0 | DCE/HFIP (V:V=4:1) |
38 |
| 6 | Pd(PPh3)2Cl2 | 2.0 | DCE/HFIP (V:V=4:1) |
27 |
| 7c | Pd(OAc)2 | 2.0 | DCE/HFIP (V:V=4:1) |
58 |
| 8d | Pd(OAc)2 | 2.0 | DCE/HFIP (V:V=4:1) |
75 |
| 9 | Pd(OAc)2 | 2.0 | DCE | 86 |
| 10 | Pd(OAc)2 | 2.0 | HFIP | 69 |
| 11 | Pd(OAc)2 | 2.0 | DMF | 85 |
| 12 | Pd(OAc)2 | 2.0 | DCM | 62 |
| 13 | Pd(OAc)2 | 2.0 | TCM | 21 |
| 14 | Pd(OAc)2 | 1.5 | DCE | 76 |
| 15 | Pd(OAc)2 | 2.5 | DCE | 93 |
| 16 | Pd(OAc)2 | 3.0 | DCE | 92 |
| 17e | Pd(OAc)2 | 2.5 | DCE | 78 |
| 18f | Pd(OAc)2 | 2.5 | DCE | 90 |
| a Reaction conditions: 1b (74.1 mg, 0.3 mmol), NIS, catalyst (10 mol%), solvent (3 mL, 0.1 mol·L-1), 100 ℃, 4 h; b NMR yield using CH2Br2 as the internal standard; c Pd(OAc)2 (5 mol%); d Pd(OAc)2 (15 mol%); e DCE (0.05 mol·L-1); f DCE (0.15 mol·L-1); NIS=N-Iodosuccinimide; DCE=1, 2-Dichloroethane; HFIP=Hexafluoroisopropanol; DMF=N, N-Dimethylfor- mamide; DCM=Dichloromethane; TCM=Trichloromethane; DMSO=Dimethyl sulfoxide. | ||||
With the optimized conditions for mono-selective C(sp2)—H bond activation/iodination established, we set about testing the generality of this methodology (Table 3). The iodination reaction with NIS catalyzed by Pd(OAc)2 was found to be perfectly compatible with a wide range of functional groups, including OMe (4e, 4l, 4u), Me (4f, 4n, 4t), Et (4m), F (4c, 4g, 4o, 4v, 4w), Cl (4h, 4p), Br (4i, 4q, 4s) and CF3 (4j, 4r).
However, most ortho-substituted 4-arylthiazoles failed to generate corresponding products, only with the exception of sterically less hindered F-substituted substrate (4b~4d). It is speculated that steric hinderance between the ortho-substituents and the C-5-methyl group on thiazole ring impedes the free rotation of benzene ring, thus preventing the formation of five-member cyclo- palladium intermediate. meta-Substituents were well tole- rated, and the desired products were obtained with up to 96% yield (4f). All iodination took place at the sterically less hindered ortho-positions (4e~4j). Notebly, substrate containing NO2 group performed no react activity (4k). This can be attributed to the strong coordination of NO2 to Pd center, which blocked the subsequent C—H bond activation[18]. Not surprisingly, di-iodination was not observed when subjecting para-substituted substrates to this catalytic system, due to aforementioned steric-hinde- rance theory.
Mono-iodinated thiohpene (4s), furan (4t) and naphthalene (4u) derivatives were sucessfully obtained in moderate to excellent yields. Furthermore, this protocol was also applicable in the cases of disubstituted masked 4-benzothiazoles (4v, 4w).
To further expand the versatility of this newly developed C—H bond functionalization reaction, iodination product 4a was successfully converted to different functionalized 4-arylthiazoles through a series of reactions. As demon- strated in Scheme 2, compounds with various functional groups, including aryl, cyano and thiol were conveniently prepared in moderate to good yields.
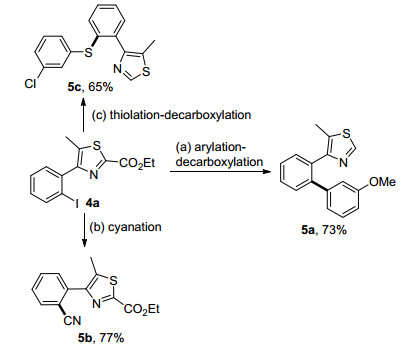
Reaction conditions: (a) 4a (111.9 mg, 0.30 mmol), 3-methoxylben-zoboric acid (59.3 mg, 0.39 mmol), I2 (15.2 mg, 0.06 mmol), K2CO3 (82.9 mg, 0.60 mmol), PEG, 140 ℃, 24 h; (b) 4a (111.9 mg, 0.30 mmol), DDQ (68.1 mg, 0.30 mmol), Cu(OAc)2 (60.0 mg, 0.30 mmol), Ag2O (36.9 mg, 0.30 mmol), NMP, 120 ℃, 22 h; (c) 4a (111.9 mg, 0.30 mmol), 3-chlorothiophenol (56.2 mg, 0.39 mmol), CuI (1.4 mg, 2.5 mol%), K2CO3 (45.6 mg, 0.33 mmol), 100 ℃, 16 h; b Isolated yields
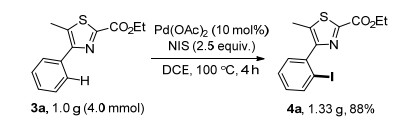
This mono-selective iodination reaction was also performed on gram scale in 88% yield (Scheme 3), illustra- ting that this protocol is still efficient on larger scale in the aspect of selectivity and productivity.
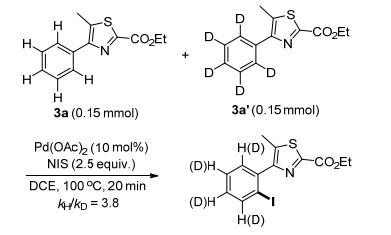
To gain insight into the mechanism of this catalytic system, a deuterium labeling isotope experiment was carried out to probe the kinetics of Pd-catalyzed C(sp2)—H iodination (Scheme 4). The intermolecular competition reactions between 3a and 3a' with NIS in one vessel performed the kinetic isotopic effect (KIE) value of 3.8, suggesting that the C(sp2)—H bond cleavage might be involved in the rate-limiting step.
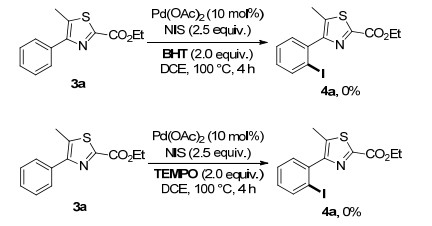
The radical inhibition experiment was then carried out to further understand the reaction path way. The use of 2, 2, 6, 6-tetramethylpiperidinooxy (TEMPO) or 2, 6-di-tert- butyl-4-methylphenol (BHT) as radical scavengers comp- letely restrained the formation of the desired product 4a (Scheme 5), implying radical may generated in this catalytic system. However, some control experiments showed that NIS reacted with TEMPO and BHT when no substrates were subjected into the reaction system. And for the difficulty of isolating, the products of radical inhibition experiments were failed to be obtained. Consequently, the oxidative addition/reductive elimination path still can not be excluded.
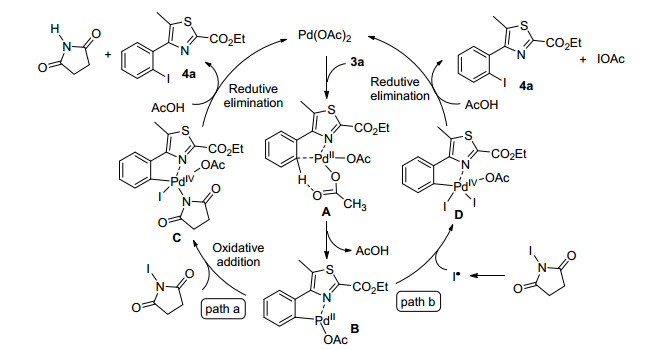
Based on these experimental investigations and the well-documented reports[10c, 10g, 14, 19], a plausible mechanism was proposed to account for this palladium-catalyzed mono-selective iodination reaction (Scheme 6). Firstly, Pd(OAc)2 coordinates with nitrogen atom on thiazole ring to generate cyclopalladium intermediate A. Acetate then participates in aromatic proton abstraction to generate PdII intermediate B. There are two possible pathways for the reaction of intermediate B. The first pathway proceeds via oxidative addition of NIS to intermediate B to form PdIV intermediate C. The subsequent reductive elimination of C furnishes the ortho-iodinated product 4a and regenerates the PdII catalyst. The second pathway b involves the generating of iodine radical, radical attack to intermediate. B and the forming of PdIV intermediate D, which undergoes reductive elimination to produce 4a and the PdII catalyst.
In summary, the first Pd-catalyzed non-fused thiazole- directed C(sp2)—H bond activation/ iodination reaction was realized. Various 4-(2-iodoaryl)thiazoles were obtained with excellent ortho- and mono-selectivities with up to 94% yield. The broad substrate scope and the subsequent diverse convenitent transformations of iodination products both increase the value of this metho- dology. A series of control experiments revealed a plausible PdII/PdIV mechanism.
All reactions were carried out in oven-dried glassware and monitored by thin layer chromatography (TLC, pre-coated silica gel plates containing HF254). NMR spectra were determined on a Bruker AV400 in CDCl3 with TMS as internal standard for 1H NMR (400 MHz) and 13C NMR (100 MHz), respectively. HRMS were measured on a QSTAR Pulsar I LC/TOF MS mass spectrometer or Micromass GCTTM gas chromatograph-mass spectrometer.
A mixture of substrate 1 (0.3 mmol), Pd(OAc)2 (6.7 mg, 10 mol%), NIS (135.0 mg, 0.6 mmol) and DCE/HFIP (V:V=4:1, 3 mL) was stirred at 100 ℃ for 12 h. Upon completion of the reaction (monitored by TLC), the mixture was transferred into saturated brine (20 mL), then extracted with ethyl acetate (20 mL×3). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo to provide a crude product, which was purified via a column chromatography on silica gel (eluents: petroleum ether/ethyl acetate) to supply the desired product 2, which was identified by 1H NMR, 13C NMR and HRMS.
Ethyl 4-(2-iodophenyl)-5-methylthiazole-2-carboxylate (2b): 85 mg, 76% yield. Yellow solid, m.p. 71~72 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.89 (dd, J=8.0, 0.9 Hz, 1H), 7.38 (td, J=7.5, 1.1 Hz, 1H), 7.30 (dd, J=7.6, 1.7 Hz, 1H), 7.07 (td, J=7.8, 1.8 Hz, 1H), 4.45 (q, J=7.1 Hz, 2H), 2.35 (s, 3H), 1.40 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.3, 156.3, 154.1, 139.2, 139.1, 137.6, 131.1, 130.23, 128.1, 99.6, 62.5, 14.3, 12.8; HRMS (EI) calcd for C13H12127INO2S: 372.9633, found 372.9635.
Ethyl 5-ethyl-4-(2-iodophenyl)thiazole-2-carboxylate (2c): 86 mg, 74% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.92 (d, J=7.8 Hz, 1H), 7.40 (t, J=7.4 Hz, 1H), 7.31 (d, J=7.4 Hz, 1H), 7.09 (t, J=7.2 Hz, 1H), 4.48 (q, J=6.9 Hz, 2H), 2.72 (q, J=7.3 Hz, 2H), 1.43 (t, J=7.0 Hz, 3H), 1.27 (t, J=7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.2, 155.2, 154.0, 145.4, 139.5, 139.0, 131.0, 130.2, 128.0, 99.9, 62.4, 21.0, 16.1, 14.4; HRMS (EI) calcd for C14H14127INO2S: 386.9790, found 386.9791.
4-(2-Iodophenyl)-2, 5-dimethylthiazole (2d): 9 mg, 9% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.92 (dd, J=8.0, 1.0 Hz, 1H), 7.39 (td, J=7.4, 1.0 Hz, 1H), 7.31 (dd, J=7.6, 1.7 Hz, 1H), 7.06 (td, J=7.7, 1.7 Hz, 1H), 2.69 (s, 3H), 2.25 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 161.9, 153.0, 140.3, 139.1, 131.0, 129.7, 129.3, 128.0, 99.8, 19.2, 12.2; HRMS (EI) calcd for C11H10127INS: 314.9579, found 314.9581.
4-(2-iodophenyl)-5-methylthiazole (2e'): 33 mg, 36% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 8.60 (s, 1H), 7.88 (dd, J=8.0, 1.1 Hz, 1H), 7.34 (td, J=7.5, 1.2 Hz, 1H), 7.24 (dd, J=7.6, 1.7 Hz, 1H), 7.02 (ddd, J=7.9, 7.5, 1.8 Hz, 1H), 2.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 154.4, 149.2, 139.9, 139.3, 131.0, 130.0, 129.9, 128.1, 99.6, 12.1; HRMS (EI) calcd for C10H8127INO2S: 300.9422, found 300.9424.
A mixture of substrate 3 (0.3 mmol), Pd(OAc)2 (6.7 mg, 10 mol%), NIS (167.8 mg, 0.75 mmol) and DCE (3 mL) was stirred at 100 ℃ for 4 h. Upon completion of the reaction (monitored by TLC), the mixture was transferred into saturated brine (20 mL), then extracted with ethyl acetate (20 mL×3). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo to provide a crude product, which was purified via a column chromatography on silica gel (eluents: petroleum ether/ethyl acetate, V:V=75:1) to supply the desired product 4.
Ethyl 4-(2-iodophenyl)-5-methylthiazole-2-carboxylate (4a): 97 mg, 87% yield. Yellow solid, m.p. 71~72 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.89 (dd, J=8.0, 0.9 Hz, 1H), 7.38 (td, J=7.5, 1.1 Hz, 1H), 7.30 (dd, J=7.6, 1.7 Hz, 1H), 7.07 (td, J=7.8, 1.8 Hz, 1H), 4.45 (q, J=7.1 Hz, 2H), 2.35 (s, 3H), 1.40 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.3, 156.3, 154.1, 139.2, 139.1, 137.6, 131.1, 130.23, 128.1, 99.6, 62.5, 14.3, 12.8; HRMS (EI) calcd for C13H12127INO2S: 372.9633, found 372.9635.
Ethyl 4-(2-fluoro-6-iodophenyl)-5-methylthiazole-2-car- boxylate (4c): 43 mg, 37% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.73 (dd, J=6.6, 2.4 Hz, 1H), 7.18~7.09 (m, 2H), 4.48 (q, J=7.1 Hz, 2H), 2.37 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.2 (t, 1JCF=126.0 Hz), 154.8, 150.2, 139.3, 134.7 (d, 4JCF=3.6 Hz), 131.9 (d, 3JCF=8.7 Hz), 130.5 (d, 3JCF=8.2 Hz), 127.3 (d, 2JCF=18.4 Hz), 124.2 (d, 4JCF=3.6 Hz), 115.7 (d, 2JCF=22.5 Hz), 62.5, 14.3, 12.3; HRMS (EI) calcd for C13H11F127INO2S: 390.9539, found 390.9543.
Ethyl 4-(2-iodo-5-methoxyphenyl)-5-methylthiazole-2- carboxylate (4e): 88 mg, 73% yield. Yellow solid, m.p. 101~103 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.76 (d, J=8.8 Hz, 1H), 6.90 (d, J=3.0 Hz, 1H), 6.71 (dd, J=8.8, 3.0 Hz, 1H), 4.48 (q, J=7.1 Hz, 2H), 3.79 (s, 3H), 2.39 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.2, 159.7, 156.2, 154.1, 140.0, 139.6 (2C), 137.6, 117.0, 116.7, 88.0, 62.5, 55.5, 12.8; HRMS (EI) calcd for C14H14127INO3S: 402.9739, found 402.9740.
Ethyl 4-(2-iodo-5-methylphenyl)-5-methylthiazole-2- carboxylate (4f): 111 mg, 96% yield. White solid, m.p. 81~82 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.73 (d, J=8.1 Hz, 1H), 7.13 (d, J=1.8 Hz, 1H), 6.88 (dd, J=8.1, 1.8 Hz, 1H), 4.43 (q, J=7.1 Hz, 2H), 2.35 (s, 3H), 2.27 (s, 3H), 1.39 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 159.2, 155.4, 152.9, 137.8, 137.7, 137.1, 136.4, 131.0, 130.2, 94.3, 61.4, 19.8, 13.3, 11.8; HRMS (EI) calcd for C14H14127INO2S: 386.9790, found 386.9789.
Ethyl 4-(5-fluoro-2-iodophenyl)-5-methylthiazole-2- carboxylate (4g): 89 mg, 76% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.86 (dd, J=8.7, 5.5 Hz, 1H), 7.10 (dd, J=8.8, 3.0 Hz, 1H), 6.88 (td, J=8.5, 3.0 Hz, 1H), 4.48 (q, J=7.1 Hz, 2H), 2.40 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.62 (d, 1JCF=249.3 Hz), 160.1, 155.1, 154.5, 141.0 (d, 3JCF=7.9 Hz), 140.4 (d, 3JCF=7.9 Hz), 138.0, 118.6 (d, 2JCF=22.5 Hz), 117.8 (d, 2JCF=21.7 Hz), 92.6 (d, 4JCF=3.4 Hz), 62.6, 14.3, 12.8; HRMS (EI) calcd for C13H11F127INO2S: 390.9539, found 390.9543.
Ethyl 4-(5-chloro-2-iodophenyl)-5-methylthiazole-2- carboxylate (4h): 100 mg, 82% yield. Yellow solid, m.p. 69~71 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.83 (d, J=8.5 Hz, 1H), 7.35 (d, J=2.5 Hz, 1H), 7.11 (dd, J=8.5, 2.5 Hz, 1H), 4.48 (q, J=7.1 Hz, 2H), 2.40 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.0, 154.9, 154.5, 140.8, 140.1, 138.0, 134.4, 131.2, 130.5, 96.7, 62.6, 14.3, 12.8; HRMS (EI) calcd for C13H1137Cl127INO2S: 406.9244, found 406.9241.
Ethyl 4-(5-bromo-2-iodophenyl)-5-methylthiazole-2- carboxylate (4i): 128 mg, 94% yield. Yellow solid, m.p. 87~89 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.77 (d, J=8.5 Hz, 1H), 7.49 (d, J=2.4 Hz, 1H), 7.24 (dd, J=8.5, 2.4 Hz, 1H), 4.48 (q, J=7.1 Hz, 2H), 2.40 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.0, 154.8, 154.5, 141.1, 140.3, 138.1, 134.0, 133.4, 122.3, 97.6, 62.6, 14.3, 12.9; HRMS (EI) calcd for C13H1181Br127INO2S: 452.8718, found 452.8721.
Ethyl 4-(2-iodo-5-(trifluoromethyl)phenyl)-5-methyl- thiazole-2-carboxylate (4j): 85 mg, 64% yield. Yellow solid, m.p. 70~72 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.1 (d, J=8.3 Hz, 1H), 7.6 (s, 1H), 7.4 (d, J=7.2 Hz, 1H), 4.5 (q, J=7.1 Hz, 2H), 2.4 (s, 3H), 1.4 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.0, 154.8, 154.7, 140.2, 139.8, 138.2, 130.8 (q, 2JCF=33.2 Hz), 127.8 (q, 3JCF=3.6 Hz), 126.7 (dd, 3JCF=7.4, 3.6 Hz), 126.4 (q, 1JCF=270.6 Hz), 104.1 (d, 4JCF=1.3 Hz), 62.7, 14.3, 12.8; HRMS (EI) calcd for C14H11F3127INO2S: 440.9507, found 440.9509.
Ethyl 4-(2-iodo-4-methoxyphenyl)-5-methylthiazole-2- carboxylate (4l): 52 mg, 43% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.37 (d, J=2.6 Hz, 1H), 7.15 (d, J=8.5 Hz, 1H), 6.87 (dd, J=8.5, 2.6 Hz, 1H), 4.40 (q, J=7.1 Hz, 2H), 3.75 (s, 3H), 2.30 (s, 3H), 1.35 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 159.3, 158.9, 155.2, 152.7, 136.7, 130.4, 130.4, 123.1, 113.1, 98.7, 61.4, 54.6, 13.3, 11.8; HRMS (EI) calcd for C14H14127INO3S: 402.9739, found 402.9740.
Ethyl 4-(4-ethyl-2-iodophenyl)-5-methylthiazole-2-car- boxylate (4m): 88 mg, 73% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.76 (s, 1H), 7.23 (d, J=0.9 Hz, 2H), 4.47 (q, J=7.1 Hz, 2H), 2.64 (q, J=7.6 Hz, 2H), 2.38 (s, 3H), 1.42 (t, J=7.1 Hz, 3H), 1.24 (t, J=7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.2, 156.4, 153.8, 146.7, 138.4, 137.5, 136.4, 130.8, 127.7, 99.6, 62.4, 28.1, 15.4, 14.3, 12.8; HRMS (EI) calcd for C15H16127INO2S: 400.9946, found 400.9950.
Ethyl 4-(2-iodo-4-methylphenyl)-5-methylthiazole-2- carboxylate (4n): 93 mg, 79% yield. Yellow solid, m.p. 70~72 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.69 (s, 1H), 7.13 (s, 2H), 4.40 (q, J=7.1 Hz, 2H), 2.30 (s, 3H), 2.28 (s, 3H), 1.35 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.3, 156.4, 153.9, 140.4, 139.5, 137.5, 136.2, 130.7, 128.9, 99.5, 62.4, 20.7, 14.3, 12.8; HRMS (EI) calcd for C13H11F127INO2S: 390.9539, found 390.9540.
Ethyl 4-(4-fluoro-2-iodophenyl)-5-methylthiazole-2- carboxylate (4o): 99 mg, 84% yield. Yellow solid, m.p. 65~67 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.65 (dd, J=8.1, 2.6 Hz, 1H), 7.31 (dd, J=9.2, 5.2 Hz, 1H), 7.14 (td, J=8.3, 2.6 Hz, 1H), 4.48 (q, J=7.1 Hz, 2H), 2.38 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.0 (d, 1JCF=253.4 Hz), 160.1, 155.3, 154.1, 137.9, 135.4 (d, 4JCF=3.6 Hz), 131.9 (d, 3JCF=8.4 Hz), 126.0 (d, 2JCF=23.8 Hz), 115.3 (d, 2JCF=21.2 Hz), 99.2 (d, 3JCF=8.2 Hz), 62.5, 14.3, 12.7; HRMS (EI) calcd for C13H11F127INO2S: 390.9539, found 390.9540.
Ethyl 4-(4-chloro-2-iodophenyl)-5-methylthiazole-2-car- boxylate (4p): 79 mg, 65% yield. Yellow solid, m.p. 93~95 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.90 (d, J=2.1 Hz, 1H), 7.37 (dd, J=8.2, 2.1 Hz, 1H), 7.23 (d, J=8.2 Hz, 1H), 4.45 (q, J=7.1 Hz, 2H), 2.35 (s, 3H), 1.40 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.1, 155.1, 154.3, 138.4, 137.4, 137.9, 135.1, 131.7, 128.4, 99.6, 62.5, 14.3, 12.8; HRMS (EI) calcd for C13H1137Cl127INO2S: 406.9244, found 406.9243.
Ethyl 4-(4-bromo-2-iodophenyl)-5-methylthiazole-2- carboxylate (4q): 103 mg, 76% yield. Yellow solid, m.p. 103~104 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.08 (d, J=1.9 Hz, 1H), 7.55 (dd, J=8.2, 1.9 Hz, 1H), 7.20 (d, J=8.2 Hz, 1H), 4.47 (d, J=7.1 Hz, 2H), 2.38 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.0, 155.1, 154.3, 141.1, 138.2, 137.9, 132.0, 131.3, 123.2, 100.1, 62.5, 14.3, 12.8; HRMS (EI) calcd for C13H1181Br127INO2S: 450.8739, found 452.8720.
Ethyl 4-(2-iodo-4-(trifluoromethyl)phenyl)-5-methyl- thiazole-2-carboxylate (4r): 99 mg, 75% yield. Yellow solid, m.p. 80~82 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.18 (s, 1H), 7.68 (dd, J=8.0, 1.0 Hz, 1H), 7.46 (d, J=7.9 Hz, 1H), 4.48 (q, J=7.1 Hz, 2H), 2.40 (s, 3H), 1.44 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.0, 154.8 (d, 3JCF=14.9 Hz), 143.0, 138.0, 135.9 (q, 4JCF=3.8 Hz), 132.2 (q, 2JCF=33.0 Hz), 131.4, 129.1, 125.0 (q, 4JCF=3.6 Hz), 123.9(q, 1JCF=271.3Hz), 99.4, 62.6, 14.3, 12.7; HRMS (EI) calcd for C14H11F13127INO2S: 440.9507, found 440.9509.
Ethyl 4-(5-bromo-3-iodothiophen-2-yl)-5-methylthia- zole-2-carboxylate (4s): 77 mg, 56% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 7.23 (s, 1H), 4.47 (q, J=7.1 Hz, 2H), 2.51 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 159.8, 154.7, 146.4, 140.6, 137.2, 136.8, 115.3, 82.0, 62.7, 14.3, 13.5; HRMS (EI) calcd for C11H981Br127INO2S2: 458.8282, found 458.8285.
Ethyl 4-(3-iodo-5-methylfuran-2-yl)-5-methylthiazole- 2-carboxylate (4t): 70 mg, 62% yield. White solid, m.p. 92~94 ℃; 1H NMR (400 MHz, CDCl3) δ: 6.21 (s, 1H), 4.46 (q, J=7.1 Hz, 2H), 2.63 (s, 3H), 2.35 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.0, 154.6, 154.1, 146.7, 143.9, 139.2, 115.2, 66.8, 62.5, 14.3, 13.6, 13.6; HRMS (EI) calcd for C12H12127INO3S: 376.9583, found 372.9584.
Ethyl 4-(3, 5-diiodo-6-methoxynaphthalen-2-yl)-5-me- thylthiazole-2-carboxylate (4u): 154 mg, 89% yield. Yellow solid, m.p. 123~125 ℃; 1H NMR (400 MHz, CDCl3) δ: 8.72 (s, 1H), 7.73 (d, J=8.9 Hz, 1H), 7.70 (s, 1H), 7.19 (d, J=8.8 Hz, 1H), 4.46 (q, J=7.0 Hz, 2H), 4.00 (s, 3H), 2.37 (s, 3H), 1.41 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.2, 157.6, 155.8, 154.0, 141.6, 138.2, 136.9, 134.2, 130.6, 130.3, 128.7, 113.8, 99.8, 85.4, 62.5, 57.2, 14.4, 13.0; HRMS (EI) calcd for C18H15127I2NO3S: 578.8862, found 578.8861.
Ethyl 4-(2, 4-difluoro-6-iodophenyl)-5-methylthiazole- 2-carboxylate (4v): 48 mg, 39% yield. White solid, m.p. 62~64 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.23 (q, 1H), 7.17~7.09 (m, 1H), 4.48 (q, J=7.1 Hz, 2H), 2.37 (s, 3H), 1.43 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 160.1, 154.5, 154.3 (d, 3JCF=1.9 Hz), 151.4 (dd, 1, 3JCF=245.0, 13.7 Hz), 149.7 (dd, 1, 3JCF=253.4, 15.1 Hz), 138.1, 136.3 (d, 3JCF=4.0 Hz), 126.6 (dd, 2, 2JCF=6.5, 3.7 Hz), 117.1 (d, 2JCF=18.0 Hz), 88.9 (d, 2JCF=21.7 Hz), 62.6, 14.3, 12.7; HRMS (EI) calcd for C13H10F2127INO2S: 408.9445, found 408.9446.
Ethyl 4-(3, 5-difluoro-2-iodophenyl)-5-methylthiazole- 2-carboxylate (4w): 90 mg, 73% yield. White solid, m.p. 71~73 ℃; 1H NMR (400 MHz, CDCl3) δ: 6.96 (d, J=8.4 Hz, 1H), 6.90 (td, J=8.3, 2.6 Hz, 1H), 4.47 (q, J=7.1 Hz, 2H), 2.38 (s, 3H), 1.42 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 162.9 (dd, 1, 3JCF=249.6, 12.2 Hz), 162.1 (dd, 1, 3JCF=246.2, 12.6 Hz), 160.0, 154.7, 153.8, 142.1 (dd, 3, 3JCF=9.4, 3.1 Hz), 138.1, 114.4 (dd, 2, 4JCF=22.3, 3.4 Hz), 104.3 (dd, 2, 2JCF=28.5, 25.8 Hz), 81.2 (dd, 2, 4JCF=25.9, 4.3 Hz), 62.6, 14.3, 12.7; HRMS (EI) calcd for C13H10F2127INO2S: 408.9445, found 408.9448.
A mixture of 4a (111.9 mg, 0.3 mmol), 3-boronoanisole (59.3 mg, 0.39 mmol), I2 (82.9 mg, 0.06 mmol), K2CO3 (82.9 mg, 0.06 mmol) and PEG (3 mL) was stirred in a sealed tube at 140 ℃ for 24 h. Upon completion of the reaction (as monitored by TLC), the mixture was transferred into saturated brine (20 mL) then extracted with ethyl acetate (20 mL×3). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo to provide a crude product, which was purified via a column chromatography on silica gel (eluents: petroleum ether/ethyl acetate, V:V=100:1) to supply the desired product 4-(3'-methoxy-[1, 1'-biphenyl]-2-yl)-5-methylthia- zole (5a), 62 mg, 73% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 8.61 (s, 1H), 7.54~7.39 (m, 4H), 7.16 (t, J=7.9 Hz, 1H), 6.83~6.74 (m, 2H), 6.63 (dd, J=2.4, 1.7 Hz, 1H), 3.64 (s, 3H), 1.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 159.1, 152.0, 149.1, 142.7, 141.7, 133.2, 131.1, 130.0, 129.3, 129.1, 128.7, 127.4, 121.5, 114.1, 113.0, 55.1, 11.6; HRMS (EI) calcd for C17H15NOS 281.0874, found 281.0872.
A mixture of 4a (111.9 mg, 0.3 mmol), DDQ (68.1 mg, 0.3 mmol), Cu(OAc)2 (60 mg, 0.3 mmol), AgO (36.9 mg, 0.3 mmol) and NMP (3 mL) was stirred in a sealed tube at 120 ℃ for 22 h. Upon completion of the reaction (as monitored by TLC), the mixture was transferred into saturated brine (20 mL), then extracted with ethyl acetate (20 mL×3). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo to provide a crude product, which was purified via a column chromatography on silica gel (eluents: petroleum ether/ethyl acetate, V:V=30:1) to supply the desired product ethyl 4-(2-cyanophenyl)-5-methylthiazole-2-carboxylate (5b), 63 mg, 77% yield. White solid, m.p. 100~101 ℃; 1H NMR (400 MHz, CDCl3) δ: 7.73 (dd, J=7.8, 0.9 Hz, 1H), 7.64 (td, J=7.7, 1.3 Hz, 1H), 7.57 (dd, J=7.8, 0.9 Hz, 1H), 7.48 (td, J=7.6, 1.3 Hz, 1H), 4.44 (q, J=7.1 Hz, 2H), 2.50 (s, 3H), 1.39 (t, J=7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ: 159.9, 155.1, 150.7, 138.9, 137.6, 133.1, 132.8, 131.3, 129.0, 117.8, 113.0, 62.6, 14.3, 12.9; HRMS (EI) calcd for C14H12N2O2S: 272.0619, found 272.0620.
A mixture of 4a (111.9 mg, 0.3 mmol), 3-chlorothio- phenol (56.2 mg, 0.39 mmol), CuI (1.4 mg, 0.0075 mmol), K2CO3 (45.6 mg, 0.33 mmol) and NMP (3 mL) was stirred in a sealed tube at 100 ℃ for 16 h. Upon completion of the reaction (as monitored by TLC), the mixture was transferred into saturated brine (20 mL), then extracted with ethyl acetate (20 mL×3). The combined organic layers were dried over anhydrous MgSO4 and concentrated in vacuo to provide a crude product, which was purified via a column chromatography on silica gel (eluents: petroleum ether/ethyl acetate, V:V=100:1) to supply the desired product 5-methyl-4-(2-(phenylthio)phenyl)thiazole (5c), 52 mg, 55% yield. Yellow oil. 1H NMR (400 MHz, CDCl3) δ: 8.65 (s, 1H), 7.34 (dd, J=5.9, 3.3 Hz, 1H), 7.29 (dd, J=6.4, 2.9 Hz, 3H), 7.25~7.17 (m, 4H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 151.1, 149.3, 136.9, 135.4, 133.7, 133.5, 133.4 (2C), 131.3, 131.0, 130.4, 129.3 (2C), 129.1, 126.9, 12.1; HRMS (EI) calcd for C16H1237ClNS3: 319.0070, found 319.0072.
Gribble, G. W. Acc. Chem. Res. 1998, 31, 141. doi: 10.1021/ar9701777
Lindley, J. Tetrahedron 1984, 40, 1433. doi: 10.1016/S0040-4020(01)91791-0
Suzuki, A. J. Organomet. Chem. 1999, 576, 147. doi: 10.1016/S0022-328X(98)01055-9
Crisp, T. G. Chem. Soc. Rev. 1998, 27, 427. doi: 10.1039/a827427z
Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525. doi: 10.1039/C3CS60289C
Merkushev, E. B. Synthesis 1988, 923.
(a) Li, B.; Liu, B.; Shi, B. F. Chem. Commun. 2015, 51, 5093.
(b) Pal, P.; Singh, H.; Panda, A. B.; Ghosh, S. C. Asian J. Org. Chem. 2015, 4, 879.
(c) Zhan, B. B.; Liu, Y. H.; Hu, F.; Shi, B. F. Chem. Commun. 2016, 52, 4934.
(d) Aihara, Y.; Chatani, N. ACS Catal. 2016, 6, 4323.
(e) Khan, B.; Kant, R.; Koley, D. Adv. Synth. Catal. 2016, 358, 2352.
(f) Kommagalla, Y.; Yamazaki, K.; Yamaguchi, T.; Chatani, N. Chem. Commun. 2018, 54, 1359.
(g) Singh, H.; Sen, C.; Sahoo, T.; Ghosh, S. C. Eur. J. Org. Chem. 2018, 34, 4748.
(h) Du, Y.; Liu, Y. Y.; Wan, J. P. J. Org. Chem. 2018, 83, 3403.
Kalyani, D.; Dick, A. R.; Anani, W. Q.; Sanford, M. S. Org. Lett. 2006, 8, 2523. doi: 10.1021/ol060747f
(a) Giri, R.; Chen, X.; Yu, J. Q. Angew. Chem., Int. Ed. 2005, 44, 2112.
(b) Li, J. J.; Mei, T. S.; Yu, J. Q. Angew. Chem., Int. Ed. 2008, 47, 6452.
(c) Mei, T. S.; Giri, R.; Maugel, N.; Yu, J. Q. Angew. Chem., Int. Ed. 2008, 47, 5215.
(d) Mei, T. S.; Wang, D. H.; Yu, J. Q. Org. Lett. 2010, 12, 3140.
(e) Nack, W. A.; Wang, X.; Wang, B.; He, G.; Chen, G. Beilstein J. Org. Chem. 2016, 12, 1243.
(f) Zhu, R. Y.; Liu, L. Y.; Yu, J. Q. J. Am. Chem. Soc. 2017, 139, 12394.
(g) Zhu, R. Y.; Saint-Denis, T. G.; Shao, Y.; He, J.; Sieber, J. D.; Senanayake, C. H.; Yu, J. Q. J. Am. Chem. Soc. 2017, 139, 5724.
(a) Dudnik, A. S.; Chernyak, N.; Huang, C.; Gevorgyan, V. Angew. Chem., Int. Ed. 2010, 49, 8729.
(b) Du, B.; Jiang, X.; Sun, P. J. Org. Chem. 2013, 78, 2786.
(c) Sadhu, P.; Alla, S. K.; Punniyamurthy, T. J. Org. Chem. 2013, 78, 6104.
(d) Pascanu, V.; Carson, F.; Solano, M. V.; Su, J.; Zou, X.; Johansson, M. J.; Martin-Matute, B. Chem.-Eur. J. 2016, 22, 3729.
(e) Testa, C.; Gigot, E.; Genc, S.; Decreau, R.; Roger, J.; Hierso, J. C. Angew. Chem., Int. Ed. 2016, 55, 5555.
(f) Yang, X.; Sun, Y.; Sun, T. Y.; Rao, Y. Chem. Commun. 2016, 52, 6423.
(g) Das, R.; Kapur, M. J. Org. Chem. 2017, 82, 1114.
(h) Dubost, E.; Babin, V.; Benoist, F.; Hebert, A.; Barbey, P.; Chollet, C.; Bouillon, J. P.; Manrique, A.; Pieters, G.; Fabis, F.; Cailly, T. Org. Lett. 2018, 20, 6302.
(i) Tang, R. J.; Milcent, T.; Crousse, B. J. Org. Chem. 2018, 83, 930.
(a) Chu, L.; Wang, X. C.; Moore, C. E.; Rheingold, A. L.; Yu, J. Q. J. Am. Chem. Soc. 2013, 135, 16344.
(b) Wang, X. C.; Hu, Y.; Bonacorsi, S.; Hong, Y.; Burrell, R.; Yu, J. Q. J. Am. Chem. Soc. 2013, 135, 10326.
(c) Chu, L.; Xiao, K. J.; Yu, J. Q. Science 2014, 346, 451
(a) Lu, C.; Zhang, S. Y.; He, G.; Nack, W. A.; Chen, G. Tetrahedron 2014, 70, 4197.
(b) Chu, L.; Shang, M.; Tanaka, K.; Chen, Q.; Pissarnitski, N.; Streckfuss, E.; Yu, J. Q. ACS Cent. Sci. 2015, 1, 394.
(c) Sun, X.; Yao, X.; Zhang, C.; Rao, Y. Chem. Commun. 2015, 51, 10014.
(d) Fan, X. M.; Guo, Y.; Li, Y. D.; Yu, K. K.; Liu, H. W.; Liao, D. H.; Ji, Y. F. Asian J. Org. Chem. 2016, 5, 499.
(a) Das, R.; Kapur, M. Asian J. Org. Chem. 2018, 7, 1524.
(b) Liao, G.; Shi, B. F. Acta Chim. Sinica. 2015, 73, 1283 (in Chinese).
(廖港, 史炳烽, 化学学报, 2015, 73, 1283.)
Santra, S. K.; Banerjee, A.; Khatun, N.; Samanta, A.; Patel, B. K. RSC Adv. 2015, 5, 11960. doi: 10.1039/C4RA15461D
(a) Al-Ghorbani, M.; Alghamdi, H. A.; Khanum, S. A. Eur. J. Biomed. Pharm. Sci. 2018, 5, 1.
(b) Attri, C.; Bhatia, P.; Kumar, P. J. Mod. Chem. Chem. Technol. 2018, 9, 19.
(c) Liaras, K.; Fesatidou, M.; Geronikaki, A. Molecules 2018, 23, 685/1.
(d) de Siqueira, L. R. P.; de Moraes Gomes, P. A. T.; de Lima Ferreira, L. P.; de Melo Rego, M. J. B.; Leite, A. C. L. Eur. J. Med. Chem. 2019, 170, 237.
(e) Scarim, C. B.; Jornada, D. H.; Machado, M. G. M.; Ferreira, C. M. R.; dos Santos, J. L.; Chung, M. C. Eur. J. Med. Chem. 2019, 162, 378.
Yu, K. K.; Guo, Y.; Hu, Y. H.; Xu, Z.; Liu, H. W.; Liao, D. H.; Ji, Y. F. Asian J. Org. Chem. 2016, 5, 1219. doi: 10.1002/ajoc.201600304
Bergstr m, M.; Suresh, G.; Naidu, V. R.; Unelius, C. R. Eur. J. Org. Chem. 2017, 2017, 3234. doi: 10.1002/ejoc.201700173
Qiao, H. J.; Yang, F.; Wang, S. W.; Leng, Y. T.; Wu, Y. J. Tetrahedron 2015, 71, 9258. doi: 10.1016/j.tet.2015.10.035
Qiu, F. C.; Yang, W. C.; Chang, Y. Z.; Guan, B. T. Asian J. Org. Chem. 2017, 6, 1361. doi: 10.1002/ajoc.201700238

Scheme 2 Diversification of the iodination products
Reaction conditions: (a) 4a (111.9 mg, 0.30 mmol), 3-methoxylben-zoboric acid (59.3 mg, 0.39 mmol), I2 (15.2 mg, 0.06 mmol), K2CO3 (82.9 mg, 0.60 mmol), PEG, 140 ℃, 24 h; (b) 4a (111.9 mg, 0.30 mmol), DDQ (68.1 mg, 0.30 mmol), Cu(OAc)2 (60.0 mg, 0.30 mmol), Ag2O (36.9 mg, 0.30 mmol), NMP, 120 ℃, 22 h; (c) 4a (111.9 mg, 0.30 mmol), 3-chlorothiophenol (56.2 mg, 0.39 mmol), CuI (1.4 mg, 2.5 mol%), K2CO3 (45.6 mg, 0.33 mmol), 100 ℃, 16 h; b Isolated yields
Table 2. Optimization of the reaction conditionsa
| ||||
| Entry | Catalyst | NIS (equiv.) | Solvent | Yieldb/% |
| 1 | None | 2.0 | DCE/HFIP (V:V=4:1) |
0 |
| 2 | Pd(OAc)2 | 2.0 | DCE/HFIP (V:V=4:1) |
76 |
| 3 | Pd(CH3CN)2Cl2 | 2.0 | DCE/HFIP (V:V=4:1) |
64 |
| 4 | Pd(TFA)2 | 2.0 | DCE/HFIP (V:V=4:1) |
57 |
| 5 | PdCl2 | 2.0 | DCE/HFIP (V:V=4:1) |
38 |
| 6 | Pd(PPh3)2Cl2 | 2.0 | DCE/HFIP (V:V=4:1) |
27 |
| 7c | Pd(OAc)2 | 2.0 | DCE/HFIP (V:V=4:1) |
58 |
| 8d | Pd(OAc)2 | 2.0 | DCE/HFIP (V:V=4:1) |
75 |
| 9 | Pd(OAc)2 | 2.0 | DCE | 86 |
| 10 | Pd(OAc)2 | 2.0 | HFIP | 69 |
| 11 | Pd(OAc)2 | 2.0 | DMF | 85 |
| 12 | Pd(OAc)2 | 2.0 | DCM | 62 |
| 13 | Pd(OAc)2 | 2.0 | TCM | 21 |
| 14 | Pd(OAc)2 | 1.5 | DCE | 76 |
| 15 | Pd(OAc)2 | 2.5 | DCE | 93 |
| 16 | Pd(OAc)2 | 3.0 | DCE | 92 |
| 17e | Pd(OAc)2 | 2.5 | DCE | 78 |
| 18f | Pd(OAc)2 | 2.5 | DCE | 90 |
| a Reaction conditions: 1b (74.1 mg, 0.3 mmol), NIS, catalyst (10 mol%), solvent (3 mL, 0.1 mol·L-1), 100 ℃, 4 h; b NMR yield using CH2Br2 as the internal standard; c Pd(OAc)2 (5 mol%); d Pd(OAc)2 (15 mol%); e DCE (0.05 mol·L-1); f DCE (0.15 mol·L-1); NIS=N-Iodosuccinimide; DCE=1, 2-Dichloroethane; HFIP=Hexafluoroisopropanol; DMF=N, N-Dimethylfor- mamide; DCM=Dichloromethane; TCM=Trichloromethane; DMSO=Dimethyl sulfoxide. | ||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们