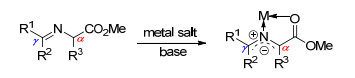
图式 1.
金属化亚甲胺叶立德
Scheme 1.
Structure of metallated azomethine ylides

氨基酸是构成生命体蛋白质并同生命活动息息相关的最基本的物质.根据其存在形式, 氨基酸可分为天然氨基酸和非天然氨基酸两大类.自然界中天然存在的氨基酸大约有180多种[1].非天然氨基酸则是通过人工合成的氨基酸, 结构与天然氨基酸存在着差异, 有的是在天然氨基酸中引入一些官能团, 用以改善其性能, 有的则与天然氨基酸结构完全不同, 是分子设计合成的结果.
具有光学活性的非天然α-氨基酸是一些新的生物活性肽的手性合成单元, 研究其合成也是探索蛋白质结构和功能及功能性多肽的一种有效工具[2].同时非天然α-氨基酸及其衍生物还是合成许多药物、添加剂和精细化学品的重要中间体[3].近年来随着其越来越多的用途被发现, 对氨基酸特别是具有特殊结构的光学纯α-氨基酸的需求量越来越大, 非天然α-氨基酸的不对称合成已经成为一个非常活跃的研究领域[4].
在众多合成手性非天然α-氨基酸的方法中, 金属化的亚甲胺叶立德参与的α-官能团化反应是最直接、最高效的方法之一[5].金属化的亚甲胺叶立德结构如Scheme 1所示, 由简便易得的天然氨基酸酯衍生的席夫碱在金属盐和碱的作用下去质子化得到.在该结构中, 金属盐的作用包括: (1)通过与席夫碱上N, O原子配位提高其α位的酸性, 使其更易脱质子; (2)通过配位形成的刚性结构, 提高了亚甲胺叶立德的稳定性.金属化亚甲胺叶立德主要含有α位和γ位两个反应位点, 在大多数反应中, 由叶立德的α位启动进攻.金属化亚甲胺叶立德除应用于不对称1,3-偶极环加成反应合成四氢吡咯衍生物外, 还被广泛用于不对称Michael加成、Mannich和亲核取代等反应中, 合成了多种类型的手性非天然氨基酸.本文着重介绍了近年来金属化的亚甲胺叶立德在非天然氨基酸合成中的应用, 并对其进行了分类讨论, 为读者了解该方向的研究动态提供帮助.
金属化亚甲胺叶立德的前体通常为天然氨基酸衍生的席夫碱.目前应用较多的亚甲胺叶立德前体依据结构和合成方法可分为三类, 如Scheme 2所示[5].
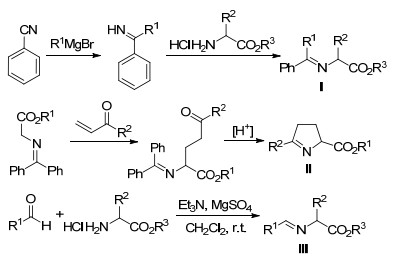
由于Ⅰ型和Ⅱ型席夫碱的α-H酸性较弱、结构较稳定, 发生α-官能团后的产物不易或无法消旋化, 因此在早期的相关研究中, 大多选择Ⅰ型和Ⅱ型席夫碱作为金属化的亚甲胺叶立德前体.如2007年, Kobayashi课题组[6]报道的Ca(Ⅱ)/Box络合物催化Ⅰ型亚甲胺叶立德参与的迈克尔加成反应, 实现了对末端缺电子烯烃的1, 4-迈克尔加成反应, 并取得了中等到优秀的立体选择性(61:39~91:9 dr, 78%~99% ee).当他们采用该催化体系, 以巴豆酸酯、丙烯酰二甲胺等缺电子烯烃为反应组分时, 只能获得1,3-偶极环加成产物(Scheme 3).随后, Kobayashi、Carretero、Zajac和Hou等[6, 7]又相继报道了金属化的Ⅰ型亚甲胺叶立德与缺电子烯烃的1, 4-共轭加成反应, 合成了多种光学纯的非天然α-氨基酸.金属化的Ⅱ型亚甲胺叶立德参与的不对称α-官能团化反应也由Fukuzawa和我们课题组[8]等先后报道.尽管Ⅰ型和Ⅱ型亚甲胺叶立德在非天然α-氨基酸合成中已有较多应用, 但都存在一个共同的弊端, 即其合成较为复杂.如Scheme 2所示, Ⅰ型亚甲胺叶立德前体合成由苯甲腈出发, 与格氏试剂反应得到亚胺中间体, 再与氨基酸酯发生胺交换反应得到目标化合物.该方法步骤较长, 亚胺中间体也不稳定, 且由于需要使用有机金属试剂, 对反应条件要求严格, 同时很多官能团在该条件下也无法耐受.而Ⅱ型亚甲胺叶立德前体更需要使用Ⅰ型亚甲胺叶立德前体作为起始原料, 先与缺电子烯烃反应, 再经水解/缩合等多步反应才能获得, 十分不经济.由于在原料合成上的诸多弊端.很显然, 使用Ⅰ型和Ⅱ型亚甲胺叶立德合成非天然α-氨基酸不具有很强的实用价值.而与Ⅰ型和Ⅱ型亚甲胺叶立德相比, Ⅲ型亚甲胺叶立德前体的合成, 仅需在室温条件下使用氨基酸酯和各种醛类化合物脱水缩合即可得到.该方法原料简便易得, 步骤简单, 条件温和且适用于各种含不同官能团的原料.除获取方法更简便外, Ⅲ型亚甲胺叶立德在合成应用中也更具有优势.如(1)反应活性更高, 可参与的反应类型相对更广泛; (2)底物使用范围更广, 如烷基醛、芳基醛、杂芳基醛以及含不同官能团的氨基酸所衍生的亚胺都能用于合成Ⅲ型亚甲胺叶立德叶立德, 并参与后续的应用转化; (3)反应产物更易实现后续的衍生转化.因此, 发展Ⅲ型亚甲胺叶立德参与的α-官能团化反应合成非天然氨基酸具有更大的研究意义.本文主要对使用Ⅲ型亚甲胺叶立德合成非天然α-氨基酸的例子进行介绍, 少量介绍Ⅰ型和Ⅱ型亚甲胺叶立德参与的例子.
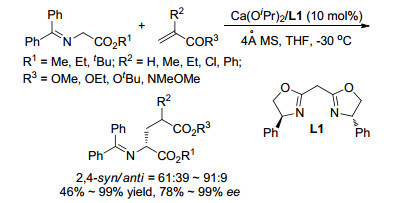
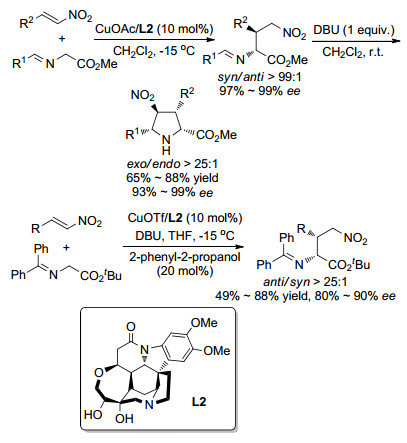
Ⅲ型亚甲胺叶立德作为1,3-偶极子广泛应用于催化不对称1,3-偶极环加成反应研究中, 相比之下该类型的叶立德作为亲核试剂参与的催化不对称合成的报道比较少. 2011年, 我们课题组[9]报道了首例Ⅲ型亚甲胺叶立德参与的不对称Michael加成反应.在之前的工作中, 我们发现甘氨酸衍生的亚甲胺叶立德与亚烷基丙二酸酯在我们的发展的催化不对称1,3-偶极环加成体系中, 完全给出环加成反应产物; 基于我们催化体系中1,3-环加成反应机理的理解与验证, 设计了位阻较大的亚烷基丙二磷酸酯作为反应组分.在进行了一系列条件筛选后发现, 采用本课题组发展的Cu(Ⅰ)/TF-BiphamPhos络合物作为催化剂, 亚甲氨叶立德与亚烷基丙二磷酸酯的反应可以在30 min内完成, 并以很高的收率获得了相应的迈克尔加成产物(70%~95%), 实现了优异的非对映选择性(syn/anti>99:1)和对映选择性(89%~99% ee)控制(Scheme 4)[10].该催化体系的底物适用范围很广, 对β位是芳基、烷基、杂芳基取代以及没有取代基的亚烷基丙二磷酸酯都有很好的兼容性与不对称催化效果.利用该方法, 可以得到一系列在医药和生物活性研究中具有重要作用的手性α-氨基磷酸酯.同时, 通过控制实验和化学计算, 提出了可能的催化机制和过渡态模型[10, 11].
在上述工作基础之上, 设计了以简便易得的Morita-Baylis-Hillman (MBH)碳酸酯为亲电组分, 借助其β-离去基团高效实现了Ⅲ型亚甲胺叶立德参与的不对称Michael加成/消除串联反应, 得到了含有季碳手性中心的焦谷氨酸衍生物[12].含有季碳手性中心的焦谷氨酸衍生物是很多天然生物碱和活性大分子的主要结构片段, 如salinosporamide A存在于热带海洋放线菌的发酵液中, 而oxazolomycin A和lactacystin都是从链霉菌的发酵液提取获得, 如Scheme 5所示[13].之前的报道中, 多数构建含季碳手性中心的焦谷氨酸衍生物的方法是采用全合成的策略, 直接采用不对称催化的方式并成功获得该类化合物的方法几乎没有.当采用Cu(MeCN)4BF4/(S)-BINAP作为催化剂, 碳酸铯为碱时, α-取代亚甲胺叶立德可以与MBH碳酸酯高效地发生Michael加成反应, 该产物在酸性条件下水解并经加热发生分子内酰胺化, 以较高的收率和优异的对映选择性得到含季碳中心的焦谷氨酸衍生物.
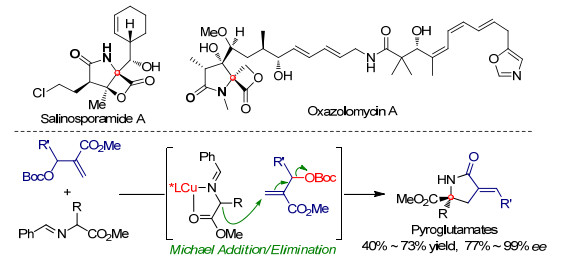
随后, 在2012年我们[14]还进一步实现了高丝氨酸衍生的Ⅲ型亚甲胺叶立德与溴代MBH产物之间的不对称Michael加成反应, 如Scheme 6所示.含有γ-丁内酯或γ-丁内酰胺的螺环结构片段广泛存在于生物活性分子和天然大分子中, 这类螺环结构还是很多药物中间体的关键组成片段[15].我们以Cu(Ⅰ)/(S)-TF-Biphamphos络合物作为催化剂, 碳酸钾为碱, 室温下即可以优异的收率、区域选择性和立体选择性得到相应的Michael加成产物.同时, 该产物还可以在对甲苯磺酸作用下转化为同时含有γ-丁内酯和γ-丁内酰胺的螺环化合物.该化合物因其结构在生物医药的合成中存在潜在的应用价值.值得一提的是, 该催化条件同样适用于甘氨酸衍生的亚甲胺叶立德.
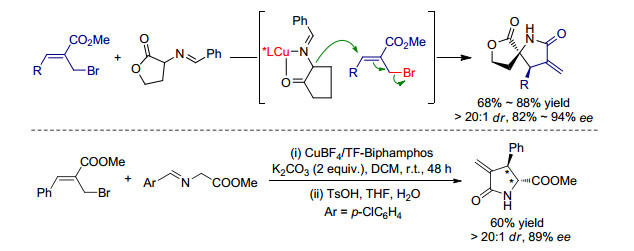
除我们课题组之外, 2011年Oh课题组[16]使用自己发展的催化体系尝试了Ⅲ型亚甲胺叶立德参与的不对称Michael加成反应, 如Scheme 7所示.利用CuOAc/L2作为催化剂, 二氯甲烷作为溶剂, -15 ℃条件下亚甲胺叶立德与硝基烯烃的反应可以很好地停在第一步的Michael加成反应, 以较高的产率和十分优秀的立体选择性得到syn-选择性加成产物.该反应中使用的金属盐CuOAc对于反应的进程有重要的作用, 除作为金属路易斯酸活化亚甲胺叶立德前体, CuOAc的醋酸根阴离子还作为弱碱脱质子, 同时较弱的碱性也不会促进加成产物发生后续的分子内环化反应.该作用在后续的实验中得到了验证, 将第一步加成产物室温下溶解于二氯甲烷中, 并加入碱性较强的DBU, 加成产物可以继续发生分子内环化反应得到一系列四氢吡咯衍生物.值得一提的是, 作者还进一步尝试合成了anti-选择性加成产物.在加入2-苯基-2-丙醇作为添加剂的条件下, 以CuOTf/L2作为催化剂, Ⅰ型亚甲胺叶立德可以与硝基烯烃反应以较高的收率和立体选择性得到anti-选择性加成产物, 同时该产物也可在一定条件下继续发生环化反应得到相应的四氢吡咯衍生物.
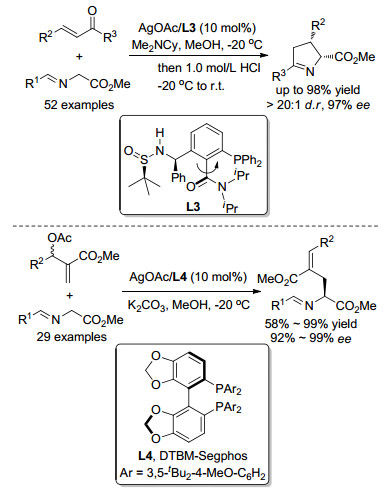
2016年, Xu课题组[17]采用AgOAc/L3络合物作为催化剂, 报道了Ⅱ型亚甲胺叶立德与查尔酮之间的不对称Michael加成.该反应所得到的加成产物并不稳定, 在酸性条件下亚胺片段水解, 并随之发生分子内脱水缩合反应得到手性二氢吡咯衍生物.随后, Xu课题组[18]还采用AgOAc/DTBM-Segphos络合物作为催化剂, 同样实现了Ⅲ型亚甲胺叶立德与MBH产物的不对称Michael加成/消除串联反应, 以较为优异的收率和对映选择性得到了一系列非天然氨基酸衍生物, 如Scheme 8所示.
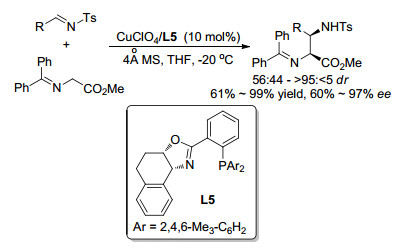
亚甲胺叶立德参与的不对称Mannich反应是合成β-氨基酸衍生物和乙二胺衍生物的重要途径, 因而受到有机化学家的关注.该类反应在已有的报道中多使用Ⅰ型亚甲胺叶立德进行研究. 2003年, Jørgensen课题组[19]首次实现了二苯酮衍生的Ⅰ型亚甲胺叶立德对Ts保护的醛亚胺的Mannich反应(Scheme 9).该报道采用Cu(Ⅰ)/L5络合物作为催化剂, 能够顺利催化反应, 但只能获得中等到高的非对映选择性(56~44~>95~5)和对映选择性(60%~97% ee).
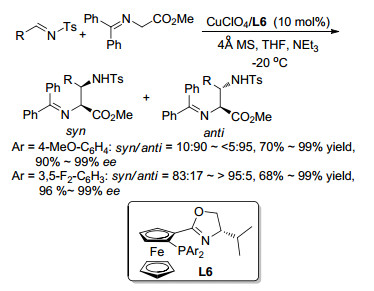
2008年, Hou课题组[20]用Cu(Ⅰ)/L6络合物催化同样实现了二苯酮衍生的Ⅰ型亚甲胺叶立德与亚胺间的Mannich反应(Scheme 10).与之前报道不同的是, 该催化体系可以通过改变配体磷原子上的取代基电性来选择性获得syn或anti构型的加成产物.当取代基为对位供电子性的甲氧基苯基时, 主要获得anti构型的产物(syn/anti=9:91~<5:95), 而当取代基为吸电子性的3, 5-二氟苯基时, 可以获得syn构型的产物(83:17~>95:5).
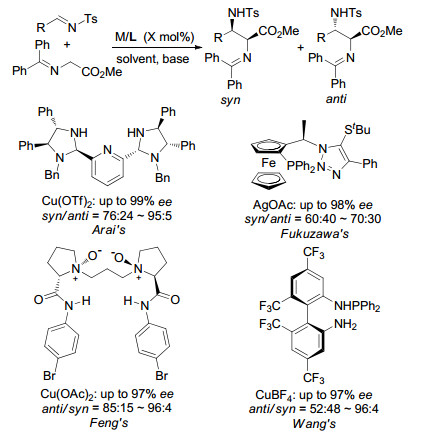
此后几年, Arai, Fukuzawa, Feng课题组和我们[21]都分别采用各自的催化体系报道了金属化的Ⅰ型亚甲胺叶立德对N-Ts亚胺的不对称Mannich反应, 并都取得了不错的效果(Scheme 11).
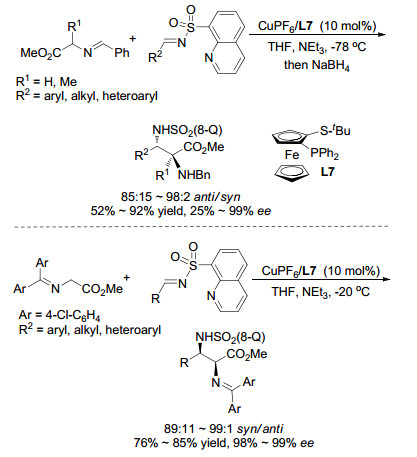
与Ⅰ型亚甲胺叶立德相比, Ⅲ型亚甲胺叶立德参与的不对称Mannich反应报道较少. 2008年, Carretero课题组[22]使用Cu(Ⅰ)/L7络合物作为催化剂实现了首例Ⅲ型亚甲胺叶立德参与的不对称Mannich反应, 如Scheme 12所示.该反应采用特殊的8-喹啉磺酰基胺与醛缩合而成的亚胺作为Mannich反应受体, 8-喹啉基在反应中起到了非常重要的作用.该基团上的N原子可以与金属路易斯酸产生配位效果, 从而影响了反应的过渡态, 使得该反应可以顺利进行并得到了优异的立体选择性.随后, 该课题组还进一步报道了Ⅰ型亚甲胺叶立德与8-喹啉磺酰亚胺的Mannich反应, 和Ⅲ型亚甲胺叶立德反应的anti-选择性不同, Ⅰ型亚甲胺叶立德反应可以得到syn-选择性的加成产物.
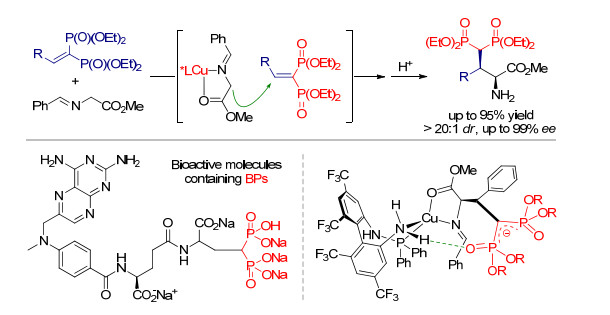
氨基酸酯衍生的二苯甲酮亚胺类化合物参与的不对称烯丙基取代反应可以得到含有烯丙基取代基的手性非天然α-氨基酸, 该类氨基酸特别是α,α-双取代α-氨基酸不但本身可以作为酶抑制剂, 也可以作为关键原料用于多种模拟肽的合成[23].同时烯丙基取代基可以进行多种后续的官能团转化, 因此在天然产物和药物合成中有着重要的作用.由于上述原因, 实现高效的亚甲胺叶立德不对称α-烯丙基化反应有着十分重要的意义.在早期的报道中, 实现该转化的催化策略主要有两种, 如Scheme 13所示.第一种是使用手性钯络合物作为催化剂[24], 第二种则是使用非手性的钯络合物同时结合手性相转移催化来实现立体控制的效果[25].但迄今为止, 这两种方法都存在明显的不足.例如, 第一种策略产物的对映选择性都较低, 这可能是由于该催化体系中反应的立体中心与催化中心距离较远导致[26]; 而对于第二种策略, 尽管产物的对映选择性较高, 但只能合成α-单取代α-氨基酸而无法获得α,α-双取代α-氨基酸, 并且两种方法都需使用Ⅰ型亚甲胺叶立德前体.
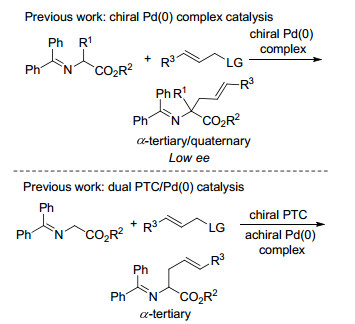
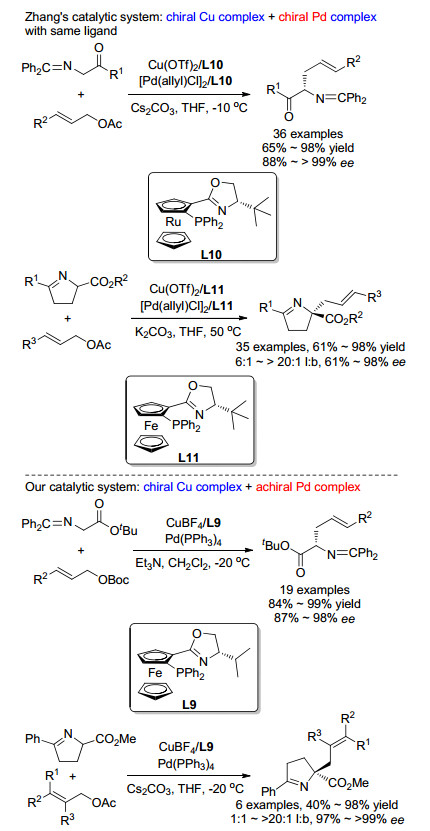
最近, 我们和Zhang课题组[27]几乎同时使用双金属协同催化策略解决了上述两种方法所存在的不足. Zhang课题组[28]的报道中采用了手性钯催化剂和手性铜催化剂实现了该反应的协同催化(Scheme 14).通过一系列控制实验, 他们证实了在该催化体系中的两种金属盐必须使用相同的手性配体L8, 这可能是由于其Cu(Ⅱ)/Pd(0)体系中不同配体之间会出现交叉配位的现象, 从而导致产物的对映选择性降低.利用该催化剂体系, Zhang课题组不仅实现了简单亚甲胺叶立德的不对称烯丙基化反应, 还完成了多种肽类化合物的α-烯丙基化, 并都取得了十分优异的结果.
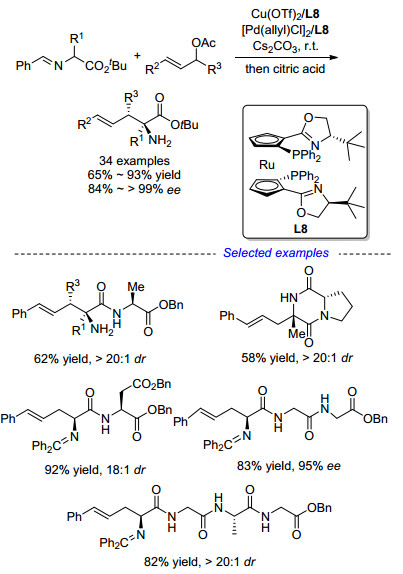
在我们的研究工作中, 使用手性铜催化剂与亚甲胺叶立德前体在碱性条件下配位得到铜(Ⅰ)稳定的Ⅰ型亚甲胺叶立德作为亲核反应组分, 使用非手性钯催化剂与碳酸酯现场生成活性π-烯丙基-Pd中间体作为亲电反应组分, 两种中间体在-20 ℃下反应即可以非常优异的收率和对映选择性得到一系列非天然α,α-双取代α-氨基酸衍生物, 如Scheme 15所示[29].我们还可以通过该方法合成多种PLG模拟肽[30], 体现了该方法的实用性.
控制实验表明两种催化剂在该反应中都必不可少.由于采用了两种不同的金属盐和配体, 我们还对配体之间可能出现的交换进行了实验验证.通过在线31P NMR实验确认, 体系中的配体不会发生明显交叉配位.随后, 用甘氨酸衍生的席夫碱作为起始原料, 与两分子不同种类的烯丙基碳酸酯发生两次烯丙基取代反应, 得到结构多样性的手性α,α-双烯丙基-α-氨基酸衍生物.这类化合物由于其取代基差别极小, 在以往的报道中还未有不对称合成的例子.研究工作揭示, 通过两步烯丙基化反应中碱的调节对于抑制副反应是至关重要的, 顺利实现了由相同的起始原料与相同的催化剂出发, 仅改变两种烯丙基碳酸酯的加料顺序, 就可以得到目标产物的两种对映异构体, 如Scheme 16所示.
随后, 我们课题组[31]和Zhang课题组[32]还将各自的催化体系进一步应用于Ⅰ型和Ⅱ型亚甲胺叶立德参与的不对称烯丙基化反应, 合成了一系列结构多样、光学纯度高的非天然氨基酸衍生物, 大大拓宽了该策略的适用范围, 如Scheme 17所示.
众所周知, 手性化合物的不同对映体在生理活性和光学活性等方面往往表现出较大的差别.例如, 非天然α,α-双取代-α-氨基酸由于其刚性立体结构和不会消旋化等特点[33], 常用于合成高生理活性的非天然多肽和蛋白质, 同时也被作为手性源设计合成手性催化剂和手性配体, 在多个领域有着重要的应用.但氨基酸的不同异构体往往在上述的应用中存在不同的效果.在传统的不对称催化策略中, 对于只含有一个手性中心的化合物, 通过改变催化剂的构型, 可以得到目标手性化合物的两种对映异构体[34].而对于含有多个手性中心的目标化合物, 只能获得单一对映异构体或者多种非对映异构体的混合物.改变催化剂的构型也只能获得一对对映异构体, 无法合成其他非对映异构体[35].由于不同的异构体在理化性质和生理活性上的差别, 使得它们在各个领域具有不同的应用, 立体多样性合成, 即从同一组起始原料出发合成目标化合物所有异构体, 成为不对称催化领域急需攻克的难题.目前仅有的几例立体多样性合成的报道大多通过改变反应的催化剂类型和反应条件来实现这一目标[20, 36]. 2013年, Carriera课题组[37]利用手性铱络合物和手性胺作为协同催化剂, 报道了醛类化合物的α-烯丙基化反应, 并且通过简单地使用两种手性催化剂的不同构型的组合, 在起始原料和反应条件相同的情况下合成目标产物的所有异构体, 首次实现了真正意义上的立体多样性合成.在此之后, Carriera、List、Dong、Zhang等课题组[37, 38]又相继利用类似方法实现多种α-烯丙基羰基化合物的立体多样性合成.然而, 尽管立体多样性合成目前已有所发展, 非天然氨基酸的立体多样性合成仍未有成熟的方法出现.
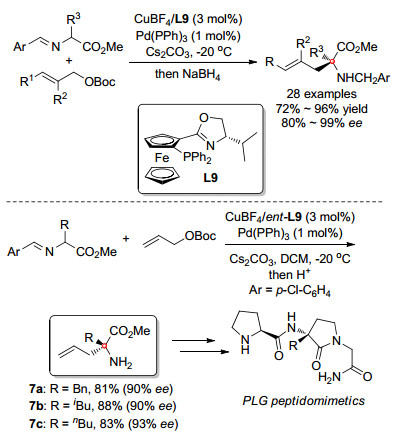
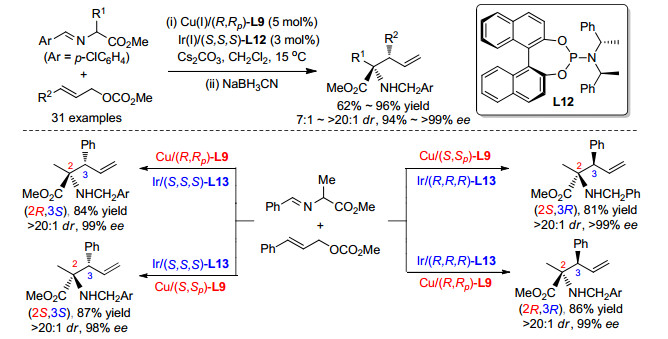
2018年, 我们课题组[39]在之前的工作的基础上, 采用金属铜络合物和金属铱络合物进行协同催化, 通过亚甲胺叶立德参与的不对称烯丙基化首次实现了非天然氨基酸的立体多样性的不对称催化合成, 如Scheme 18所示.在该策略中, 手性铱催化剂与烯丙基碳酸酯现场生成手性π-烯丙基-Ir中间体, 并控制烯丙位所产生的立体中心的构型.于此同时, 铜催化剂则与之前的报道中类似, 控制亚甲胺叶立德α-位所产生的季碳中心的构型, 两种催化剂独立工作, 互补干扰, 31P NMR实验也证明了该催化体系中的两种手性配体L9和L12不会在反应中出现配体交叉的情况.因此, 采用该催化体系, 可以通过改变两种催化剂所使用手性配体的构型, 即可改变相应手性中心的构型, 从而使用同一组反应原料在相同反应条件下合成目标氨基酸的所有异构体.实验结果也表明, 对于每一种异构体, 该方法都能得到十分优异的结果.进一步研究表明, 该体系对于甘氨酸衍生的亚甲胺叶立德同样适用, 只需将碱由碳酸铯换为碱性较弱的三乙胺即可得到高收率、高立体选择性的产物.
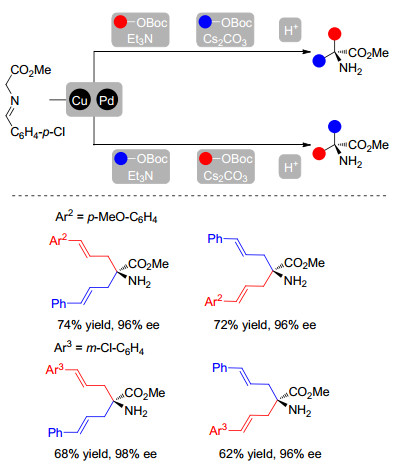
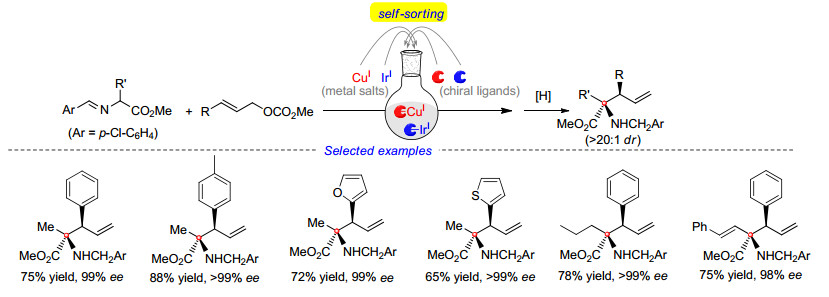
在对该反应进行研究时, 我们意识到双金属催化策略的一大弊端, 即相比于传统的单金属催化, 反应的操作较为繁琐, 两种不同催化剂需要在独立的反应容器中先行制备, 以避免可能的配体交叉.基于Hartwig[40]和Helmchen课题组[41]对反应机理的研究, 我们了解到金属铱与配体L12会通过C—H键活化得到稳定的环铱络合物, 该络合物在碱性条件下不易解离.因此在随后的实验中, 我们将两种金属及两种配体同时加入同一容器中, 金属铱盐与L12发生C—H键活化后得到的稳定络合物会使铜盐与L9留在体系中, 并进一步配位得到相应的手性铜催化剂, 从而通过“金属/配体自分类”[42]的方法同时获得两种活性催化剂(Scheme 19).实验结果也表明, 该方法十分可靠, 使用该操作方法得到的一系列产物与之前的方法相比, 结果未出现任何降低.
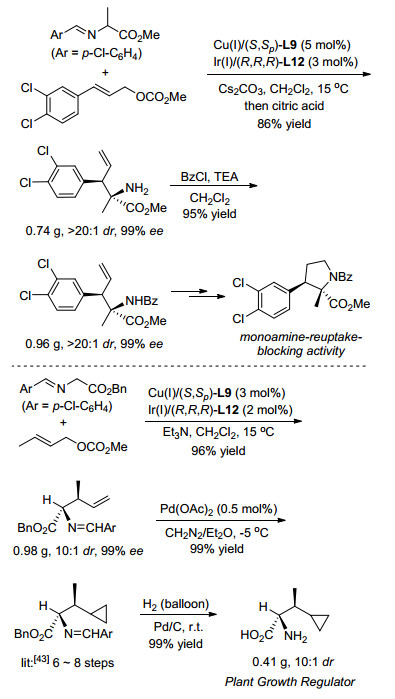
最后, 利用该方法学, 不但可以很方便地合成具有一元胺再吸收阻断活性的四氢吡咯化合物的关键中间体, 更能以3步共95%的收率合成植物生长抑制剂, 而之前的方法由手性原料出发合成该化合物至少需要6~8步[43], 如Scheme 20所示.
Zhang课题组[44]几乎在同一时间报道了使用Cu(OTf)2/L10与[Ir(COD)Cl]2/L12作为协同催化剂的反应.通过改变手性配体的构型, 该策略同样可以实现氨基酸衍生物的立体多样性合成.同时, 除去简单Ⅲ型亚甲胺叶立德, 该方法也适用于Ⅱ型以及肽类化合物衍生的亚甲胺叶立德, 都能得到非常优异的结果, 如Scheme 21所示.
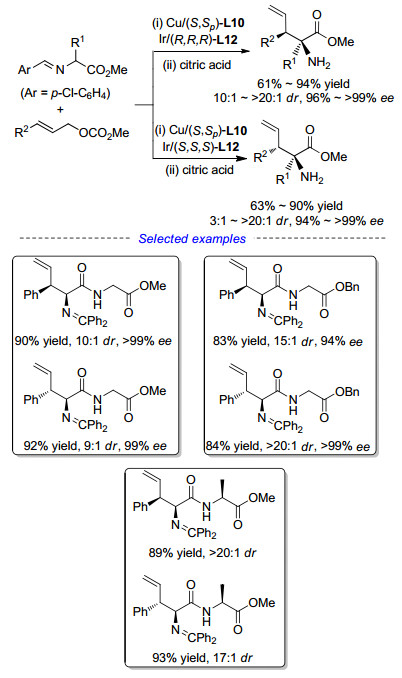
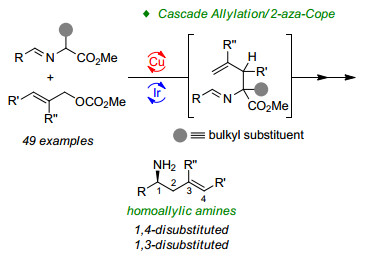
在对金属铜络合物和金属铱络合物协同催化的烯丙基化反应进行研究时, 亚甲胺叶立德发生烯丙基化反应后的产物具有特殊的烯丙基-氮杂烯丙基结构, 我们认为增大亚甲胺叶立德α-位取代基的位阻, 有可能迫使该中间体通过重排反应来释放位阻得到更稳定的高烯丙胺衍生物.随后通过对该反应醛亚胺酯的位阻进行调控研究, 成功实现了不对称烯丙基化与3, 3-sigmatropic重排串联反应, 以优异的立体选择性和收率获得在合成中应用广泛的高烯丙胺类化合物[45], 从而实现了首例亚甲胺叶立德作为起始原料合成手性胺的研究工作, 如Scheme 22所示[46].利用该策略, 使用不同的催化剂组合即可以较高的收率和优异的对映选择性得到一系列1, 4-和1,3-取代高烯丙胺.基于协同催化策略, 我们以亚甲胺叶立德和烯丙基碳酸酯作为原料, 不但能实现非天然氨基酸的立体多样性合成, 还可以合成高烯丙胺衍生物, 大大提高了铜/铱协同催化反应体系的适用范围和实用性.
近年来, 有关金属化的亚甲胺叶立德参与的不对称α-官能团化反应的新方法、新策略层出不穷, 大量结构新颖、潜在应用价值高的非天然氨基酸衍生物通过这些方法被合成.但目前仍有部分反应类型的催化体系活性较低, 或立体控制效果较差, 利用协同催化策略, 将较为成熟的金属化亚甲胺叶立德的催化体系与其他的金属催化体系或有机小分子催化体系结合, 可能是比较好的解决方式.目前关于协同催化体系的研究仅限于Cu/Pd和Cu/Ir协同催化, 该领域需要更进一步的探索.于此同时产物的应用转化研究还不够全面, 仍需更深入的研究.
吴梧桐, 生物化学, 人民卫生出版社, 北京, 2004, pp. 39~42.Wu, W. T. Biochemistry, People's Medical Publishing House, Beijing, 2004, pp. 39~42(in Chinese).
(a) Bellier, B.; McCort-Tranchenpain, I.; Ducos, B.; Danascimento, S.; Meudal, H.; Noble, F.; Garbay, C.; Roques, B. P. J. Med. Chem. 1997, 40, 3947.
(b) Dery, O.; Josien, H.; Grassi, J.; Chassaing, G.; Couraud, J. Y.; Lavielle, S. Biopolymers 1996, 39, 67.
(c) Benedetti, E.; Gavuzzo, E.; Santini, A.; Kent, D. R.; Zhu, Y.-F.; Zhu, Q.; Mahr, C.; Goodman, M. J. Pept. Sci. 1995, 1, 349.
(d) Schiller, P. W.; Weltrowska, G.; Nguyen, T. M.-D.; Lemieux, C.; Chung, N. N.; Marsden, B. J.; Wilkes, B. C. J. Med. Chem. 1991, 34, 3125.
(a) Koert, U. Nachr. Chem. Technol. Lab. 1995, 43, 347.
(b) Yano, S.; Nakanishi, Y.; Ikuina, Y.; Ando, K.; Yoshida, M.; Saitoh, Y.; Matsuda, Y.; Bando, C. J. Antibiot. 1997, 50, 992.
(c) Kende, A. S.; Liu, K.; Jos Brands, K. M. J. Am. Chem. Soc. 1995, 117, 10597.
(a) Ohfune, Y.; Shinada, T. Eur. J. Org. Chem. 2005, 5127.
(b) Vogt, H.; Brase, S. Org. Biomol. Chem. 2007, 5, 406.
(c) Bera, K.; Namboothiri, I. N. N. Asian J. Org. Chem. 2014, 3, 1234.
(a) Adrio, J.; Carretero, J. C. Chem. Commun. 2014, 50, 12434.
(b) O'Donnell, M. J. Acc. Chem. Res. 2004, 37, 506.
(c) Taggi, A. E.; Hafez, A. M.; Lectka, T. Acc. Chem. Res. 2003, 36, 10.
(d) Lygo, B.; Andrews, B. I. Acc. Chem. Res. 2004, 37, 518.
(e) Maruoka, K.; Ooi, T.; Kano, T. Chem. Commun. 2007, 1487.
(f) Kazmaier, U. Org. Chem. Front. 2016, 3, 1541.
(f) Tang, S.; Zhang, X.; Sun, J.; Niu, D.; Chruma, J. J. Chem. Rev. 2018, 118, 10393.
(a) Saito, S.; Tsubogo, T.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 5364.
(b) Tsubogo, T.; Saito, S.; Seki, K.; Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2008, 130, 13321.
(a) Li, Q.; Ding, C.-H.; Hou, X.-L.; Dai, L.-X. Org. Lett. 2010, 12, 1080.
(b) Strohmeier, M.; Leach, K.; Zajac, M. A. Angew. Chem., Int. Ed. 2011, 50, 12335.
(c) Hernández-Toribio, J.; Arrayás, R. G.; Carretero, J. C. Chem. Eur. J. 2011, 17, 6334.
(d) He, F.-S.; Jin, J.-H.; Yang. Z.-T.; Yu, X.; Fessey, J. S.; Deng, W.-P. ACS Catal. 2016, 6, 652.
(e) Konno, T.; Watanabe, S; Takahashi, T.; Tokoro, Y.; Fukuzawa, S. Org. Lett. 2013, 15, 4418.
(f) Imae, K.; Konno, T.; Ogata, K.; Fukuzawa, S. Org. Lett. 2012, 13, 4410.
(a) Koizumi, A.; Kimura, M.; Arai, Y.; Tokoro, Y.; Fukuzawa, S. J. Org. Chem. 2015, 80, 10883;
(b) Matsuda, Y.; Koizumi, A.; Haraguchi, R.; Fukuzawa, S. J. Org. Chem. 2016, 81, 7939;
(c) Koizumi, A.; Matsuda, Y.; Haraguchi, R.; Fukuzawa, S. Tetrahedron: Asymmetry 2017, 28, 428;
(d) Xue, Z.-Y.; Song, Z.-M.; Wang, C.-J. Org. Biomol. Chem. 2015, 13, 5460.
Xue, Z.-Y.; Liu, T.-L.; Lu, Z.; Huang, H.; Tao, H.-Y.; Wang, C.-J. Chem. Commun. 2010, 46, 1727. doi: 10.1039/b919625k
Xue, Z.-Y.; Li, Q.-H.; Tao, H.-Y.; Wang, C.-J. J. Am. Chem. Soc. 2011, 133, 11757. doi: 10.1021/ja2043563
Wang, M.; Wang, C.-J. Lin, Z. Organometallics 2012, 31, 7870. doi: 10.1021/om300435s
Teng, H.-L.; Luo, F.-L.; Tao, H.-T.; Wang, C.-J. Org. Lett. 2011, 13, 5600. doi: 10.1021/ol202326j
(a) Panday, S. K.; Prasad, J.; Dikshit, D. K. Tetrahedron: Asymmetry 2009, 20, 1581.
(b) Smith, M. B. Alkaloids: Chem. Biol. Perspect. 1998, 12, 229.
(c) Benoit, R.; Pascal, C.; Dominique, F.; Francois, S. Trends Heterocycl. Chem. 1991, 2, 155.
Teng, H.-L.; Huang, H.; C.-J. Chem. Eur. J. 2012, 18, 12614. doi: 10.1002/chem.201201475
(a) Mori, T.; Takahashi, K.; Kashiwabara, M.; Uemura, D. Tetrahedron Lett. 1985, 26, 1073;
(b) Omura, S.; Fujimoto, T.; Otoguro, K.; Matsuzaki, K.; Moriguchi, R.; Tanaka, H.; Sasaki, Y. J. Antibiot. 1991, 44, 113;
(c) S. Omura, K.; Matsuzaki, T.; Fujimoto, K.; Kosuge, T.; Furuya, S.; Fujita, A. J. Antibiot. 1991, 44, 117.
Kim, H. Y.; Li, J.-Y.; Kim, S.; Oh, K. J. Am. Chem. Soc. 2011, 133, 20750. doi: 10.1021/ja2100356
Bai, X.-F.; Li, L.; Xu, Z.; Zheng, Z.-J.; Xia, C.-G.; Xu, L.-W. Chem. Eur. J. 2016, 18, 10339.
Yuan, Y.; Yu, B.; Bai, X.-F.; Xu, Z.; Zheng, Z.-J.; Cui, Y.-M.; Cao, J.; Xu, L.-W. Org. Lett. 2017, 19, 4896. doi: 10.1021/acs.orglett.7b02378
Bernardi, L.; Gothelf, A. S.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 2003, 68, 2583. doi: 10.1021/jo026766u
Yan, X.-X.; Peng, Q.; Li, Q.; Zhang, K.; Yao, J.; Hou, X.-L.; Wu, Y.-D. J. Am. Chem.Soc. 2008, 130, 14362. doi: 10.1021/ja804527r
(a) Arai, T.; Mishiro, A.; Matsumura, E.; Awata, A.; Shirasugi, M. Chem. Eur. J. 2012, 18, 11219.
(b) Imae, K.; Shimizu, K.; Ogata, K.; Fukuzawa, S. J. Org. Chem. 2011, 76, 3604.
(c) Shang, D. J.; Liu, Y. L.; Zhou, X.; Liu, X. H.; Feng, X. M. Chem. Eur. J. 2009, 15, 3678.
(d) Liang, G.; Tong, M.-C.; Tao, H. Y.; Wang, C.-J. Adv. Synth. Catal. 2010, 352, 1851.
(a) Hernández-Toribio, J.; Arrayás, R. G.; Carretero, J. C. J. Am. Chem. Soc. 2008, 130, 16150.
(b) Hernández-Toribio, J.; Arrayás, R. G.; Carretero, J. C. Chem. Eur. J. 2010, 16, 1153.
(a) Paradisi, M. P.; Torrini, I.; Zecchini, G. P.; Lucente, G.; Gavuzzo, E.; Mazza, F.; Pochetti, G. Tetrahedron 1995, 51, 2379.
(b) Burgess, K.; Ho, K.-K.; Pal, B. J. Am. Chem. Soc. 1995, 117, 3808.
(c) Giannis, A.; Kolter, T. Angew. Chem., Int. Ed. 1993, 32, 1244.
(d) Balaram, P. Curr. Opin. Struct. Biol. 1992, 2, 845.
(a) You, S.-L.; Hou, X.-L.; Dai, L.-X.; Cao, B.-X.; Sun, J. Chem. Commun. 2000, 1933.
(b) Kazmaier, U.; Zumpe, F. L. Angew. Chem., Int. Ed. 1999, 38, 1468.
(c) Trost, B. M.; Ariza, X. J. Am. Chem. Soc. 1999, 121, 10727.
(d) Kazmaier, U.; Maier, S.; Zumpe, F. L. Synlett 2000, 1523.
(e) Genet, J. P.; Juge, S.; Achi, S.; Mallart, S.; Ruiz Montes, J.; Levif, G. Tetrahedron 1988, 44, 5263.
(f) Baldwin, I. C.; Williams, J. M. J.; Beckett, R. P. Tetrahedron: Asymmetry 1995, 6, 1515.
(a) Cheng, G.; Deng, Y.; Gong, L.; Mi, A.; Cui, X.; Jiang, Y.; Choi, M. C. K.; Chan, A. S. C. Tetrahedron: Asymmetry 2001, 12, 1567.
(b) Kanayama, T.; Yoshida, K.; Miyabe, H.; Takemoto, Y. Angew. Chem., Int. Ed. 2003, 42, 2054.
(c) Kanayama, T.; Yoshida, K.; Miyabe, H.; Kimachi, T.; Takemoto, Y. J. Org. Chem. 2003, 68, 6197.
(a) Malleron, J. L.; Fiaud, J. C.; Legros, J. Y. Handbook of Palladium-Catalyzed Organic Reactions, Academic Press, San Diego, 1997.
(b) Trost, B. M.; Van Vranken, D. L. Chem. Rev. 1996, 96, 395.
(c) Trost, B. M. Acc. Chem. Res. 1996, 29, 355.
(d) Williams, J. M. J. Synlett 1996, 705.
(e) Tsuji, J. Palladium Reagents and Catalysts, Wiley, Chichester, 1995.
(a) Allen, A. E.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 633.
(b) Du, Z.; Shao, Z. Chem. Soc. Rev. 2013, 42, 1337.
(c) Butt, N. A.; Zhang, W. Chem. Soc. Rev. 2015, 44, 7929.
(d) Inamdar, S. M.; Shinde, V. S.; Patil, N. T. Org. Biomol. Chem. 2015, 13, 8116.
Huo, X.; He, R.; Fu, J.; Zhang, J.; Yang, G.; Zhang, W. J. Am. Chem. Soc. 2017, 139, 9819. doi: 10.1021/jacs.7b05460
Wei, L.; Xu, S.-M.; Zhu, Q.; Che, C.; Wang, C.-J. Angew. Chem., Int. Ed. 2017, 56, 12312. doi: 10.1002/anie.201707019
Dolbeare, K.; Pontoriero, G. F.; Gupta, S. K.; Mishra, R. K.; Johnson, R. L. J. Med. Chem. 2003, 46, 727. doi: 10.1021/jm020441o
Wei, L.; Xiao, L.; Wang, C.-J. Adv. Synth. Catal. 201, 360, 4715.
(a) Huo, X.; Fu, J.; He, X.; Chen, J.; Xie, F.; Zhang, W. Chem. Commun. 2018, 54, 599.
(b) Liu, P.; Hou, X.; Li, B.; He, R.; Zhang, J.; Wang, T.; Xie, F.; Zhang, W. Org. Lett., 2018, 20, 6564.
(a) Paradisi, M. P.; Torrini, I.; Zecchini, G. P.; Lucente, G.; Gavuzzo, E.; Mazza, F.; Pochetti, G. Tetrahedron 1995, 51, 2379.
(b) Burgess, K.; Ho, K.-K.; Pal, B. J. Am. Chem. Soc. 1995, 117, 3808.
(c) Giannis, A.; Kolter, T. Angew. Chem., Int. Ed. 1993, 32, 1244.
(d) Balaram, P. Curr. Opin. Struct. Biol. 1992, 2, 845.
Jacobsen, E. N.; Pfaltz, A.; Yamamoto, H. Comprehensive Asymmetric Catalysis, Vol. Ⅰ~Ⅲ, Suppl. Ⅰ~Ⅱ, Springer, New York, 1999.
Hoveyda, A. H.; Evans, D. A.; Fu, G. C. Chem. Rev. 1993, 93, 1307. doi: 10.1021/cr00020a002
(a) Luparia, M.; Oliveira, M. T.; Audisio, D.; Frébault, F.; Goddard, R.; Maulide, N. Angew. Chem., Int. Ed. 2011, 50, 12631.
(b) McInturff, E. L.; Yamaguchi, E.; Krische, M. J. J. Am. Soc. Chem. 2012, 134, 20628.
(c) Morgen, M.; Bretzke, S.; Li, P.; Menche, D. Org. Lett. 2010, 12, 4494.
(d) Nojiri, A.; Kumagai, N.; Shibasaki, M. J. Am. Soc. Chem. 2009, 131, 3779.
(e) Tian, X.; Cassani, C.; Liu, Y.; Moran, A.; Urakawa, A.; Galzerano, P.; Arceo, E.; Melchiorre, P. J. Am. Soc. Chem. 2011, 133, 17934.
(f) Wang, B.; Wu, F.; Wang, Y.; Liu, X.; Deng, L. J. Am. Soc. Chem. 2007, 129, 768.
(a) Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065.
(b) Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020.
(c) Sandmeier, T.; Krautwald, S.; Zipfel, H. F.; Carreira, E. M. Angew. Chem., Int. Ed. 2015, 54, 14363.
(a) Nӕsborg, L.; Halskov, K. S.; Tur, F.; Mønsted, S. M. N.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2015, 54, 10193.
(b); Huo, X.; He, R.; Zhang, X.; Zhang, W. J. Am. Chem. Soc. 2016, 138, 11093.
(c) Cruz, F. A.; Dong, V. M. J. Am. Chem. Soc. 2017, 139, 1029.
(d) Jiang, X.; Beiger, J. J.; Hartwig, J. F. J. Am. Chem. Soc. 2017, 139, 87.
(f) Zheng, H.; Wang, Y.; Xu, C.; Xu, X.; Lin, L.; Liu, X.; Feng, X. Nat. Commun. 2018, 9, 1968.
Wei, L.; Zhu, Q.; Xu, S.-M.; Chang, X.; Wang, C.-J. J. Am. Chem. Soc. 2018, 140, 1508. doi: 10.1021/jacs.7b12174
Kiener, C. A.; Shu, C.; Incarvito, C.; Hartwig, J. H. J. Am. Chem. Soc. 2003, 125, 14272. doi: 10.1021/ja038319h
(a) Raskatov, J. A.; Spiess, S.; Gnamm, C.; Brö dner, K.; Rominger, F.; Helmchen, G. Chem. Eur. J. 2010, 16, 6601.
(b) Spiess, S.; Welter, C.; Franck, G.; Taquet, J.-P.; Helmchen, G. Angew. Chem., Int. Ed. 2008, 47, 7652.
(a) Wu, A.; Chakraborty, A.; Fettinger, J. C.; Flowers Ii, R. A.; Isaacs, L. Angew. Chem., Int. Ed. 2002, 41, 4028.
(b) Safont-Sempere, M. M.; Fernández, G.; Würthner, F. Chem. Rev. 2011, 111, 5784.
(c) He, Z.; Jiang, W.; Schalley, C. A. Chem. Soc. Rev. 2015, 44, 779.
(a) Morimoto, Y.; Takaishi, M.; Kinoshita, T.; Sakaguchi, K.; Shibata, K. Chem. Commun. 2002, 42.
(b) Spangenberg, T.; Schoenfelder, A.; Breit, B.; Mann, A. Org. Lett. 2007, 9, 3881.
Huo, X.; Zhang, J.; Fu, J.; He, R.; Zhang, W. J. Am. Chem. Soc. 2018, 140, 2080. doi: 10.1021/jacs.8b00187
(a) Nugent, T. C. Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH, Weinheim, 2010.
(b) Puentes, C. O.; Kouznetsov, V. J. Heterocycl. Chem. 2002, 39, 595.
(c) Przheval'skii, N. M.; Grandberg, I. I. Usp. Khim. 1987, 56, 814.
(d) Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626.
(e) Yus, M.; González-Gómez, J. C.; Foubelo, F. Chem. Rev. 2011, 111, 7774.
(f) Yus, M.; González-Gómez, J. C.; Foubelo, F. Chem. Rev. 2013, 113, 5595.
Wei, L.; Zhu, Q.; Xiao, L.; Tao, H.-Y.; Wang, C.-J. Nat. Commun. 2019, 10, 1594. doi: 10.1038/s41467-019-09563-6

图式 2 常见亚甲胺叶立德前体的合成
Scheme 2 Synthetic procedures for commonly-used azomethine ylides precursors

图式 3 Ca(Ⅱ)/Box络合物催化Ⅰ型亚甲胺叶立德的迈克尔加成反应
Scheme 3 Ca(Ⅱ)/Box complex catalyzed Michael addition of type-Ⅰ azomethine ylides

图式 4 Cu(Ⅰ)/TF-BiphamPhos催化亚甲胺叶立德对烷叉丙二磷酸酯的迈克尔加成反应及机理推测
Scheme 4 Cu(Ⅰ)/TF-BiphamPhos catalyzed Michael addition of azomethine ylides to alkylidene biphosphates and proposed mechanism

图式 5 含有季碳焦谷氨酸片段的天然产物及Cu(Ⅰ)/BINAP催化亚甲胺叶立德对MBH碳酸酯的迈克尔加成/消除串联反应
Scheme 5 Nature products contain pyroglutamates bearing a quaternary stereogenic center and Cu(Ⅰ)/BINAP catalyzed cascade Michael addition/elimination of azomethine ylides to MBH carbonates

图式 6 Cu(Ⅰ)/TF-Biphamphos催化环状亚甲胺叶立德对溴代MBH产物的迈克尔加成/消除串联反应
Scheme 6 Cu(Ⅰ)/TF-biphamphos catalyzed cascade Michael addition/elimination of azomethine ylides to MBH bromide

图式 7 Cu(Ⅰ)/L2催化亚甲胺叶立德对硝基烯烃的迈克尔加成反应
Scheme 7 Cu(Ⅰ)/L2 catalyzed Michael addition of azomethine ylides to nitroalkenes

图式 8 手性铜络合物催化亚甲胺叶立德与查尔酮的Michael加成反应及与MBH羧酸酯的Michael加成/消除反应
Scheme 8 Chiral copper complex catalyzed Michael addition of azomethine ylides to chalcones and MBH carbonates

图式 9 Cu(Ⅰ)/L5催化亚甲胺叶立德对N-Ts亚胺的Mannich反应
Scheme 9 Cu(Ⅰ)/L5 catalyzed Mannich reaction of azomethine ylides to N-Ts imines

图式 10 不同电性的配体对于亚甲胺叶立德参与的不对称Mannich反应的立体选择型影响
Scheme 10 The electron effect of ligand on the stereochemistry of azomethine ylides involved Mannich reaction

图式 11 其他催化体系催化Ⅰ型亚甲胺叶立德的不对称Mannich反应
Scheme 11 Asymmetric Mannich reaction of type-Ⅰ azomethine ylides catalyzed by other catalytic system

图式 12 Cu(Ⅰ)/L7催化Ⅰ型/Ⅲ型亚甲胺叶立德参与的不对称Mannich反应
Scheme 12 Cu(Ⅰ)/L7 catalyzed asymmetric Mannich reaction of type-Ⅰ/Ⅲ azomethine ylides

图式 13 传统方法报道的Ⅰ型亚甲胺叶立德前体参与的不对称烯丙基取代反应
Scheme 13 Previous investigation on asymmetric allylic alkylation of benzophenone Schiff base

图式 14 手性铜/手性钯协同催化亚甲胺叶立德及肽参与的不对称烯丙基取代反应
Scheme 14 Synergistic catalysis for asymmetric allylic alkylation of azomethine ylides and peptides with chiral copper complex and chiral palladium complex

图式 15 手性铜/非手性钯协同催化亚甲胺叶立德参与的不对称双烯丙基取代反应及其应用
Scheme 15 Synergistic catalysis for asymmetric allylic alkylation of azomethine ylides with chiral copper complex and achiral palladium complex

图式 16 手性铜/非手性钯协同催化亚甲胺叶立德参与的不对称双烯丙基取代反应
Scheme 16 Synergistic Cu/Pd catalysis for asymmetric double allylic alkylation of azomethine ylides

图式 17 Cu/Pd协同催化Ⅰ型/Ⅱ型亚甲胺叶立德参与的不对称烯丙基取代反应
Scheme 17 Synergistic Cu/Pd catalysis for asymmetric allylic alkylation of type Ⅰ/Ⅱ azomethine ylides

图式 18 Cu/Ir协同催化亚甲胺叶立德参与的不对称烯丙基取代反应及立体多样性合成
Scheme 18 Synergistic Cu/Ir catalysis for stereodivergent allylic alkylation azomethine ylides

图式 19 金属/配体自分类策略合成Cu/Ir双金属催化剂的催化效率考察
Scheme 19 Preparation and evaluation of dual Cu/Ir catalysts using self-sorting strategy

图式 20 Cu/Ir协同催化亚甲胺叶立德烯丙基取代反应的合成应用
Scheme 20 Synthetic application of Cu/Ir catalyzed allylic alkylation of azomethine ylides

图式 21 Cu/Ir协同催化亚甲胺叶立德及肽参与的烯丙基取代反应
Scheme 21 Synergistic Cu/Ir catalyzed allylic alkylation of azomethine ylides and peptides

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
