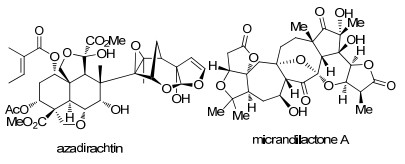
图 1.
印楝素和micrandilactone A的结构
Figure 1.
Structures of azadirachtin and micrandilactone A

降三萜(nortriterpenoid)是一类具有重要生物活性的三萜类植物次级代谢产物, 主要分为柠檬苦素类和五味子降三萜类等[1a, 1b].具有代表性的化合物有作为杀虫剂[12]在农业中已有广泛应用的柠檬苦素类化合物印楝素(azadirachtin)以及由孙汉董课题组[1c]报道的首个具有抗1型艾滋病毒(HIV-1)活性的五味子降三萜micrandilactone A (图 1).它们在生源合成途径上有着密切的关系, 主要从四环三萜tirucallane (euphane)骨架或cycloartane骨架开始, 经过氧化、降解、裂环和重排等过程, 形成了这一类化合物环系复杂、高氧化态的结构特点[1a, 1b].由于其具有重要的生物活性与复杂的结构, 受到合成化学家们的极大关注, 并展开了深入的合成研究.杨震、李昂、汤平平、Anderson和Ley小组都分别完成了多个降三萜类化合物的不对称全合成[2].
结构骨架类型与上述两类不同, 但在生源合成上有一定关联的四降三萜类天然产物solanoeclepin A(图 2), 是由Mulder等[3a]从茄科植物马铃薯根部分离得到.研究发现, 该天然产物对造成土豆大量减产的土豆包囊线虫(PCN, Globodera rostochiensis and G. pallida)的孵化过程具有极强的诱导活性(0.3 g/ha土地).其化学结构由Schenk等[3b]于1999年通过X射线单晶衍射等方法确证.
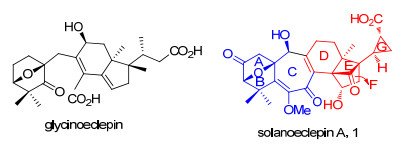
Solanoeclepin A具有非常独特的化学结构, 其主要特征为: (1)含有三至七元的各种大小不同的环, (2)具有高张力的三环[5.2.1.01, 6]癸烯核心骨架(DEF环系), (3)含有高度官能团化的ABC环系.同时, 所包含的立体化学也极具特点, 共有9个手性中心, 其中DEFG环系具有6个连续的手性碳原子(3个为全碳季碳中心).并且solanoeclepin A在pH小于2或大于7的溶液中以及温度高于35 ℃时均显示不稳定[3a].
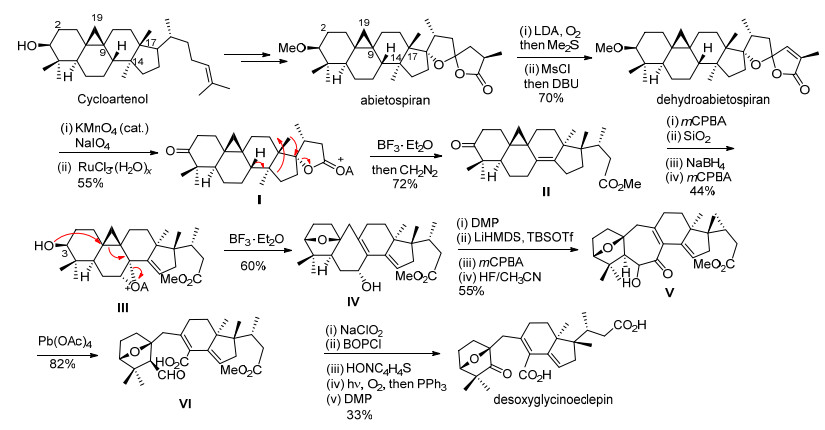
结合solanoeclepin A的骨架结构和立体化学特征, 我们期望提出其可能的主要生源合成途径.通过查阅文献发现, Masamune小组[3c]从豆科植物芸豆(kidney beans)中分离得到的天然产物glycinoeclepin, 其结构与solanoeclepin A有一定的相关性, 并且对大豆孢囊线虫(Heterodera glycines)的孵化过程具有极强的诱导活性(10-12 g/mL).该天然产物潜在的应用价值受到了化学家们的广泛关注, 先后以不同的合成策略完成了该天然产物的全合成[3d~3f].其中, 最引人注意的是Corey等[3g]为阐明glycinoeclepin的生物合成途径, 以环阿屯醇(Cycloartenol)类型的四环三萜化合物abietospiran为起始原料, 首先通过酯羰基α位氧化后脱水得到dehydroabietospiran, 再经氧化降解合成了化合物Ⅰ, Ⅰ在三氟化硼乙醚的作用下发生内酯开环形成叔碳正离子, 串联发生Wagner-Meerwein重排反应得到化合物Ⅱ.接着, Ⅱ通过4步常规的氧化、还原等反应得到环氧化合物Ⅲ, Ⅲ在三氟化硼乙醚的作用下, 发生环氧开环串联环丙烷开环反应, 所形成的碳正离子被C-3位β-羟基捕获, 形成氧桥环化合物Ⅳ, 再经氧化反应等4步转化得到α-羟基酮化合物Ⅴ, Ⅴ在四醋酸铅的作用下, 切断碳碳键得到化合物Ⅵ.最后, Ⅵ通过Barton自由基脱羧等6步反应, 实现了desoxyglycinoeclepin的仿生合成(Scheme 1).
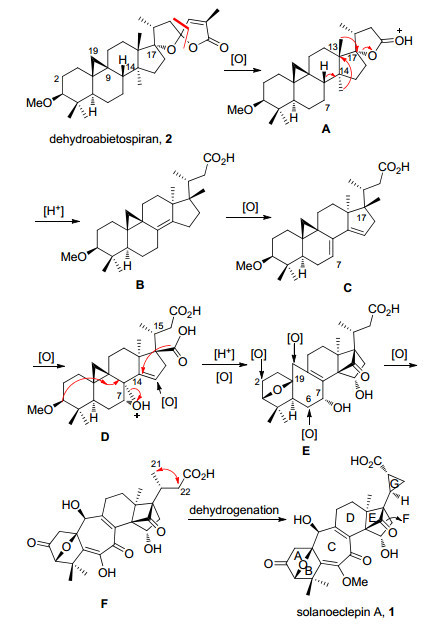
基于Corey等的仿生合成工作, 我们认为环阿屯醇(Cycloartenol)类型的四环三萜化合物abietospiran同样可能成为solanoeclepin A生物合成的起始原料(Scheme 2).其主要的生物合成途径可能包括: (1)化合物2 (dehydroabietospiran)的内酯环通过氧化降解四个碳原子形成化合物A; (2)化合物A在酸性条件下发生内酯开环反应, 形成C-17位叔碳正离子, 随后发生串联的Wagner-Meerwein重排反应, C-13位β-甲基重排至C-17位, C-14位α-甲基重排至C-13位, 最后发生β-氢消除形成双键, 得到化合物B; (3)化合物B通过氧化脱氢作用转化为共轭双烯化合物C; (4)化合物C的C-7位双键与C-17位甲基通过氧化反应分别形成环氧、羧酸化合物D; (5)首先, 化合物D中新形成的羧酸与C-14位双键发生Prins类型的反应得到高张力的环丁酮结构, 而C-15位则被氧化为α-羟基, 形成solanoeclepin A独有的三环[5.2.1.01, 6]癸烯核心骨架(DEF环系).然后, C-7位环氧在酸的作用下, 依次串联发生环氧开环反应、环丙烷开环反应及甲氧基的分子内环化反应形成氧桥环结构单元(ABC环系), 得到化合物E; (6)化合物E通过C-2, C-6, C-19和C-7位的氧化反应引入氧化态形成F, F的C-21和C-22位发生脱氢反应得到环丙烷(G环), 最终形成solanoeclepin A.
具有生物活性的天然产物的全合成和结构修饰始终是有机合成化学家们热衷的前沿研究领域, 这在新药的发展过程中起着至关重要的作用.由于solanoeclepin A独特的化学结构、极具应用价值的生物活性以及有限的来源, 多个合成小组开展了全合成研究工作.虽然Hiemstra[4]、Isobe[5]、Adachi和Nishikawa[6]、李闯创[7]、李卫东和邱发洋小组[8]分别报道了各自关于该天然产物的合成研究工作, 但到目前为止, 仅Tanino与Miyashita小组[9]以52步反应和0.18%的总收率, 于2011年完成了solanoeclepin A的不对称全合成.因此, 为能够突破该天然产物的来源限制, 实现进一步的生物活性研究, 发展新型的合成方法学和高效的合成策略显得尤为重要.本文详细总结了关于solanoeclepin A的合成研究的策略与方法.
2011年, Tanino小组和Miyashita小组[9]合作, 首次完成了solanoeclepin A的不对称全合成.其中所采用的关键合成策略(图 3)包括: (1)通过碱诱导的分子内环化反应构建张力较大的E环, 完成了三环[5.2.1.01, 6]癸烷骨架的构建(DEF环系); (2)通过3-甲氧基呋喃(双烯体)与不饱和酮(亲双烯体)的分子内Diels-Alder反应一步构建ABC环系; (3)通过Charette手性配体诱导的不对称Simmons-Smith环丙烷化反应合成G环.
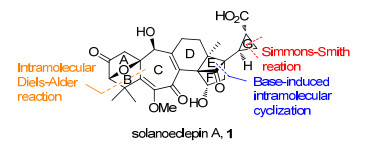
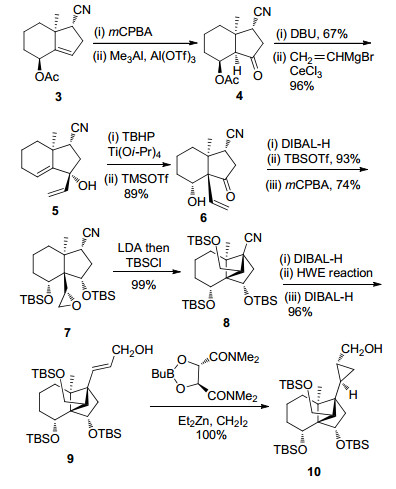
DEFG环系的合成见Scheme 3.以已知中间体3为起始原料, 通过底物控制的不对称环氧化及路易斯酸催化的Meinwald重排反应得到化合物4, 再经乙酰氧基消除并与乙烯基溴化镁发生1, 2-加成反应得到化合物5.然后, 通过羟基导向的不对称环氧化反应、TMSOTf催化的环氧开环串联pinacol重排反应得到化合物6. 6通过羰基选择性还原、叔丁基二甲基硅基(TBS)保护以及底物控制的不对称环氧化得到环化反应前体7, 再在强碱二异丙基氨基锂(LDA)的作用下发生分子内环化反应, 构建了高张力的三环[5.2.1.01, 6]癸烯核心骨架8, 其绝对立体化学通过X射线单晶衍射得到确证. 8经过氰基还原及Horner-Wadsworth-Emmons (HWE)反应延长碳链等三步反应得到化合物9, 再采用Charette配体诱导的不对称Simmons-Smith环丙烷反应, 以94:6的dr值合成了化合物10, 完成了solanoeclepin A的DEFG环系的构建.
分子内Diels-Alder反应构建ABC环系见Scheme 4.化合物10通过保护基转换和Sharpless烯合成反应等11步常规反应合成了化合物11. 11与呋喃基锂衍生物发生1, 2-加成反应得到化合物12, 再经Heck反应引入亲双烯体等3步转化以94%的总收率合成了Diels-Alder反应前体13.最终通过二甲基氯化铝催化的分子内Diels-Alder反应一步完成了ABC环系的构建.在构建solanoeclepin A所有环系的基础上, 进一步引入官能团, 完成了solanoeclepin A的不对称全合成.
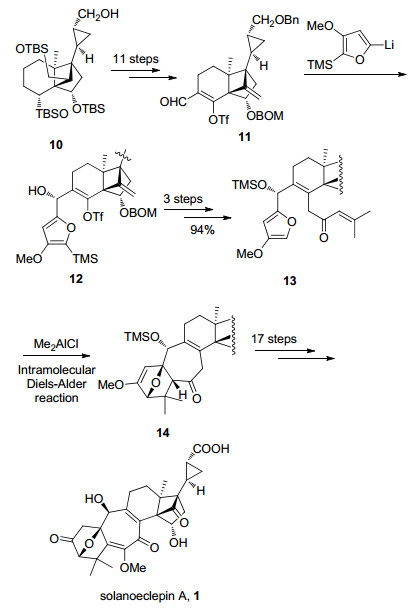
至此, Tanino以已知化合物3为起始原料, 采用线性合成策略, 以LDA介导的分子内环化和Charette手性配体诱导的不对称Simmons-Smith环丙烷化反应为关键步骤, 通过14步反应完成了DEFG环系的构建.并以此为基础, 经15步转化得到化合物13, 再采用路易斯酸催化的分子内Diels-Alder反应一步构建了ABC环系.通过进一步的官能团转化最终以52步反应(以3-甲基环己烯酮为起始原料计算)实现了solanoeclepin A的不对称全合成.
纵观Tanino的合成工作, 其中DEFG环系的合成过程中采用了较多的官能团化操作和氧化态转换, 使得合成路线冗长.但通过分子内Diels-Alder反应一步构建了ABC环系, 这为高效合成solanoeclepin A提供了重要的合成策略借鉴.作为solanoeclepin A的第一例也是目前为止唯一一例全合成, 开启了该天然产物全合成研究的篇章.
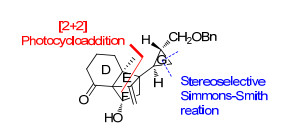
继Tanino小组完成了solanoeclepin A的全合成之后, Hiemstra小组[4k]于2016年完成了该天然产物的形式合成(Tanino小组报道的已知中间体)(图 4).其中所采用的关键合成策略包括: (1)以钯催化的丙二烯衍生物不对称双硼化反应建立整个分子的绝对立体化学; (2)运用自己发展的光催化的分子内[2+2]环加成反应[4]构建DEF三环骨架核心; (3)采用Charette手性配体诱导的不对称Simmons-Smith环丙烷化反应的方法实现G环不对称合成.
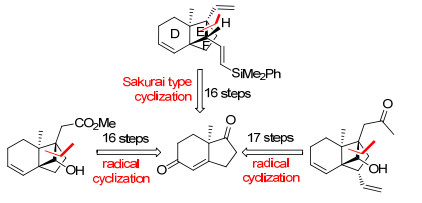
Solanoeclepin A的合成路线见Scheme 5. Hiemstra小组以丙二烯化合物15为起始原料, 通过钯与配体17催化的不对称双硼化反应合成了烯丙基硼衍生物16.16与醛19发生1, 2-加成反应后, 再经羟基保护, 以91%的ee值得到环加成反应前体20.化合物20在365 nm光照射的条件下发生分子内的[2+2]环加成反应, 快速地成功构建了DEF环系. Hiemstra小组从已知化合物15和18出发, 仅6步反应即完成了高张力的DEF环系的合成, 是目前已报道的合成路线中最为简洁高效的.另外, 作者还采用流动化学的合成方法使得光催化的[2+2]环加成反应规模达到20 g.
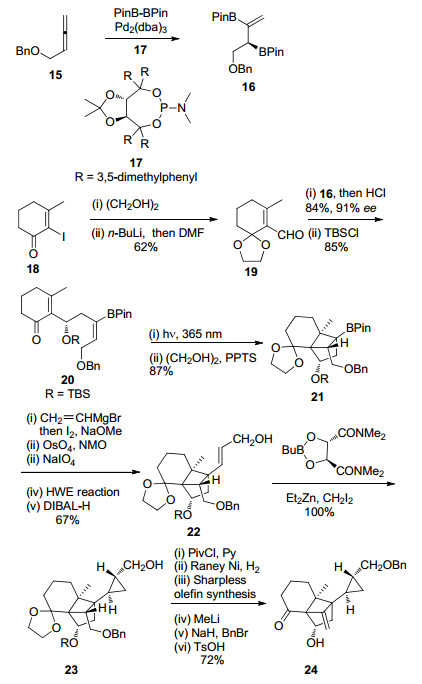
化合物21通过乙烯基置换BPin、切断末端双键、HWE反应延长碳链等5步反应得到化合物22. Hiemstra小组采用与Tanino小组同样的G环构建方法, 即Charette配体诱导的不对称Simmons-Smith环丙烷化反应, 以99%的ee值高产率地得到了化合物23.再经保护基变换、Sharpless烯合成反应等6步反应, 合成了Tanino小组报道的已知中间体24.
Hiemstra小组采用结构简单易得的化合物15和18为起始原料, 仅以5步反应不对称合成了关键反应前体化合物20, 减少了保护基的使用和官能团转换, 再通过光催化的环加成反应实现了高张力骨架核心的大量合成.以此为基础, 再经13步反应完成了solanoeclepin A的形式合成(DEFG环系), 相比于Tannio小组报道的化合物24的合成路线精简了8步反应.此合成路线的优势在于运用光催化的分子内[2+2]环加成反应高效地构建了DEF环系以及采用Charette配体诱导的不对称Simmons-Smith环丙烷化反应构建了G环.
总结目前已报道的Solanoeclepin A的ABC环系的关键合成策略和方法, 主要集中于手性辅基诱导的分子内不对称Diels-Alder反应和I(collidine)2PF6介导的分子内碘醚化反应.
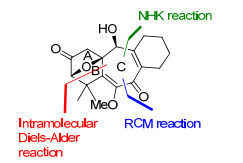
Hiemstra小组[4]一直致力于solanoeclepin A的全合成研究, 先后报道了大量的研究工作.关于该天然产物最早的研究报道也是由其完成的. 2000年, Hiemstra小组[4a]首次报道了solanoeclepin A的ABC环系的合成研究工作, 其中所采用的关键合成策略包括: (1)通过手性辅基诱导的分子内不对称Diels-Alder反应构建AB桥环; (2)采用镍催化的Nozaki-Hiyama-Kishi (NHK)反应与RCM反应合成C环(图 5).
Diels-Alder反应构建AB环系见Scheme 6.以糠醛和3-甲基巴豆酰氯为起始原料, R-苯甘氨醇为手性辅基, 通过还原胺化和酰化等4步反应得到化合物25, 在正丁基氯化镁催化作用下发生分子内不对称Diels-Alder反应, 以7:1的dr值合成了氧桥环化合物26.化合物26 通过手性辅基脱除、酰胺重氮化后碱开环等7步反应转化为化合物27, 然后经过硼氢化条件筛选, 实现了区域选择性的硼氢化氧化反应, 得到化合物28, 再通过羟基保护、Wittig反应延长碳链、羟基氧化等7步反应合成了具备所需官能团的AB环系, 为继续构建七元C环做好准备.
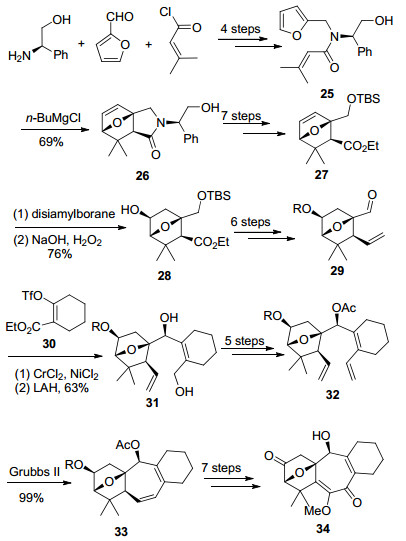
化合物29与已知化合物30在镍的催化作用下发生Nozaki-Hiyama-Kishi (NHK)反应, 以2.3:1的dr值得到两个片段偶联的化合物31.通过保护基变换、Wittig反应等5步反应得到双末端烯烃化合物32, 然后在Grubbs Ⅱ催化剂的作用下发生RCM反应成功地构建了C环.化合物33再通过7步反应引入C环的官能团, 完成了ABC环系的合成.
Hiemstra小组以糠醛和3-甲基巴豆酰氯为起始原料, R-苯甘氨醇为手性辅基, 最终以32步反应完成了solanoeclepin A的ABC环系的合成, 合成路线中较多的保护基使用以及氧化态转换使得合成路线冗长.但分子内不对称Diels-Alder反应构建AB桥环、中间体29及30的偶联反应及RCM合成七元环等合成策略的成功运用具有重要意义.其中, Tanino与Miyashita小组正是运用了呋喃衍生物的分子内Diels-Alder反应一步完成了ABC环系的合成, 在此基础上完成了solanoeclepin A的全合成.
自2005年以来, Isobe小组[5]也一直致力于solano- eclepin A的全合成研究, 并陆续发展了多种solanoeclepin A片段的合成方法.
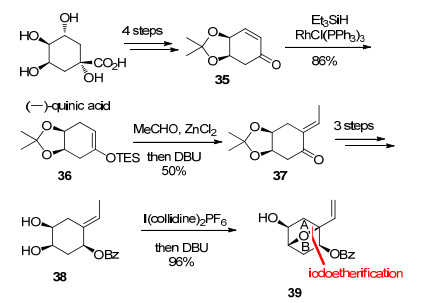
2005年, Isobe小组[5a]首次以天然产物(-)-quinic acid为起始原料, 采用I(collidine)2PF6介导的分子内碘醚化反应为关键步骤构建了AB环系(Scheme 7).
以(-)-quinic acid为起始原料, 按已知方法合成了化合物35, 然后通过Wilkinson催化剂催化的还原反应得到烯醇硅醚化合物36.化合物36与乙醛发生Mukaiyama aldol反应, 再消除羟基得到不饱和羰基化合物37, 再经底物手性控制的Luche还原反应、羟基保护和脱除缩酮保护三步反应得到碘醚化反应前体化合物38.化合物38在I(collidine)2PF6的作用下发生分子内醚化反应, 以DBU消除碘取代基得到化合物39, 成功地构建了AB桥环.
Isobe小组[5d]于2014年更加系统地研究报道了solanoeclepin A的ABC环系的合成.其中所采用的关键合成策略包括: (1)通过手性辅基诱导的分子内不对称Diels-Alder反应构建AB桥环, 这一策略与Hiemstra小组报道的合成方法有一定程度的相似性, 但更为简洁高效; (2)由炔-Co2(CO)6配合物辅助, 利用路易斯酸催化的Prins-ene反应实现C环的构建(Scheme 8).
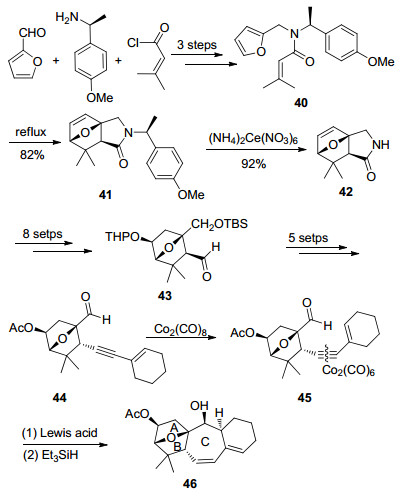
以糠醛和3-甲基巴豆酰氯为起始原料, α-甲基苄胺为手性辅基, 经三步反应合成了化合物40, 其在加热的条件下发生分子内Diels-Alder反应, 以82%的产率转化为化合物41, 快速地完成了AB环系的构建.化合物41通过硝酸铈铵脱除手性辅基等9步反应得到化合物43, 再经Bestman-Ohira反应及保护基转换等5步反应得到炔基化合物44. 44与Co2(CO)8反应得到炔-Co2(CO)6配合物45, 再经路易斯酸催化的分子内Prins-ene反应和还原脱钴反应得到化合物46, 最终完成ABC环系的合成.
Isobe小组以糠醛和3-甲基巴豆酰氯为起始原料, 经22步反应完成了solanoeclepin A的ABC环系的合成.其主要优势在于采用了更易脱除的手性辅基诱导的不对称Diels-Alder反应以及发展了以炔-羰基钴配合物辅助, 路易斯酸催化的Prins-ene反应构建C环的合成方法.
针对化学结构复杂的solanoeclepin A, Adachi和Nishikawa小组[6b]期望采用汇聚式合成策略完成该天然产物的全合成, 并于2015年报道了solanoeclepin A的ABC环系的合成工作.主要的合成策略包括: (1)采用与Isobe小组相同的合成方法, 即I(collidine)2PF6介导的分子内碘醚化反应构建AB环系; (2)通过RCM反应构建C环(图 6).
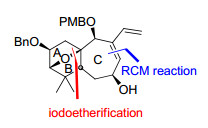
ABC环系的合成路线见Scheme 9.以D-泛酰内酯为起始原料, 根据文献已报道的方法合成了醛基化合物47. 47与炔丙基溴在锌粉的作用下, 通过底物手性诱导, 发生非对映选择性(dr值>20:1)的1, 2-加成反应得到了化合物48, 再经过保护基变换、HWE反应延长碳链等5步反应转化为化合物49. 49通过锡介导的6-exo-trig自由基环化和对甲苯磺酸脱锡化反应, 以7:1的dr值得到环化产物50, 再经I(collidine)2PF6介导的分子内碘醚化反应, 构建了AB桥环.化合物51通过常规的7步反应转化为RCM反应前体52, 再在Grubbs Ⅱ催化剂的作用下, 发生关环反应分别得到了差向异构体53与54, 完成了solanoeclepin A的ABC环系的合成.
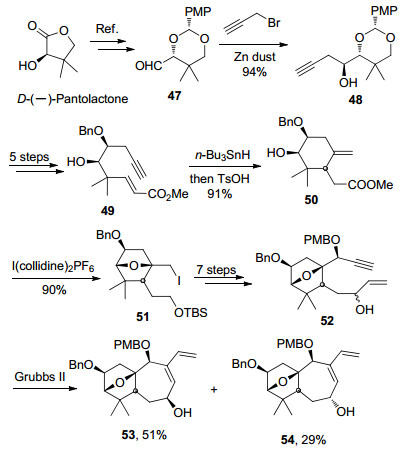
李闯创小组[7]于2017年报道了solanoeclepin A的ABC环系的合成.其中所采用的关键合成策略包括: (1)以ent-Hajos-Parrish ketone为起始原料, 通过底物手性控制来实现不对称合成; (2)通过呋喃(双烯体)与不饱和酮(亲双烯体)的分子内Diels-Alder反应一步构建了ABC环系(图 7).
Diels-Alder反应构建ABC环系见Scheme 10.以ent-Hajos-Parrish ketone为起始原料, 通过底物手性控制的化学选择性还原、TBS保护羟基和烷基化三步反应转化得到化合物55, 再经乙烯基溴化镁与不饱和酮羰基的1, 2-加成反应, 以及进一步O3切断末端双键得到醛基化合物56. 56依次与2-呋喃基锂和环丙基烯基锂发生加成反应, 合成了分子内Diels-Alder反应前体化合物58.然后, 在加热条件下发生分子内Diels-Alder反应, 再经催化氢化得到了化合物59, 完成了形式上的ABC环系的构建.
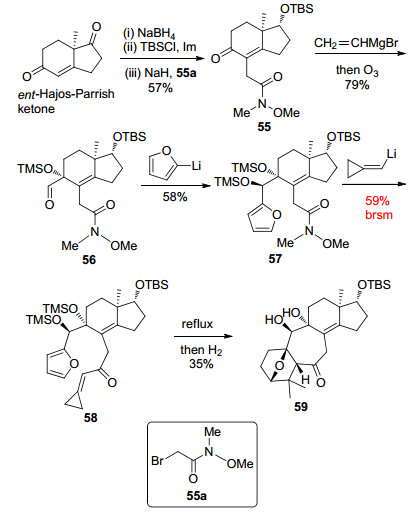
李闯创小组以ent-Hajos-Parrish ketone为起始原料, 经底物手性控制的11步反应完成了solanoeclepin A的ABC环系的不对称合成.合成路线中采用了分子内Diels-Alder反应的策略, 快速地构建了ABC环系, 这与Tanino小组的合成策略有异曲同工之处.同时, 也说明Diels-Alder反应在环系复杂的天然产物全合成中具有高效性.
总结目前已报道的solanoeclepin A的DEF环系的关键合成策略和方法, 主要包括炔-羰基钴配合物辅助的Hosomi-Sakurai类型的环化反应、SmI2介导的分子内自由基环化反应以及分子内aldol反应.
Isobe小组[5]在成功地发展了solanoeclepin A的ABC环系构建方法的同时, 也陆续报道了该天然产物DEF核心骨架的合成研究.纵观其已报道的合成研究工作, 所采用的主要合成策略是: (1)利用炔-羰基钴配合物辅助的Hosomi-Sakurai类型的环化反应[5c]; (2) SmI2介导的4-exo-trig分子内自由基环化反应[5e, g]构建E环(图 8).
炔-羰基钴配合物辅助的Hosomi-Sakurai类型的环化反应见Scheme 11.作者以ent-Hajos-Parrish ketone为起始原料, 首先化学选择性地保护不饱和酮羰基, 然后采用HWE反应延长碳链, 脱除缩酮保护得到化合物60, 再以过量的DIBAL-H同时还原酯与不饱和酮羰基, 选择性保护伯羟基, 进一步利用Mitsunobu反应翻转烯丙位羟基构型, 再与炔丙基溴发生醚化反应得到化合物61.接着, 通过三步常规反应得到烯丙基硅化合物62, 62在正丁基锂的作用下发生[2, 3]-Wittig重排反应合成了化合物63, 63脱除TMS后再与Co2(CO)8生成了稳定的配合物64.最终, 化合物64在三氟甲磺酸三甲基硅酯(TMSOTf)的作用下, 生成炔丙位碳正离子中间体64a, 64a进一步发生Hosomi-Sakurai类型环化反应, 再还原脱除Co2(CO)6, 以此构建了高张力的三环[5.2.1.01, 6]癸烯核心骨架(DEF环系).
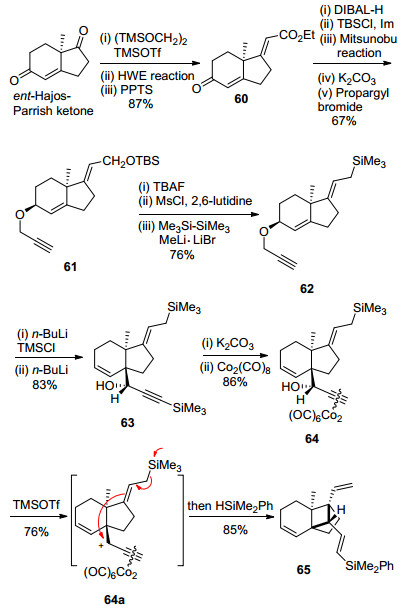
SmI2介导的4-exo-trig分子内自由基环化反应见Scheme 12.同样地, 作者以ent-Hajos-Parrish ketone为起始原料, 首先通过底物手性控制的5步常规反应合成了炔丙基醚化合物66, 66在正丁基锂的作用下, 发生[2, 3]-Wittig重排反应, 再用乙酰基保护炔丙醇得到化合物67.接着, 67通过常规的两步反应转化为68, 再经Hg(Ⅱ)介导的炔基羟汞化串联分子内aldol反应和乙酰基脱除反应, 得到α-羟基酮化合物69, 以Pb(OAc)4氧化切断碳碳键, 再消除羟基得到了环化前体70 (E:Z=3:1).最后, 70在SmI2的作用下生成羰基自由基, 再发生4-exo-trig分子内环化反应, 成功地实现了高张力的三环[5.2.1.01, 6]癸烯核心骨架的合成.
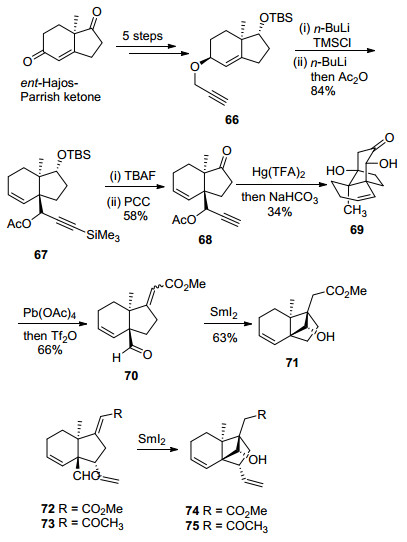
基于上述合成策略的顺利实施, Isobe小组[5g]又合成了不同官能团化的化合物74[5e]和75.纵观Isobe小组发展的DEF环系的合成路线, 由于过多的官能团操作和部分反应步骤的产率较低, 导致整个合成过程的效率下降.但是, 其极具创新性地采用炔-羰基钴配合物辅助的Hosomi-Sakurai类型的环化反应或SmI2介导的4-exo-trig分子内环化反应, 构建高张力的三环[5.2.1.01, 6]癸烯核心骨架的方法, 为solanoeclepin A的全合成提供了重要的方法学借鉴.
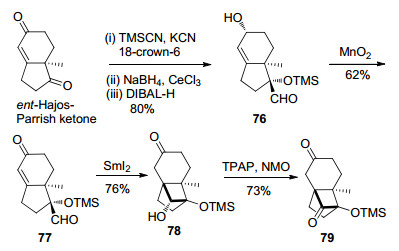
Adachi和Nishikawa小组[6a]采用了与Isobe小组构建DEF环系相似的合成方法, 即采用了SmI2介导的4-exo-trig分子内自由基环化反应.合成路线见Scheme 13.以ent-Hajos-Parrish ketone为起始原料, 通过底物手性控制的3步已知反应[6b]转化为化合物76, 再经二氧化锰氧化得到不饱和酮77.化合物77在SmI2的作用下发生4-exo-trig分子内自由基环化反应, 再经Ley氧化反应, 成功构建了高张力的三环[5.2.1.01, 6]癸烯核心骨架79.
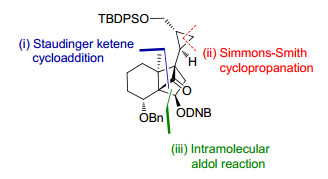
基于前人关于solanoeclepin A的合成研究工作, 李卫东小组与邱发洋小组[8]合作于2019年报道了solanoe-clepin A的DEFG环系的合成(图 9).其中所采用的关键合成策略包括: (1)底物手性诱导的不对称Staudinger烯酮环加成反应; (2)分子内aldol缩合反应; (3) Charette手性配体诱导的不对称Simmons-Smith环丙烷化反应.
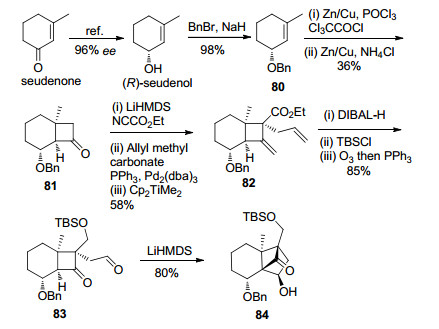
作者以商品可得的seudenone为起始原料, 通过文献报道的不对称还原的方法制备了ee值为96%的(R)-seudenol, 并以此为基础控制了整个DEF环系的立体化学(Scheme 14).化合物80与原位产生的二氯烯酮发生Staudinger烯酮环加成反应, 再还原脱除氯取代基得到化合物81. 81通过强碱拔氢形成的动力学烯醇锂盐与Mander’s试剂反应, 得到α-羰基酯化合物, 再经底物手性控制的Tsuji-Trost烯丙基化反应引入季碳中心, 接着,在二甲基二茂钛的作用下将环丁酮羰基转化为末端双键. 82通过酯基还原、羟基TBS保护、O3切断两个末端双键得到了环化前体83, 83在位阻强碱LiHMDS的作用下, 发生分子内aldol反应, 顺利构建了DEF环系, 化合物84的绝对立体化学通过衍生物的X射线单晶衍射进行了确证.
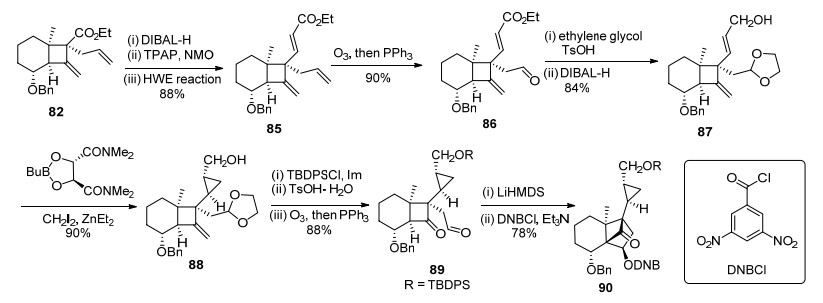
在顺利地通过分子内aldol反应构建了DEF环系的基础上, 作者进一步完成了G环的合成(Scheme 15).化合物82通过酯基还原、氧化和HEW反应延长碳链三步反应, 得到反式不饱和酯化合物85. 85通过化学选择性的O3化反应和醛基保护反应, 再还原不饱和酯得到烯丙醇化合物87.最后采用Charette手性配体诱导的不对称Simmons-Smith环丙烷化反应, 化学选择性地实现了G环的不对称合成, 得到化合物88.接着, 叔丁基二苯基硅基(TBDPS)保护伯羟基、缩醛水解、切断末端双键得到aldol反应前体89.然后, 在LiHMDS的作用下发生分子内环化, 以3, 5-二硝基苯甲酰氯(DNBCl)保护新生成的羟基, 防止发生retro-aldol反应.
降三萜类天然产物自发现至今已有非常悠久的历史, 主要包括柠檬苦素类、五味子属降三萜、solanoeclepin A及glycinoeclepin.这一类天然产物具有骨架结构复杂、高度氧化和高度官能团化等特点, 并且具有重要的生物活性, 例如抗肿瘤[10]、抗HIV-1[10a, 11]、杀虫[12]等, 无论在新药研究还是农业应用中都具有潜在价值.由于这一类活性天然产物的来源限制, 阻碍了进一步的活性以及构效关系研究, 因此, 受到了合成化学家们的广泛关注, 并且展开了深入的全合成研究.
其中, solanoeclepin A化学结构中同时包含了三至七元的各种环系, 这在已知的萜类化合物中是非常独特的.其复杂的分子结构、重要的生物活性以及有限来源引起了全球多个合成小组关注.自2000年后, 随着合成方法学的发展, Hiemstra[4]、Isobe[5]、Adachi[6]、李闯创[7]、李卫东与邱发洋小组[8]陆续报道了关于该化合物片段的合成研究, Tanino与Miyashita小组[9]采用线性合成策略于2011年最终以52步反应完成了不对称全合成(Scheme 16).
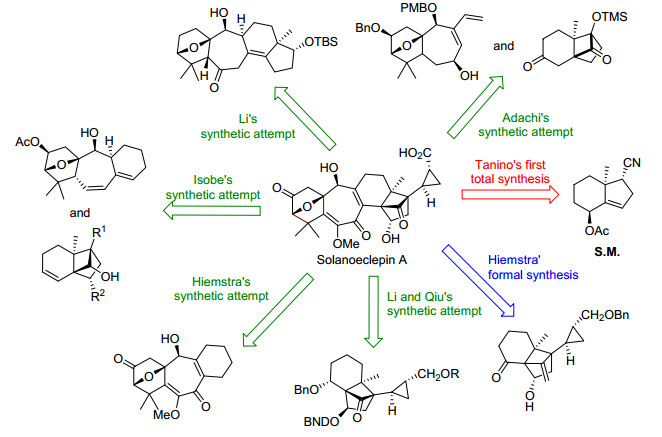
纵观目前已报道的相关全合成研究工作, 虽然对确证和深入理解这一类天然产物的化学结构与性质有着非常重要的意义, 但由于合成方法学的局限性, 导致步骤冗长, 效率较低, 并未解决活性天然产物来源限制这一重要问题.因此, 结合目前已报道的合成研究工作及Corey教授早期有关glycinoeclepin仿生合成途径的探索和启示, 将来的全合成研究需进一步通过solanoeclepin A及其它具有重要生物活性的降三萜类化合物的仿生策略的设计和探索, 发展简洁、高效的目标合成方法及类似物分子的合成, 用于生物活性的系统性研究.
(a) Tan, Q. G.; Luo, X. D. Chem. Rev. 20011, 111, 7437.
(b) Xiao, W. L.; Li, R. T.; Huang, S. X.; Pu, J. X.; Sun, H. D. Nat. Prod. Rep. 2008, 25, 871.
(c) Li, R. T.; Zhao, Q. S.; Li, S. H.; Han, Q. B.; Sun, H. D.; Lu, Y.; Zhang, L. L.; Zheng, Q. T. Org. Lett. 2003, 5, 1023.
(a) Xiao, Q.; Ren, W. W.; Chen, Z. X.; Sun, T. W.; Li, Y.; Ye, Q. D.; Gong, J. X.; Meng, F. K.; You, L.; Liu, Y. F.; Zhao, M. Z.; Xu, L. M.; Shan, Z. H.; Tang, Y. F.; Chen, J. H.; Yang, Z. Angew. Chem., Int. Ed. 2011, 50, 7373.
(b) You, L.; Liang, X. T.; Xu, L. M.; Wang, Y. F.; Zhang, J. J.; Su, Q.; Li, Y. H.; Zhang, B.; Yang, S. L.; Chen, J. H.; Yang Z. J. Am. Chem. Soc. 2015, 137, 10120.
(c) Li, J.; Yang, P.; Yao, M.; Deng, J.; Li, A. J. Am. Chem. Soc. 2014, 136, 16477.
(d) Kleinnijenhuis, R. A.; Timmer, B. J. J.; Lutteke, G.; Smits, J. M. M.; de Gelder, R.; van Maarseveen, J. H.; Hiemstra, H. Chem.-Eur. J. 2008, 14, 10683.
(e) Wang, L.; Wang, H. T.; Li, Y. H.; Tang, P. P. Angew. Chem., Int. Ed. 2015, 54, 5732.
(f) Goh, S. S.; Chaubet, G.; Gockel, B.; Cordonnier, M. A.; Baars, H.; Phillips, A. W.; Anderson, E. A. Angew. Chem., Int. Ed. 2015, 54, 12618.
(a) Mulder, J. G.; Diepenhorst, P.; Brügge mann-Rotgans, I. E. M. WO 93020083, 1993[Chem. Abstr. 1993, 118, 185844z].
(b) Schenk, H.; Driessen, R. A. J.; de Gelder, R.; Goubitz, K.; Nieboer, H.; Brüggemann Rotgans, I. E. M.; Diepenhorst, P. Croat. Chem. Acta 1999, 72, 593.
(c) Fukuzawa, A.; Furusaki, A.; Ikura, M.; Masamune, T. J. Chem. Soc., Chem. Commun. 1985, 222.
(d) Murai, A.; Tanimoto, N.; Sakamoto, N.; Masamune, T. J. Am. Chem. Soc. 1988, 10, 1985.
(e) Mori, K.; Watanabe, H. Pure Appl. Chem. 1989, 61, 543.
(f) Corey, E. J.; Houpis, I. N. J. Am. Chem. Soc. 1990, 112, 8997.
(g) Corey, E. J.; Hong, B. C. J. Am. Chem. Soc. 1994, 116, 3149.
(a) Blaauw, R. H.; Brière, J.-F.; de Jong, R.; Benningshof, J. C. J.; van Ginkel, A. E.; Rutjes, F. P. J. T.; Fraanje, J.; Goubitz, K.; Schenk, H.; Hiemstra, H. Chem. Commun. 2000, 1463.
(b) Benningshof, J. C. J.; Blaauw, R. H.; van Ginkel, A. E. J. H.; Rutjes, F. P. J. T.; Fraanje, J.; Goubitz, K.; Schenk, H.; Hiemstra, H. Chem. Commun. 2000, 1465.
(c) Blaauw, R. H.; Brière, J. F.; de Jong, R.; Benningshof, J. C. J.; van Ginkel, A. E.; Fraanje, J.; Goubitz, K.; Schenk, H.; Rutjes, F. P. J. T.; Hiemstra, H. J. Org. Chem. 2001, 66, 233.
(d) Brière, J.-F.; Blaauw, R. H.; Benningshof, J. C. J.; van Ginkel, A. E.; van Maarseveen, J. H.; Hiemstra, H. Eur. J. Org. Chem. 2001, 2371.
(e) Blaauw, R. H.; Benningshof, J. C. J.; van Ginkel, A. E.; van Maarseveen, J. H.; Hiemstra, H. J. Chem. Soc., Perkin Trans. 1 2001, 2250.
(f) Benningshof, J. C. J.; Blaauw, R. H.; van Ginkel, A. E.; van Maarseveen, J. H.; Rutjes, F. P. J. T.; Hiemstra, H. J. Chem. Soc., Perkin Trans. 1 2002, 1693.
(g) Benningshof, J. C. J.; IJsselstijn, M.; Wallner, S. R.; Koster, A. L.; Blaauw, R. H.; van Ginkel, A. E.; Brière, J.-F.; van Maarseveen, J. H.; Rutjes, F. P. J. T.; Hiemstra, H. J. Chem. Soc., Perkin Trans. 1, 2002, 1701.
(h) Hue, B. T. B.; Dijkink, J.; Kuiper, S.; Larson, K. K.; Guziec, Jr. F. S.; Goubitz, K.; Fraanje, J.; Schenk, H.; van Maarseveen, J. H.; Hiemstra, H. Org. Biomol. Chem. 2003, 1, 4364.
(i) Hue, B. T. B.; Dijkink, J.; Kuiper, S.; van Schaik, S.; van Maarseveen, J. H.; Hiemstra, H. Eur. J. Org. Chem. 2006, 127.
(j) Lutteke, G.; Kleinnijenhuis, R. A.; Jacobs, I.; Wrigstedt, P. J.; Correia, A. C. A.; Nieuwenhuizen, R.; Hue, B. T. B.; Goubitz, K.; Peschar, R.; van Maarseveen, J. H.; Hiemstra, H. Eur. J. Org. Chem. 2011, 3146.
(k) Kleinnijenhuis, R. A.; Timmer, B. J. J.; Lutteke, G.; Smits, J. M. M.; de Gelder, R.; van Maarseveen, J. H.; Hiemstra, H. Chem.-Eur. J. 2016, 22, 1266.
(a) Tojo, S.; Isobe, M. Synthesis 2005, 1237.
(b) Adachi, M.; Yamauchi, E.; Komada, T.; Isobe, M. Synlett 2009, 1157.
(c) Tsao, K. W.; Cheng, C. Y.; Isobe, M. Org. Lett. 2012, 14, 5274.
(d) Chuang, H. Y.; Isobe, M. Org. Lett. 2014, 16, 4166.
(e) Lin, Y. T.; Lin, F. Y.; Isobe, M. Org. Lett. 2014, 16, 5948.
(f) Chuang H. Y.; Isobe, M. Tetrahedron 2017, 73, 2705.
(g) Chuang, H. Y.; Isobe, M. J. Org. Chem. 2017, 82, 2045.
(a) Komada, T.; Adachi, M.; Nishikawa, T. Chem. Lett. 2012, 41, 287.
(b) Adachi, M.; Komada, T.; Nishikawa, T. J. Org. Chem. 2011, 76, 6942.
(c) Adachi, M.; Torii, M.; Nishikawa, T. Synlett 2015, 26, 965.
Liu, G.; Han, J. C.; Li, C. C. Tetrahedron 2017, 73, 3629. doi: 10.1016/j.tet.2017.03.090
Sun, M.; Li, W. D.; Qiu, F. Y. Org. Lett. 2019, 21, 644. doi: 10.1021/acs.orglett.8b03742
Tanino, K.; Takahashi, M.; Tomata, Y.; Tokura, H.; Uehara, T.; Miyashita, M. Nat. Chem. 2011, 3, 484. doi: 10.1038/nchem.1044
(a) Chang, J. B.; Reiner, J.; Xie, J. X. Chem. Rev. 2005, 105, 4581.
(b) Kuo, Y. H.; Kuo, L. Y.; Chen, C. F. J. Org. Chem. 1997, 62, 3242.
(a) Chen, D. F.; Zhang, S. X.; Wang, H. K.; Zhang, S. Y.; Sun, Q. Z.; Cosentino, M.; Lee, K. H. J. Nat. Prod. 1999, 62, 94.
(b) Sun, H. D.; Qiu, S. X.; Lin, L. Z.; Wang, Z. Y.; Lin, Z. W.; Pengsuparp, T.; Pezzuto, J. M.; Fong, H. H. S.; Cordell, C. A.; Farnsworth, N. R. J. Nat. Prod. 1996, 59, 525.
(a) Mordue, A. J.; Blackwell, A. J. Insect Physiol. 1993, 39, 903.
(b) Simmonds, M. S. J.; Blaney, W. M. Entomol. Exp. Appl. 1996, 80, 23.
(c) Karnavar, G. K. Proc.-Indian Acad. Sci., Anim. Sci. 1987, 96, 341.

图 1 印楝素和micrandilactone A的结构
Figure 1 Structures of azadirachtin and micrandilactone A

图 2 Glycinoeclepin和solanoeclepin A的结构
Figure 2 Structures of glycinoeclepin and solanoeclepin A

图式 2 Solanoeclepin A的主要生物合成途径
Scheme 2 Proposed major biosynthesis routes of solanoeclepin A

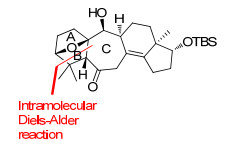
图 3 Tanino与Miyashita小组的关键合成策略
Figure 3 Tanino and Miyashitakey's key synthetic strategy

图式 4 分子内Diels-Alder反应构建ABC环系
Scheme 4 Construction of ABC ring system via intramolecular Diels-Alder reaction

图式 5 Hiemstra小组关于solanoeclepin A的形式合成
Scheme 5 Hiemstra's formal synthesis of solanoeclepin A

图式 6 Hiemstra小组关于solanoeclepin A的ABC系的合成
Scheme 6 Hiemstra's synthesis of the ABC ring system of solanoeclepin A

图式 8 Isobe小组关于solanoeclepin A的ABC环系的合成
Scheme 8 Isobe's synthesis of the ABC ring system of solanoeclepin A

图式 9 Adachi和Nishikawa小组关于solanoeclepin A的ABC环系的合成
Scheme 9 Adachi and Nishikawa's synthesis of the ABC ring system of solanoeclepin A

图式 10 Diels-Alder反应构建ABC环系
Scheme 10 Construction of ABC ring system via Diels-Alder reaction

图式 11 钴介导的Hosomi-Sakurai类型的环化反应
Scheme 11 Cobalt-mediated Hosomi-Sakurai type cyclization

图式 12 SmI2介导的4-exo-trig分子内环化反应
Scheme 12 SmI2-mediated 4-exo-trig intramolecular cyclization

图式 13 Adachi和Nishikawa小组关于DEF环系的合成
Scheme 13 Adachi and Nishikawa's synthesis of DEF ringsystem

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:











